

Abstract
We previously showed that inositol hexakisphosphate kinase 2 (IHPK2) functions as a growth-suppressive and apoptosis-enhancing kinase during cell stress. Overexpression of IHPK2 sensitized ovarian carcinoma cell lines to the growth-suppressive and apoptotic effects of interferon β (IFN-β), IFN-α2, and γ-irradiation. Expression of a kinase-dead mutant abrogated 50% of the apoptosis induced by IFN-β. Because the kinase-dead mutant retained significant response to cell stressors, we hypothesized that a portion of the death-promoting function of IHPK2 was independent of its kinase activity. We now demonstrate that IHPK2 binds to tumor necrosis factor (TNF) receptor-associated factor (TRAF) 2 and interferes with phosphorylation of transforming growth factor β-activated kinase 1 (TAK1), thereby inhibiting NF-κB signaling. IHPK2 contains two sites required for TRAF2 binding, Ser-347 and Ser-359. Compared with wild type IHPK2-transfected cells, cells expressing S347A and S359A mutations displayed 3.5-fold greater TAK1 activation following TNF-α. This mutant demonstrated a 6–10-fold increase in NF-κB DNA binding following TNF-α compared with wild type IHPK2-expressing cells in which NF-κB DNA binding was inhibited. Cells transfected with wild type IHPK2 or IHPK2 mutants that lacked S347A and S359A mutations displayed enhanced terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling staining following TNF-α. We believe that IHPK2-TRAF2 binding leads to attenuation of TAK1- and NF-κB-mediated signaling and is partially responsible for the apoptotic activity of IHPK2.
Inositol polyphosphates play diverse biologic roles, including regulation of endocytic trafficking (1), protein phosphorylation (2), chemotaxis (3), regulation of non-homologous end joining (4, 5, 6), and apoptosis (7, 8). Inositol hexakisphosphate kinase 2 (IHPK2)2 is a kinase that catalyzes the synthesis of diphosphoinositol pentakisphosphate and bis-diphosphoinositol tetrakisphosphate. Overexpression of IHPK2 sensitizes ovarian carcinoma cell lines to the growth-suppressive and apoptotic effects of IFN-β, IFN-α2 treatment, and ionizing radiation (9). Snyder and co-workers (8) recently demonstrated that IHPK2 enhanced the cytotoxic actions of several different cell stressors.
By co-immunoprecipitation, we found that IHPK2 associated with TRAF2. The tumor necrosis factor (TNF) receptor-associated factor (TRAF) family of proteins serve as adapter proteins for the TNF-R and interleukin-1 receptor superfamilies (10, 11). The role of TRAF2 in mediating the TNF-α response is controversial. Early studies with TRAF2 and TRAF5 knock-out mice or dominant negative (DN) TRAF2 mice suggested that these proteins were not essential or played redundant roles in TNF-induced NF-κB activation (12, 13, 14). TRAF2–/– fibroblasts are severely impaired in their ability to activate c-Jun N-terminal kinase in response to TNF-α (12). Subsequent studies suggested that TRAF2–/– fibroblasts (15) or TRAF2/TRAF5 double knock-out murine fibroblasts (16) were impaired in their ability to activate NF-κB in response to TNF-α. Most recently, Aggarwal and co-workers (17) have shown that NF-κB activation in carcinomas could be disrupted by expression of DN-TRAF2 but not by DN-TRAF5, suggesting that TRAF2 plays a critical role in TNF-α-induced NF-κB activation.
Within the trimeric signaling complex TRAF2 serves to recruit IκB kinase (IKK) and RIP1 to the TNF-R1 (18, 19). Transforming growth factor β-activated kinase (TAK1) is a critical component of this cascade, functioning downstream of RIP1 and TRAF2 to activate IKKβ (20). We hypothesized that the binding of IHPK2 to TRAF2 might disrupt TNF-α signaling. The aims of this study were to identify motifs in IHPK2 required for TRAF2 binding and to determine whether disruption of IHPK2-TRAF2 binding would affect TNF-α signaling.
EXPERIMENTAL PROCEDURES
Materials—Human IFN-β (Serono, Rockland, MA)-specific activity, 2.7 × 108 units/mg, was used in these studies.
Construction of IHPK2 SXXE Mutants—IHPK2 was cloned into the pCXN2myc mammalian expression vector (21). Using PCR, point mutations were made in IHPK2. The serine residues in SXXE were changed to AXXE. Thus, the wild type sequences (in bold) SXXE1, VDIVDNSDCEPKS; SXXE2, LLENLTSRYEVPC; SXXE3, PEVVLDSDAEDLE; and SXXE4, EDLSEESADESAG were changed to SXXE1, VDIVDNADCEPKS; SXXE2, LLENLTARYEVPC; SXXE3, PEVVLDADAEDLE; and SXXE4, EDLSEEAADESAG. This was done using primers 5′-pGTGGACATTGTAGATAATGCAGACTGTGAACCAAAAAGT-3′ and 5′-pACTTTTTGGTTCACAGTCTGCATTATCTACAATGTCCAC-3′ for SXXE1, 5′-pTTACTGGAAAACCTGACTGCCCGCTATGAGGTGCCTTGT-3′ and 5′-pACAAGGCACCTCATAGCGGGCAGTCAGGTTTTCCAGTAA-3′ for SXXE2, 5′-pCCCGAAGTGGTCCTGGACGCAGATGCTGAGGATTTGGAG-3′ and 5′-pCTCCAAATCCTCAGCATCTGCGTCCAGGACCACTTCGGG-3′ for SXXE3, and 5′-pGAGGACCTGTCAGAGGAAGCAGCTGATGAGTCTGCTGGT-3′ and 5′-pACCAGCAGACTCATCAGCTGCTTCCTCTGACAGGTCCTC-3′ for SXXE4 (p indicates phosphorylated 5′-nucleotide). The PCR product was digested with EcoRI and XhoI and cloned into pCXN2myc. Mutations were confirmed by sequencing. TRAF2 cDNA was cloned into pCXN2myc in a similar manner. Constructs were transfected into NIH-OVCAR-3 cells by nucleofection with Cell Line Nucleofector kit T (Amaxa, Koeln, Germany). For transient transfection studies, cells were utilized after 48 h. In other co-immunoprecipitation studies, transfectants were selected with G418 for 4 weeks of selection, and clones were pooled. Expression of mutants was monitored by immunoblotting.
siRNA Target Sequence—The IHPK2 target sequence consisted of a 29-mer oligonucleotide representing the 21-nucleotide sequence starting at nucleotide 181 (AAAUUCACUCCCCAGUACAAA) and contained an 8-nucleotide sequence at the 3′-end complementary to the T7 promoter primer provided in the Silencer siRNA construction kit (Ambion, Austin, TX). Complementary oligonucleotides were annealed, and double-stranded RNA was prepared by in vitro transcription. The resulting double-stranded RNA was then treated with RNase A and DNase I to generate the final siRNA product. An oligonucleotide sequence that does not silence any mammalian gene (siRNA-scrambled, siSCR) was utilized as a negative control. Nucleofection was used to transiently transfect siRNAs into cells at a concentration of 40 nm.
Immunoprecipitation—Mouse monoclonal antibody (15 μg) made in our laboratory was incubated with Protein A-Sepharose beads (Amersham Biosciences) in cell lysis buffer (0.1% Nonidet P-40, 50 mm Tris, pH 8.0, 0.1 mm EDTA, 0.5 mm dithiothreitol, 75 mm NaCl, 10% glycerol, and protease inhibitors). For in vitro interaction studies, recombinant purified IHPK2 and TRAF2 (50 μg), encoded in pET32a and expressed in BL21-DE3 bacteria, were incubated in 20 mm Hepes, pH 7.9, 150 mm NaCl, 10 mm EDTA, 0.1% Nonidet P-40 (v/v), 10% glycerol, 1 mm dithiothreitol, 1 mm phenylmethanesulfonyl fluoride, 20 μg/ml leupeptin. For in vivo interaction studies, total cell protein (100 μg) was immunoprecipitated with IHPK2 monoclonal antibody. Precipitate was subjected to electrophoresis, transferred to polyvinylidene difluoride membrane, and immunoblotted with rabbit anti-mouse TRAF2 polyclonal antibody (Leinco Technologies, St. Louis, MO). Membranes were incubated with anti-mouse IgG antibody conjugated to horseradish peroxidase and developed using ECL reagents (Pierce). Converse experiments were performed by immunoprecipitating with anti-TRAF2 and probing with anti-IHPK2. Lysates were also immunoprecipitated with anti-NF-κB p50 antibody (Zymed Laboratories Inc., San Francisco, CA), anti-TAK1 (Cell Signaling Technology, Danvers, MA), or anti-DR4 (Genetex, San Antonio, TX) and probed for IHPK2.
Immunoblot Analysis—Total cell protein (50 μg) was separated on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane. Membranes were incubated with antibody raised against TAK1, or phospho-TAK1, or phospho-AKT or AKT (Cell Signaling Technology), or human XIAP (BD Transduction Laboratories, San Jose, CA), or GAPDH (Trevigen, Inc., Gaithersburg, MD). After washing, membranes were incubated with appropriate secondary antibody conjugated to horseradish peroxidase and developed using ECL reagents (Pierce). Band intensity was quantitated using densitometry (FluorChem SP imaging station; Alpha Innotech, San Leandro, CA). -Fold induction was expressed as the ratio of normalized band intensities: (TNF-treated)/(PBS-treated).
Electrophoretic Mobility Shift Assay (EMSA)—As a positive control for NF-κB induction, cells were treated with TNF-α (20 ng/ml) for 15 min. Plates were washed with cold Dulbecco's phosphate-buffered saline (Sigma). Cells were scraped from plates and resuspended in cold CellLytic M lysis reagent (Sigma) with phosphatase and protease inhibitors (Calbiochem). Lysates were incubated (4 °C, 30 min) followed by centrifugation (20,000 × g for 15 min). Supernatant protein concentrations were assessed using the Bradford method (Bio-Rad protein assay). The NF-κB consensus binding sequence oligonucleotide (5′-AGTTGAGGCGACTTTCCCAGGC-3′) (Santa Cruz Biotechnology, Santa Cruz, CA) was end-labeled with [γ32-P]dATP (PerkinElmer Life Sciences), using T4 polynucleotide kinase (Roche Applied Science). DNA binding reactions were performed (30 min, 4 °C, 20 μg of protein) in 100 mm HEPES, 3.0 mm EDTA, 50% glycerol, 5 mm dithiothreitol, 25 mm MgCl2, 20 mm Tris, pH 7.90, 5 μg of poly d(I-C), and labeled probe. Complexes were resolved on 6% non-denaturing polyacrylamide gels. Gels were dried and exposed to film. To verify that band shifts were comprised of NF-κB, lysates were stimulated for 15 min with TNF-α (20 ng/ml) and incubated with 1 μg of anti-NF-κB p50 antibody (Zymed Laboratories Inc., San Francisco, CA). Band intensity was quantitated by phosphor image analysis on a Storm-840 imager using Image Quant v 5.1 software (Amersham Biosciences). -Fold induction was expressed as the ratio of normalized band intensities: (TNF-treated)/(PBS-treated).
IKK Assay—Whole cell extracts (300 μg of protein) were supplemented with 150 μl of Buffer A (20 mm Hepes, pH 7.9, 20 mm β-glycerophosphate, 10 mm NaF, 0.1 mm orthovanadate, 5 mm para-nitrophenyl phosphate, 10 mm 2-mercaptoethanol, 0.5 mm phenylmethylsulfonyl fluoride, and protease mixture) and 2 μl of normal rabbit serum and mixed by rotation (4 °C, 1 h) (22). A 50% slurry of Protein G-Sepharose (80 μl) (Amersham Biosciences) in Buffer A (without β-mercaptoethanol or phenylmethylsulfonyl fluoride) was added and mixed by rotation at 4 °C, 1 h. Protein G-Sepharose was removed by centrifugation (800 × g, 1 min) and discarded. Anti-IKKα monoclonal antibody (0.5 μg; BD Biosciences) or anti-β-actin antibody was added to the supernatant and mixed by rotation (4 °C, 2 h). A 50% slurry of Protein G-Sepharose (60 μl) prepared in Buffer C (Buffer A plus 50 mm NaCl and 10 mm MgCl2, without β-mercaptoethanol and phenylmethylsulfonyl fluoride) was added and mixed by rotation (4 °C, 30 min). Protein G immunopellets were collected by centrifugation (800 × g, 30 s) and washed three times with Buffer B (Buffer A plus 250 mm NaCl) and once with Buffer C (Buffer A plus 50 mm NaCl and 10 mm MgCl2). Immunopellets were resuspended in 30 μl of kinase buffer with 0.1 mm orthovanadate, 50 μm unlabeled ATP, 5 μCi of [γ-32P]ATP, 2 mm dithiothreitol, and 2 μg of recombinant GST-IκBα-(1–54) and incubated (30 °C, 30 min). Reactions were stopped with 15 μl of 4× SDS-PAGE loading buffer (200 mm Tris-HCl, pH 6.8, 8% SDS, 40% glycerol, 0.2% 2-mercaptoethanol), heated (95 °C, 10 min), and resolved by SDS-PAGE on 12% acrylamide gels. Gels were rinsed, stained with Bio-Safe Coomassie (Bio-Rad) to visualize protein bands, rinsed, photographed, dried, and exposed to Kodak X-OMAT AR film (Eastman Kodak Co., Rochester, NY) to detect substrate phosphorylation. IKK activation was quantified by phosphor image analysis (GE Healthcare).
Antiproliferative Assays—Cells were treated with IFN-β during growth in RPMI 1640 (Mediatech Inc., Herndon, VA) and 5% fetal bovine serum (HyClone, Logan, UT). Cells were confirmed mycoplasma-free by PCR. Growth was monitored using a colorimetric assay (23). Each treatment group contained 8 replicates. Cells were fixed and stained with sulforhodamine B after 4 days. Bound dye was eluted from cells, and absorbance (Aexp) was measured at 570 nm. One plate was fixed 8 h after plating to determine the absorbance representing starting cell number (Aini). Absorbance with this plate and that obtained with untreated cells at the end of the growth period (Afin) were taken as 0 and 100% growth, respectively. Thus, Percent Control Growth = 00%*(Aexp – Aini)/Afin – Aini). Expressed as a percent of untreated controls, a decrease in cell number (relative to starting cell number) is a negative number on the y-axis.
TUNEL Assay—NIH-OVCAR-3 cells were exposed to TNF-α (10 ng/ml) for 16 h. Apoptotic cells were detected by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end-labeling) staining using a kit (APO-BRDU kit; BD Biosciences). Cells were processed according to the manufacturer's protocol. The percentage of fluorescein isothiocyanate-positive cells was analyzed by fluorescent-activated cell scanning (Facsvantage; BD Biosciences).
RESULTS
IHPK2 Binds TRAF2—Bacterially translated proteins were co-immunoprecipitated with antibody specific for IHPK2 (Fig. 1a, top lane), whereas antibody to the DR4 death receptor did not precipitate TRAF2 (Fig. 1a, bottom lane). Untransfected NIH-OVCAR-3 cells were treated with PBS or IFN-β (100 units/ml, 24 h); lysates were immunoprecipitated (IP) with anti-IHPK2 followed by immunoblot (IB) with anti-TRAF2 antibody (Fig. 1b, top lane). IFN-β increased the interaction of IHPK2 with TRAF2. The converse experiment (IP with anti-TRAF2 and IB with IHPK2) gave similar results (second lane). Immunoprecipitation with irrelevant antibodies (p50 and TAK1) yielded no signal.
FIGURE 1.
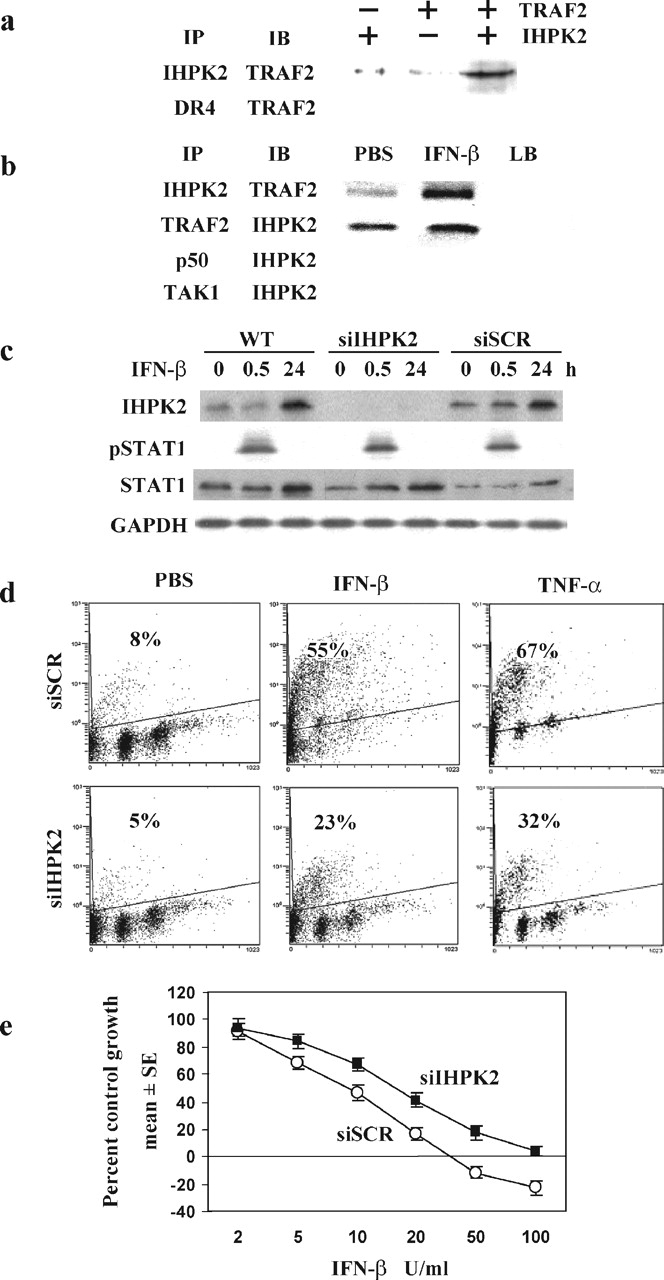
IHPK2-TRAF2 interaction.a, interaction of recombinant proteins in vitro. Presence and absence of proteins indicated by + and–, respectively. The reaction was subject to immunoprecipitation (IP) followed by immunoblot (IB) with the indicated antibodies. b, wild type NIH-OVCAR-3 cells were treated with IFN-β (100 units/ml × 24 h). Lysates were subject to immunoprecipitation followed by immunoblot with the indicated antibodies. Anti-p50 NF-κB and anti-TAK1 were included as negative controls. As additional controls, lysis buffer (LB) alone, without cell lysate, was immunoprecipitated. c, untransfected (WT) cells were treated with PBS (0h) or IFN-β as above and harvested at 0.5 and 24 h. Immunoblots indicate induction of IHPK2 and STAT1 (at 24 h) and induction of pSTAT1 (at 30 min). Cells were transiently transfected with siRNA directed against IHPK2 (siIHPK2) or scrambled siRNA (siSCR) by nucleofection. Two days later cells were treated with PBS or IFN-β (0.5 and 24 h) and subjected to immunoblot analysis. d, transiently transfected cells were treated with PBS, IFN-β (100 units/ml), or TNF-α (10 ng/ml) for 24 h and subjected to TUNEL staining. e, transiently transfected cells were grown in the presence of IFN-β (0–100 units/ml). After 4 days of growth, cells were fixed and stained, and cell number (proportional to intensity of retained SRB dye) was expressed as a percentage of PBS-treated control cells (n = 8 replicates/data point).
IHPK2 Knock Down Induces IFN-β Resistance—We previously showed that IFN-β induced IHPK2 in a post-transcriptional manner (7). This induction could be detected after IFN-β (100 units/ml, 24 h, Fig. 1c, top lane). Transient transfection with siRNA against IHPK2 (siIHPK2) efficiently suppressed IHPK2 expression, whereas scrambled siRNA (siSCR) did not. Because siRNA can potentially induce an IFN response, we examined effects of siRNA transfection on activation and phosphorylation of the transcription factor STAT1. Treatment of untransfected cells with IFN-β induced phosphorylation of STAT1 after 30 min, which was no longer detectable after 24 h. Similar activation of STAT1 was detected in siIHPK2- and siSCR-expressing cells, suggesting that down-regulation of IHPK2 did not affect IFN signaling. The expression of STAT1, an IFN-stimulated gene, was increased in untransfected, siIHPK2-transfected, and siSCR-transfected cells (Fig. 1c, third lane).
Transiently transfected NIH-OVCAR-3 cells were subjected to TUNEL analysis (Fig. 1d). Following IFN-β, cells expressing scrambled siRNA (siSCR) displayed 2-fold greater TUNEL staining (55%) compared with cells expressing siIHPK2 (23%). Similarly, siIHPK2 reduced the level of apoptosis following TNF-α from 67 to 32%. Hence, knock down of IHPK2 mRNA conferred resistance to apoptosis induced by IFN-β and by TNF-α. Inhibition of apoptosis correlated with enhanced proliferation. Anti-proliferative assays (Fig. 1e) revealed a 2-fold increase in ID50 for IFN-β in siIHPK2 cells compared with siSCR cells: 16 and 8 units/ml, respectively.
IHPK2 Mutation Inhibits IHPK2-TRAF2 Binding—Ligation of death receptors such as Fas, TNFR1, DR4, and DR5 activates the external apoptotic pathway. TNF-α binds TNFR1, inducing clustering of TRADD and receptor-interacting protein (RIP1), which leads to caspase activation. However, ligation of TNFR1 generates not only a death signal but an additional survival signal mediated by NF-κB. Apo2L/TRAIL triggers apoptosis via binding to death receptors DR4 and DR5. Immunoprecipitation of NIH-OVCAR-3 cell lysates with IHPK2 monoclonal antibody showed that there was no association between IHPK2 and DR4 or DR5 (not shown).
Treatment with IFN-β enhanced binding of IHPK2 to TRAF2 (Fig. 1b). The interaction was even stronger when TRAF2 and IHPK2 were co-transfected (Fig. 2a). When cells are transfected with only one construct (either IHPK2 or TRAF2, Fig. 2, a and b, lanes 1–2) less TRAF2 is immunoprecipitated when compared with co-transfection. IHPK2 contains four SXXE motifs, putative binding sites for TRAF2 (24). For brevity, we named these sites S1 (S102), S2 (S211), S3 (S347), and S4 (S359). The four SXXE sites were point mutated (serine to alanine) singly and in combination. NIH-OVCAR-3 cells were stably co-transfected with IHPK2 mutants and TRAF2. Cell lysates were immunoprecipitated with IHPK2 monoclonal antibody. Immunoblot analysis demonstrated that two of the SXXE mutations (S3 and S4) in combination (but not singly) reduced TRAF2 binding activity (Fig. 2a). Protein levels of IHPK2 and TRAF2 were comparable in transfected cells (Fig. 2b).
FIGURE 2.

Effect of IHPK2 mutation on binding of IHPK2 to TRAF2.a, NIH-OVCAR-3 cells were co-transfected with IHPK2 and TRAF2 by nucleofection. Pluses and minuses indicate the presence or absence of indicated construct, respectively. Total cell protein (100 μg) from each transfected cell line was immunoprecipitated with IHPK2 mouse monoclonal antibody. Precipitate was subjected to immunoblot analysis with TRAF2 polyclonal antibody (bottom row). Immunoblot analysis demonstrated that the IHPK2 S3&4 mutations in combination (but not single mutations) caused reduced TRAF2 binding when compared with the interaction of wild type IHPK2 and all other IHPK2 mutations. b, immunoblot to demonstrate protein levels of IHPK2, TRAF2, and GAPDH in lysates of transfected cells. The experiment was performed three times with similar results.
Effect of IHPK2 Mutation on TAK1 Phosphorylation—Because TAK1 plays an important role in the TNFR-mediated cascade and functions immediately downstream of TRAF2 (20), we determined whether IHPK2 mutation had any effect on phosphorylation of TAK1. Wild type untransfected and vector-transfected cells displayed a 2.5- to 3-fold induction of phospho-TAK1 (pTAK1) in response to TNF-α (Fig. 3a). TNF-α failed to activate TAK1 above base line in cells expressing IHPK2 or the S3 or S4 single mutants (<1.5-fold induction of pAKT). However, phosphorylation of TAK1 following TNF-α was restored in cells containing S3&S4 double mutants (3- to 3.5-fold induction). Hence, the S3&S4 double mutant had diminished TRAF2 binding and no longer inhibited TAK1 phosphorylation.
FIGURE 3.
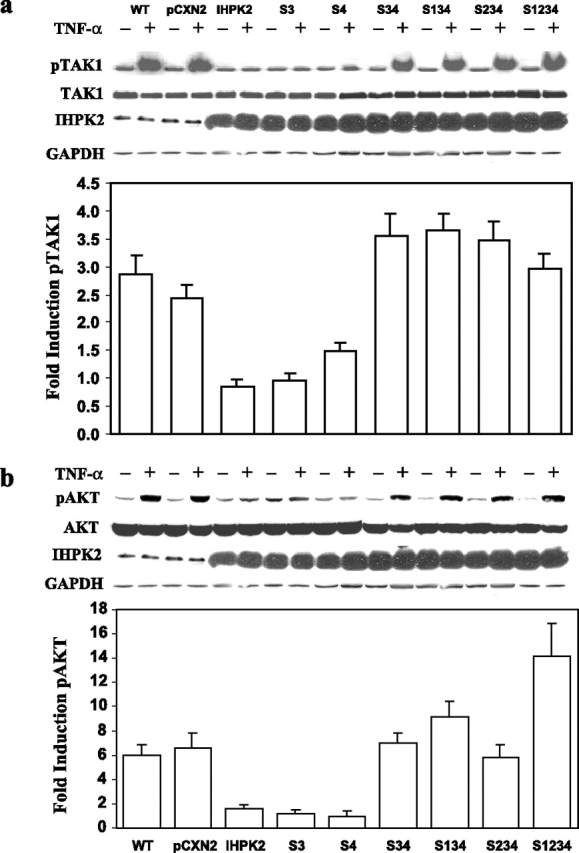
Effect of IHPK2 mutation on phosphorylation of TAK1 and AKT.a, untransfected (WT), vector-transfected (pCXN2), and mutant-transfected cells were treated with PBS (–) or TNF-α, 15 ng/ml (+) for 1 h. Lysates were subjected to immunoblot with anti-phospho-TAK1 followed by stripping and reprobing with anti-TAK1. Expression of IHPK2 transgene is indicated, and GAPDH served as loading control. Bands were quantitated by densitometry, and -fold induction was calculated (n = 3). b, using the same lysates, similar studies were performed with anti-phospho-AKT and anti-AKT.
Effect of IHPK2 Mutation on AKT Phosphorylation—Because AKT may also contribute to NF-κB activation, mainly through phosphorylation of IKKα (25), we determined whether IHPK2 mutation had any effect on activation of AKT. Wild type untransfected and vector-transfected cells displayed a 6-fold induction of phospho-AKT (pAKT) in response to TNF-α (Fig. 3b). The TNF-α response of cells expressing IHPK2 or the S3 or S4 single mutants was suppressed (1- to 1.5-fold induction of pAKT). However, the TNF-α response was restored in cells containing S3&S4 double mutants; they displayed 5- to 17-fold induction of pAKT (Fig. 3b, bars 6–9). Hence, the S3&S4 double mutant lost the ability to inhibit pAKT activation.
Effect of IHPK2 Mutation on IκBα Kinase Activity—The IKK complex, consisting of α, β, and γ subunits, is responsible for phosphorylation of IκB, leading to IκB degradation, and subsequent nuclear translocation of NF-κB. TAK1 phosphorylates IKKβ, the main catalytic subunit, whereas AKT phosphorylates IKKα. We examined IKK enzymatic activity in lysates of cells stably transfected with IHPK2 mutants. IKK was immunoprecipitated from cell extracts using anti-IKKα antibody, and kinase activity was assessed using recombinant GST-IκBα-(1–54) as a substrate. TNF-α induced IKKα activity in wild type untransfected cells, vector-transfected cells, and all transfectants (Fig. 4). Cells expressing IHPK2 S3&4 double mutants displayed similar kinase activity as wild type cells or other transfectants. Therefore, the S3&4 mutant did not appear to modulate IKK enzymatic activity in vitro.
FIGURE 4.

IKK activity. IKK activity was assessed using recombinant GST-IκBα-(1–54) and [γ-32P]ATP as substrates. The phosphorylated GST fusion protein was detected by autoradiography. a, IKK activity was determined in NIH-OVCAR-3 cells co-transfected with IHPK2 (wild type or mutants) and TRAF2, followed by TNF-α stimulation for 15 min. Cells expressing the double mutant did not show significant differences in IKK activity compared with cells expressing wild type IHPK2. Untreated cells typically exhibit 5–10% the activity of stimulated cells (31). Expression of IHPK2 transgene is indicated, and GAPDH served as loading control. The experiment was performed three times with similar results. b, Coomassie blue-stained gel demonstrated equal loading of GST-IκBα-(1–54) substrate utilized in the kinase assay.
Effect of IHPK2 Mutation upon NF-κB DNA Binding—We next determined whether IHPK2-TRAF2 binding and inhibition of TAK1 phosphorylation disrupted NF-κB signaling. In untransfected wild type NIH-OVCAR-3 cells, treatment with TNF-α resulted in robust NF-κB activation as demonstrated by EMSA (Fig. 5a). Supershift (ss) with anti-p50 confirmed the identity of NF-κB. Transfection with IHPK2 reduced the intensity of DNA binding activity in response to TNF-α (Fig. 5b, compare lanes 3–6). Quantitation of the band shift indicated a 3-fold reduction (Fig. 5b, bars 2 and 3). Hence, overexpression of IHPK2 blunted NF-κB DNA binding activity in response to TNF-α. Cells expressing two IHPK2 mutations (S3&4) demonstrated 7- to 11-fold activation of NF-κB compared with cells co-transfected with wild type IHPK2. The other IHPK2 mutants that lacked S3&4 mutations had blunted NF-κB activation (1-fold induction).
FIGURE 5.

Effect of IHPK2 mutation upon NF-κB DNA binding activity.a, wild type untransfected NIH-OVCAR-3 cells were treated with TNF-α, and EMSA was performed. Lysates were incubated with anti-GAPDH or anti-NF-κB(p50) prior to EMSA to demonstrate supershift (ss). b, cells transfected with IHPK2 S3&4 mutations (S347A and S359A) displayed enhanced NF-κB DNA binding activity induced by TNF-α. Cells transfected with wild type IHPK2 or IHPK2 mutants that lacked S3&4 mutations had suppressed NF-κB DNA binding activity. Cells were stimulated with TNF-α (20 ng/ml) for 15 min. Expression of IHPK2 transgene is indicated, and GAPDH served as loading control. EMSA band intensities were quantified with a Phosphorimager, and -fold induction was calculated. Cells expressing IHPK2 mutants S3&4, S1&3&4, S2&3&4, and S1&2&3&4 were 7- to 11-fold more effective at activation of NF-κB compared with cells transfected with wild type IHPK2, whereas cells expressing IHPK2 and single mutants had <2-fold induction. Data are expressed as mean ± S.E. of three separate experiments.
Effect of IHPK2 Mutation on XIAP—Activated NF-κB translocates to the nucleus and induces transcription of target genes such as inhibitor of apoptosis proteins (IAPs). The X-linked mammalian inhibitor of apoptosis protein (XIAP) binds to caspase 3, caspase 9, DIABLO/Smac, HtrA2/Omi, and TAB1 (26). Cells expressing IHPK2 mutants S3&4 displayed up to a 2.4-fold increase in XIAP. The IHPK2 single mutants (S3 and S4) acted like wild type IHPK2, resulting in suppressed XIAP expression (fold increase 0.1–0.9) (Fig. 6). Thus, effects of IHPK2 mutation upon XIAP induction correlated with changes in NF-κB DNA binding activity.
FIGURE 6.

Effect of IHPK2 mutation on XIAP levels. Immunoblotting for XIAP was performed with lysates harvested 24 h after TNF-α (15 ng/ml). Expression of IHPK2 transgene is indicated, and GAPDH served as loading control. Cells expressing IHPK2 mutations S3&4 had up to 2.4-fold increase in XIAP. XIAP was not induced in cells expressing IHPK2, or IHPK2 single mutations, demonstrating -fold increases of -0.1 to -0.9. -Fold increase <1.0 corresponds to decreased XIAP expression following TNF-α (n = 3).
IHPK2 Mutants Inhibit TNF-α-induced Apoptosis—The effects of IHPK2 mutation on TNF-α-induced apoptosis were analyzed by TUNEL (Fig. 7). Untransfected wild type and vector-transfected cells displayed 40 and 26% TUNEL positivity following TNF-α, respectively. Cells expressing IHPK2 or S3 or S4 single point mutations had enhanced TUNEL staining, 64, 51, 52%, respectively. Cells expressing the S3&S4 double mutation were 33% TUNEL-positive, a response that was similar to untransfected or vector-transfected cells. Therefore, the double mutant was defective in enhancing TNF-α-induced apoptosis.
FIGURE 7.

Effect of IHPK2 mutation on apoptosis induction by TNF-α. Untransfected (WT), vector-transfected (pCXN2), and mutant-transfected cells were treated with PBS or TNF-α, 15 ng/ml for 16 h. Induction of apoptosis was determined by TUNEL staining.
IHPK2 Mutants Affect Growth-suppressive Activities of IFN-β—The biological relevance of the IHPK2 mutations was determined in antiproliferative assays using pools of stable transfectants. Overexpression of IHPK2 sensitized cells to IFN-β, as the IC50 value fell from 15 to 6 units/ml in vector-transfected and IHPK2-transfected cells, respectively (Fig. 8). Constructs containing the S3&4 mutations conferred resistance to IFN-β, as the IC50 value increased from 15 units/ml in vector-transfected cells to 20–75 units/ml in double mutant-transfected cells, respectively. Cells expressing irrelevant IHPK2 mutations (IRREL, constructs lacking S3&4 mutations) displayed sensitivity to IFN-β between that of vector- and IHPK2-transfected cells. Therefore, overexpression of IHPK2 sensitized cells to IFN-β, and the S3&4 double mutation conferred resistance to IFN-β.
FIGURE 8.

Effect of IHPK2 mutation on IFN-β antiproliferative activity. NIH-OVCAR-3 cells were stably transfected with IHPK2 constructs and grown in the presence of 5–500 units/ml IFN-β. After 4 days, cells were fixed and stained with sulforhodamine B. Absorbance of bound dye was expressed as percent of untreated controls (n = 8, each data point). Overexpression of IHPK2 sensitized cells to IFN-β. Cells expressing IHPK2 mutations S3&4 in combination conferred relative resistance to IFN-β. The dose response curve of cells expressing irrelevant IHPK2 mutations (S1, S2, S3, S4, S1&2, S1&3, S1&4, S2&3, S2&4, S1&2&3, S1&2&4) lay in a narrow band between pCXN2 (vector) and IHPK2 and is represented as a single curve (IRREL).
DISCUSSION
In our previous study, overexpression of the kinase-dead IHPK2 mutant suppressed IFN-induced cell death, but the magnitude of suppression was 50%, even though transfection efficiency was 90%. Snyder and co-workers (8) recently demonstrated that transfection of IHPK2 augmented the apoptotic activities of several cell stressors. Although transfection of all three subtypes of IHPK increased cell death, suppression of IHPK2, but not IHPK1 or IHPK3, abrogated apoptosis.
Our studies and those of Snyder and co-workers suggest that kinase activity is not the sole determinant of the apoptotic activity of IHPK2. The present study provides evidence that IHPK2 is a TRAF2-interacting protein that suppresses TAK1 phosphorylation and inhibits NF-κB activation. In the absence of cell stressors such as IFN-β or TNF-α, IHPK2-TRAF2 interaction is still detected at low levels (Fig. 1b). This may be responsible for the ∼10% constitutive levels of apoptosis observed in NIH-OVCAR-3 cells. Immunoprecipitation analysis confirmed that the SXXE mutations (S347A and S359A) in combination inhibited TRAF2 binding. Therefore, these residues are important for IHPK2-TRAF2 association. Cells expressing these IHPK2 mutations were more effective at phosphorylation of TAK1 and activation of NF-κB compared with cells expressing wild type IHPK2. Similarly, the double mutants were more effective at XIAP induction and more resistant to the apoptotic effects of IFN-β.
The NF-κB pathway is activated following degradation of IκB (27). This pathway depends on the IKK complex, which contains three subunits, IKKα, IKKβ, and IKKγ. TRAF2 functions to recruit IKKα and IKKβ to the TNF-R1 receptor (18). Although initial studies utilizing TRAF2 knock-out MEFs suggested that TRAF2 did not play a role in activation of NF-κBby TNF-α (12), later studies indicated that TRAF2 was indeed required for NF-κB activation. Nakano and co-workers (16) showed that TRAF2/TRAF5 double knock-out MEFs were severely impaired in NF-κB activation following TNF-α, suggesting that TRAF2 and TRAF5 function may be redundant. In squamous carcinoma, expression of DN-TRAF2 strongly inhibited transcription of an NF-κB reporter plasmid, whereas DN-TRAF5 had no effect on NF-κB activation (17). Hence, in some carcinomas TRAF2 and TRAF5 function is not redundant.
A model for this signaling cascade is: TNFR1-(RIP1/TRAF2)-TAK1-IKKβ-NF-κB. Knock-out MEFs that lack TAK1 cannot activate NF-κB in response to TNF-α and are highly sensitive to TNF-α-induced apoptosis, indicating that the survival pathway is severely inhibited in these cells (20). Phosphorylation of IKKα, mediated by NF-κB-inducing kinase, appears to be intact in TAK1 knock-out MEFs (20). Thus, TAK1 phosphorylates IKKβ but does not appear to phosphorylate IKKα. Immunoprecipitation experiments with recombinant proteins demonstrated that IKKβ is the main catalytic subunit of the IKK complex (28).
The GST-IκBα-(1–54) fusion protein, containing glutathione and the first 54 residues of IκBα, is phosphorylated by IKK in the kinase assay. Antibodies directed against IKKβ disrupt IKK activity; therefore, antibodies against IKKα or IKKγ are generally utilized to precipitate the complex. Other proteins such as Cdc37 and Hsp90 may be required for optimal activity of the IKK complex (29). If such auxiliary proteins are not pulled down efficiently, the assay may not reflect the full extent of IκBα phosphorylation in vivo.
Mutation of IHPK2 clearly affected TAK1 phosphorylation (Fig. 3a) but did not affect IKK kinase activity in vitro (Fig. 4a). There is also cross-talk between TRAF2 and AKT in NIH-OVCAR-3 cells, as overexpression of IHPK2 inhibited AKT activation (Fig. 3b). CD40, a TNF receptor superfamily member, requires TRAF2 to activate AKT. In TRAF2 knock-out MEFs, and carcinoma cells that received siRNA-TRAF2, AKT phosphorylation was severely impaired (30). Our studies suggest that following binding to TRAF2, IHPK2 functions to inhibit TAK1 phosphorylation, AKT phosphorylation, and subsequent NF-κB activation.
Footnotes
The abbreviations used are: IHPK2, inositol hexakisphosphate kinase 2; DN, dominant negative; IFN, interferon; IκB, inhibitor of NF-κB; IKK, IκB kinase; MEF, murine embryonic fibroblast; NF-κB, nuclear factor-κB; PBS, phosphate-buffered saline; TAK1, transforming growth factor β-activated kinase 1; TNF, tumor necrosis factor; TRAF, TNF receptor-associated factor; XIAP, X-linked inhibitor of apoptosis; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling; GAPDH, glyceraldehyde-3phosphate dehydrogenase; EMSA, electrophoretic mobility shift assay; siRNA, small interfering RNA; siSCR, siRNA-scrambled; GST, glutathione S-transferase.
References
- 1.Saiardi A., Sciambi C., McCaffery J.M., Wendland B., Snyder S.H. Proc. Natl. Acad. Sci. U. S. A. 2002;99:14206–14211. doi: 10.1073/pnas.212527899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saiardi A., Bhandari R., Resnick A.C., Snowman A.M., Snyder S.H. Science. 2004;306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 3.Luo H.R., Huang Y.E., Chen J.C., Saiardi A., Iijima M., Ye K., Huang Y., Nagata E., Devreotes P., Snyder S.H. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 4.Hanakahi L.A., Bartlet-Jones M., Chappell C., Pappin D., West S.C. Cell. 2000;102:721–729. doi: 10.1016/s0092-8674(00)00061-1. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y., Lieber M.R. J. Biol. Chem. 2002;30:30. doi: 10.1074/jbc.C200030200. [DOI] [PubMed] [Google Scholar]
- 6.Byrum J., Jordan S., Safrany S.T., Rodgers W. Nucleic Acids Res. 2004;32:2776–2784. doi: 10.1093/nar/gkh592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison B.H., Bauer J.A., Kalvakolanu D.V., Lindner D.J. J. Biol. Chem. 2001;276:24965–24970. doi: 10.1074/jbc.M101161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagata E., Luo H.R., Saiardi A., Bae B.I., Suzuki N., Snyder S.H. J. Biol. Chem. 2005;280:1634–1640. doi: 10.1074/jbc.M409416200. [DOI] [PubMed] [Google Scholar]
- 9.Morrison B.H., Bauer J.A., Hu J., Grane R.W., Ozdemir A., Chawla-Sarkar M., Gong B., Almasan A., Kalvakolanu D.V., Lindner D.J. Oncogene. 2002;21:1882–1889. doi: 10.1038/sj/onc/1205265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arch R.H., Gedrich R.W., Thompson C.B. Genes Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 11.Kopp E.B., Medzhitov R. Curr. Opin. Immunol. 1999;11:13–18. doi: 10.1016/s0952-7915(99)80003-x. [DOI] [PubMed] [Google Scholar]
- 12.Yeh W.C., Shahinian A., Speiser D., Kraunus J., Billia F., Wakeham A., de la Pompa J.L., Ferrick D., Hum B., Iscove N., Ohashi P., Rothe M., Goeddel D.V., Mak T.W. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 13.Nakano H., Sakon S., Koseki H., Takemori T., Tada K., Matsumoto M., Munechika E., Sakai T., Shirasawa T., Akiba H., Kobata T., Santee S.M., Ware C.F., Rennert P.D., Taniguchi M., Yagita H., Okumura K. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9803–9808. doi: 10.1073/pnas.96.17.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S.Y., Reichlin A., Santana A., Sokol K.A., Nussenzweig M.C., Choi Y. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- 15.Devin A., Cook A., Lin Y., Rodriguez Y., Kelliher M., Liu Z. Immunity. 2000;12:419–429. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 16.Tada K., Okazaki T., Sakon S., Kobarai T., Kurosawa K., Yamaoka S., Hashimoto H., Mak T.W., Yagita H., Okumura K., Yeh W.C., Nakano H. J. Biol. Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 17.Jackson-Bernitsas D.G., Ichikawa H., Takada Y., Myers J.N., Lin X.L., Darnay B.G., Chaturvedi M.M., Aggarwal B.B. Oncogene. 2007;26:1385–1397. doi: 10.1038/sj.onc.1209945. [DOI] [PubMed] [Google Scholar]
- 18.Devin A., Lin Y., Yamaoka S., Li Z., Karin M., Liu Z. Mol. Cell. Biol. 2001;21:3986–3994. doi: 10.1128/MCB.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song H.Y., Regnier C.H., Kirschning C.J., Goeddel D.V., Rothe M. Proc. Natl. Acad. Sci. U. S. A. 1997;94:9792–9796. doi: 10.1073/pnas.94.18.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shim J.H., Xiao C., Paschal A.E., Bailey S.T., Rao P., Hayden M.S., Lee K.Y., Bussey C., Steckel M., Tanaka N., Yamada G., Akira S., Matsumoto K., Ghosh S. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinoshita S., Suzuki H., Ito K., Kume K., Shimizu T., Sugiyama Y. Pharm. Res. 1998;15:1851–1856. doi: 10.1023/a:1011953906065. [DOI] [PubMed] [Google Scholar]
- 22.DiDonato J.A. Methods Enzymol. 2000;322:393–400. doi: 10.1016/s0076-6879(00)22038-7. [DOI] [PubMed] [Google Scholar]
- 23.Skehan P., Storeng R., Scudiero D., Monks A., McMahon J., Vistica D., Warren J.T., Bokesch H., Kenney S., Boyd M.R. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 24.Park Y.C., Burkitt V., Villa A.R., Tong L., Wu H. Nature. 1999;398:533–538. doi: 10.1038/19110. [DOI] [PubMed] [Google Scholar]
- 25.Ozes O.N., Mayo L.D., Gustin J.A., Pfeffer S.R., Pfeffer L.M., Donner D.B. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 26.Silke J., Hawkins C.J., Ekert P.G., Chew J., Day C.L., Pakusch M., Verhagen A.M., Vaux D.L. J. Cell Biol. 2002;157:115–124. doi: 10.1083/jcb.200108085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Senftleben U., Cao Y., Xiao G., Greten F.R., Krahn G., Bonizzi G., Chen Y., Hu Y., Fong A., Sun S.C., Karin M. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 28.Zandi E., Chen Y., Karin M. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 29.Chen G., Cao P., Goeddel D.V. Mol. Cell. 2002;9:401–410. doi: 10.1016/s1097-2765(02)00450-1. [DOI] [PubMed] [Google Scholar]
- 30.Davies C.C., Mak T.W., Young L.S., Eliopoulos A.G. Mol. Cell. Biol. 2005;25:9806–9819. doi: 10.1128/MCB.25.22.9806-9819.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiDonato J.A., Hayakawa M., Rothwarf D.M., Zandi E., Karin M. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]