

Abstract
Previously we determined that chronic alcohol ingestion (6 wks) in rats increases lung epithelial permeability in vivo ~5-6-fold and promotes flooding of the alveolar airspaces with proteinaceous fluid in response to stresses such as sepsis. In parallel, alveolar epithelial cells isolated from alcohol-fed rats fail to form tight monolayers in vitro, even when cultured for up to 8 days in the absence of alcohol. However, the molecular mechanisms underlying alcohol-induced permeability are unknown. Claudins are key components of tight junctions that restrict the paracellular movement of water, proteins, and solutes across cellular barriers including the alveolar epithelium. In this study, we examined the expression of multiple members of the claudin protein family in the lungs of alcohol-fed vs. control-fed rats (Lieber-DeCarli liquid diet with 36% of calories as alcohol vs. maltin-dextrin for 6 wks). We determined that chronic alcohol ingestion affected the expression of multiple claudins; most striking were decreases in claudin-1 and claudin-7, and an increase in claudin-5, in the whole lung and in alveolar epithelial monolayers derived from alcohol-fed rats. In parallel, immunocytochemistry of alveolar epithelial monolayers from alcohol-fed rats revealed abnormal intracellular accumulation of claudin-7 protein and relatively decreased localization to cell membranes. Claudin-1 and claudin-7 are relatively specific to alveolar epithelial type I pneumocytes that form the vast majority of the alveolar epithelial barrier in vivo, and increases in claudin-5 have been associated with increased epithelial permeability in other systems. Therefore, these findings suggest that changes in claudin expression in the alveolar epithelium produce a “leakier” phenotype that renders the alcoholic lung susceptible to alveolar flooding during acute inflammatory stresses.
Keywords: claudin, ARDS, ethanol, type II pneumocyte, type I pneumocyte, tight junction
Introduction
Epidemiological studies performed in the past decade have revealed a strong but previously unrecognized association between alcohol abuse and acute lung injury. It is now clear that chronic alcohol abuse is an independent co-morbid factor that significantly increases the risk of the acute respiratory distress syndrome (ARDS) by as much as 2-4-fold in critically ill individuals (Licker et al., 2003; Moss et al., 1996; Moss et al., 2003; Moss and Burnham, 2003). ARDS is a severe form of lung injury that occurs in response to insults such as trauma, sepsis, gastric aspiration, and multiple blood transfusions (Ware and Matthay, 2000; Frutos-Vivar et al., 2006; Rubenfeld et al., 2005; Levitt and Matthay, 2006). A cardinal feature of ARDS is diffuse damage to the alveolar epithelium with subsequent flooding of the alveolar space with proteinaceous fluid. These events lead to respiratory failure and, in all cases, the need for prolonged mechanical ventilation in the intensive care unit. Even with current best medical practices, the mortality of ARDS is in the range of 40-50% (Ware and Matthay, 2000; Rubenfeld et al., 2005). Although the precise incidence of ARDS in the U.S. is unknown, the estimate is that as many as 150,000 new cases occur each year (Ware and Matthay, 2000). Based on the two largest series that examined this association, in which half of all the patients with ARDS had underlying alcohol abuse (Moss et al., 1996; Moss et al., 2003), it would appear that alcohol abuse contributes to tens of thousands of cases of ARDS and accounts for a significant fraction of the morbidity and mortality associated with the syndrome.
As noted, ARDS involves diffuse and severe damage to the alveolar epithelium with consequent loss of this critical barrier that is necessary for efficient exchange of oxygen and carbon dioxide between the airways and the vasculature. In fact, there is evidence that those patients with ARDS that have the most severe dysfunction of the alveolar epithelium, as reflected by the inability to remove fluid from the alveolar airspace, have a worse prognosis (Ware and Matthay, 2001). We demonstrated that chronic alcohol ingestion in rats causes significant alveolar epithelial dysfunction including increased protein permeability and decreased ability to clear a saline challenge in vivo (Guidot et al., 2000). Consistent with the clinical epidemiological studies, chronic alcohol ingestion alone, in the absence of nutritional deficiencies or other biological stresses, impairs alveolar epithelial function and renders the lung susceptible to acute edematous injury in response to stresses such as endotoxemia and sepsis (Brown et al., 2001a; Holguin et al., 1998; Velasquez et al., 2002). When alveolar epithelial cells are isolated and cultured from alcohol-fed rats, they fail to form tight monolayers and have increased paracellular leak of larger molecules such as inulin or sucrose even when cultured for up to 8 days in the absence of alcohol (Guidot et al., 2000). Although this defect in physical barrier formation appears to be somehow mediated by oxidant stress and glutathione depletion (Guidot et al., 2000), the precise molecular mechanisms by which chronic alcohol ingestion increases paracellular permeability in the alveolar epithelium are unknown.
The alveolar epithelium is composed of two distinct cell types. The alveolar type II pneumocytes are cuboidal cells that synthesize and secrete surfactant and regulate the levels of glutathione within the alveolar space. In comparison, alveolar type I pneumocytes are relatively flat cells with a much greater surface area and, even though they are present in approximately equal numbers as type II pneumocytes, comprise ~95% of the alveolar epithelial barrier in vivo. Paracellular movement of fluid, proteins, and solutes across the alveolar epithelium is carefully regulated by intercellular tight junctions. These tight junctions are sites of cell-cell contact that are formed by a combination of various proteins, including a family of transmembrane proteins named claudins. There are over 20 claudins, with distinctive distribution throughout various tissues including the lungs (Coyne et al., 2003; Daugherty et al., 2004; Wang et al., 2003). The potential effects of alcohol abuse on tight junction proteins within the lung have not been examined. Therefore, in this study we sought to determine whether or not chronic alcohol ingestion alters the expression of claudins within the lung and in particular, within the alveolar epithelium. We reasoned that significant disruptions in claudin expression in the alcoholic lung could explain, at least in part, why alcoholics are more susceptible to acute edematous lung injuries such as ARDS. To address these questions we examined the gene and protein expression of multiple claudin proteins in the whole lungs and in isolated alveolar epithelial cells in alcohol-fed vs. control-fed rats.
Materials and Methods
Animals and alcohol diet
Adult Male Sprague-Dawley rats (150-200g, Charles River Laboratory, Wilmington, MA) were fed the Lieber-DeCarli liquid diet (Research Diets, New Brunswick, NJ) containing either alcohol (36% of total calories) or an isocaloric substitution with maltin-dextrin ad lib for 6 weeks as previously published (Joshi et al., 2005; Joshi et al., 2006). All work was performed with the approval of the Institutional Care and Use of Animals Committee at the Atlanta VAMC.
Surgery and lung isolation
Rats were anesthetized with pentobarbital intraperitoneally and the ventral surface cleaned with 70% ethanol. A tracheostomy cannula was placed and secured using 2-0 ligature. After an incision was made from the pubis to the sternum exposing the abdominal and thoracic cavities, the diaphragm was isolated and cut from the chest wall, and the lungs intermittently ventilated using a sterile syringe with approximately 8 ml of air. The inferior vena cava was then severed and an incision made into the right ventricle followed by placement of a cannula into the pulmonary artery. The lungs were perfused blood-free with buffer and then removed en bloc with the distal trachea attached. At this stage, the lungs were either snap frozen in liquid nitrogen for subsequent protein and mRNA analyses, or were used for primary isolation of alveolar type II pneumocytes as described below.
Alveolar type II pneumocyte isolation and alveolar epithelial monolayer cultures
Alveolar type II pneumocytes from control- and alcohol-fed rats were isolated using an established protocol in our laboratory (Pelaez et al., 2004). Briefly, after lungs were removed en bloc following tracheostomy, alveolar macrophages were removed by lavaging the lungs with 40 ml PBS (pH 7.4) and then the lungs were filled with an elastase solution to dissociate cells. The lung tissue was minced in a solution containing DNAse I and newborn calf serum and successively filtered through 100- and 20-μM nylon mesh. The cell suspension was plated on bacteriological plastic plates coated with IgG to remove remaining alveolar macrophages. After 1 h of incubation at 37° C the non-adherent type II pneumocytes were carefully collected for experiments. Cells obtained by this method were always >95% viable by trypan blue exclusion. This established technique in our previous studies has demonstrated the following phenotype: >90% keratin positive, ~90% SP-C positive, <1% CD14 (macrophage marker) positive, <0.2% CD32 (alveolar macrophage marker) positive, and <1% vimentin (fibroblast marker) positive by flow cytometric analyses (Joshi et al., 2006). To establish alveolar epithelial monolayers in vitro, freshly isolated type II pneumocytes were re-suspended at DMEM/F-12 containing 10% serum and then plated on 6-well plates and cultured at 37°C in 90% air/10% CO2. The medium was changed every 48 hrs, and after 7 days in culture the cells form confluent monolayers that are a mixture of cuboidal type II pneumocytes and large flat cells that have a characteristic type I pneumocyte-like appearance and express type I pneumocyte markers including aquaporins (Dobbs et al., 1988; Borok et al., 1998b).
Western immunoblot
Whole lung or epithelial cell lysates were prepared as described previously [Joshi et al., 2006]. 30 - 50 μg of protein from each sample was loaded onto a 12% SDS-PAGE gel and electrophoresed at 150 volts for 75 min. The separated proteins were transferred to a 0.45 μM polyvinylidine difluoride (PVDF) membrane at 15 volts for 75 min. Membranes were blocked at RT for 1 hr in Tris-buffered saline with 0.2% Tween 20 (TBS-T) containing 5% non-fat dry milk (blocking buffer) and then incubated with primary antibodies for each specific claudin protein (Invitrogen) at 1:500 at 4°C overnight. After incubating at RT with horseradish peroxidase-labeled anti-rabbit IgG antibody for 2h, blots were exposed in ECL plus chemiluminescence reagent (Amersham, Arlington Heights, IL). Immunoblots were stripped and re-probed with anti-GAPDH antibody. The densities of both the claudin band and GAPDH band were quantified using a BioRad Imaging System.
RNA isolation and real-time PCR
Total RNA was extracted using Qiagen RNeasy Mini Kit (Valencia, CA) and treated with Qiagen DNase I to eliminate contaminating genomic DNA. Reverse transcription was performed with 1 μg of total RNA in total volume of 20 μl per reaction in Bio-Rad iScript cDNA synthesis kit (Hercules, CA). Real-time PCR was carried on the Bio-Rad iCycler. Amplification was in 25 μl containing primers 0.5 μM in Bio-Rad iQ SYBR green supermix. Aliquots of cDNA were diluted 10-10,000 fold to generate relative standard curves to which sample cDNA was compared. Standards and samples were run in triplicate. For claudin-1, forward and reverse primers were 5’-CCT CCA ATG CCG TTC TGT AT-3’ and 5’- AGG GCC TTT GCT ACA GAT GA-3’, creating a product of 118 bp. A 164 bp of claudin-5 amplicon was generated from forward primer 5’-CAC AGA GAG GGG TCG TTG AT-3’ and reverse primer 5’-ACT GTT AGC GGC AGT TTG GT’. Claudin-7 forward and reverse primers were 5’-GTC TGC TCT GGT CCT TCT GG-3’ and primer 5’-GTC CCC AGC TCA CAC GTA TT-3’ respectively, producing a 133 bp amplicon. QuantumRNA class II 18S primers were purchased from Ambion (Austin, TX). PCR amplicons from all species were normalized for the amount of 18S in the same RT sample, which was also standardized on a dilution curve from the RT samples. Dilution curve showed the real-time PCR efficiency was >95% for all genes analyzed.
Immunocytochemistry
Anti-claudin antibodies (Invitrogen) are routinely tested in our laboratory for cross-reactivity using cells transfected with claudin cDNAs (Wang et al., 2003). In this study, we compared claudin-7 localization in alveolar epithelial cell monolayers from control-fed and alcohol-fed rats. In order to perform immunocytochemistry, freshly isolated type II pneumocytes were cultured on glass coverslips for 7 days to form monolayers; the monolayers were then washed with PBS, fixed and permeabilized with MeOH/acetone 1:1 for 2 min and then washed with PBS + 0.5% Triton X-100 (PBS/TX) and PBS + 0.5% Triton X-100 + 2% goat serum (PBS/TX/GS). The cells were incubated with primary antiserum diluted into PBS/GS for 1 h at RT, washed, and then incubated with Cy2-conjugated goat anti-rabbit IgG and Cy3-conjugated goat anti-mouse IgG (Jackson Immunoresearch, Malvern, PA) for 1 h at RT. The images were captured by using an Olympus IX-70 inverted fluorescence microscope outfitted with a Hamamatzu Orca charge-coupled device and then analyzed with Image Pro software.
Statistical analyses
Data are presented as mean ± SEM and were subjected to Student t-Test test for comparison of control-fed rats with alcohol-fed rats. In this study the actual P values that were < 0.10 are provided in the figures; otherwise if not shown the P vales were ≥ 0.10. P values <0.05 were considered statistically significant.
Results
Effects of alcohol ingestion on claudin expression in the whole lung
As individual claudins are relatively expressed in tissue-specific and even cell-specific fashion, our first step was to measure the expression of multiple claudin proteins in the lungs of control-fed and alcohol-fed rats. As shown in Figure 1, alcohol ingestion significantly decreased the protein expression of claudin-1 and claudin-3 (P<0.05). Claudin-7 expression also appeared to be decreased (P=0.06). In contrast, alcohol ingestion had no effect on claudin-4 expression, and there was actually a trend toward increased expression of claudin-5 (P = 0.09). Claudin-18 protein was not detected in the whole cell lysates from either control-fed or alcohol-fed rats (ND). In each case the data shown are the mean ± SEM from at least six rats. Although chronic alcohol ingestion decreased protein expression of claudin-1 and claudin-3 (and possibly claudin-7), mRNA expression for claudin-1, -3, and -7 did not show a decrease in the whole lung level as assessed by real-time PCR (not shown). This is in contrast to our findings in alveolar epithelial monolayers, in which alcohol decreased mRNA expression for claudin-1, -3, and -7 (see below and Figure 4). Although this may be explained by the fact that there are so many different cell types within the lung tissue and the source of RNA was from its mixed population of cells, within the context of this study this remains a speculation.
Figure 1.
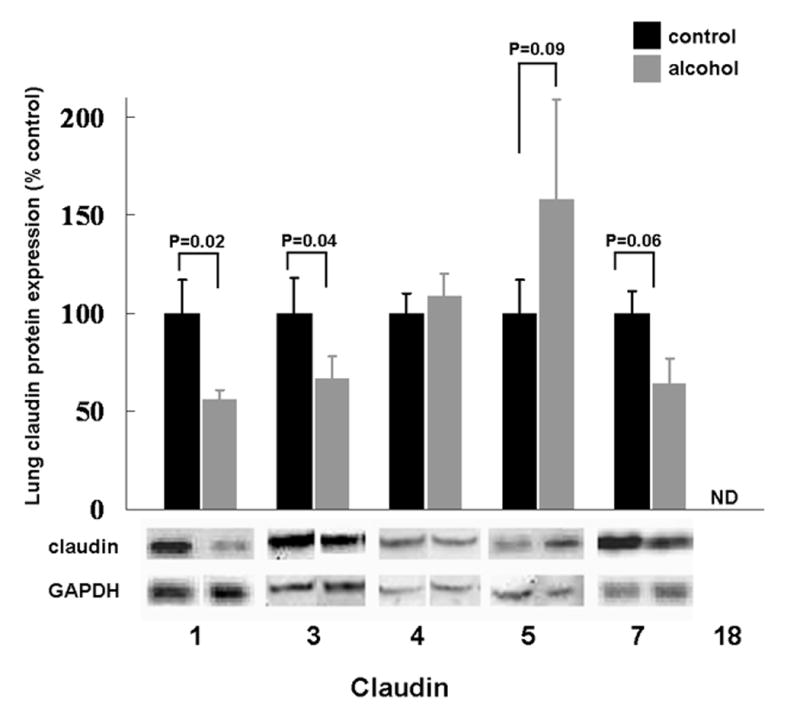
The relative protein expression, as determined by western immunoblot analysis, of claudin-1, -3, -4, -5, -7, and -18 in the lung tissue of rats fed either the Lieber-DeCarli liquid diet with 36% of the total calories as maltin-dextrin (control) or an isocaloric Lieber-DeCarli liquid diet with 36% of the total calories as ethanol (alcohol). For each specific claudin the relative amount of protein was quantified by determining the densitometry of the bands on the western immunoblots, and then normalized to the densitometry of the GAPDH band from the same sample. Each claudin in the alcohol-fed group was then expressed as a percentage of change compared to the lung tissue from control-fed rats. Claudin-18 protein was not detectable (ND) in the lung tissue from either dietary group. Each value represents the mean ± SEM of at least six individual samples, and the P values that were < 0.10 for comparisons between the control-fed and alcohol-fed rats are shown. Representative immunoreactive bands for the respective claudins are shown below the summary data.
Figure 4.
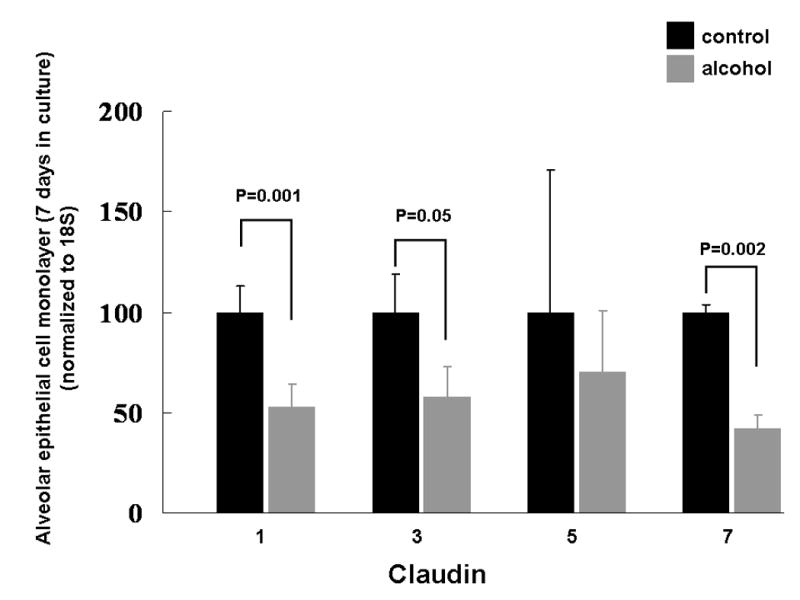
The relative gene expression of claudin-1, -3, -5, and -7, as determined by real-time PCR (see text for details), in alveolar epithelial monolayers (7 days in culture) derived from type II pneumocytes isolated from rats fed either the Lieber-DeCarli liquid diet with 36% of the total calories as maltin-dextrin (control) or an isocaloric Lieber-DeCarli liquid diet with 36% of the total calories as ethanol (alcohol). The alveolar epithelial monolayers include cells that have predominantly a type II phenotype and cells that have predominantly a type I phenotype. mRNA expression for each of the claudins was normalized to 18S and then expressed relative change to control monolayers. Each value represents the mean ± SEM of at least six rats, and the P values that were < 0.10 for comparisons between the control-fed and alcohol-fed rats are shown.
Effects of alcohol ingestion on claudin expression in freshly isolated alveolar type II pneumocytes
Based on changes in the expression of some claudin proteins in lungs of alcohol-fed rats, we next focused on whether there were changes in claudin protein expression within alveolar epithelial cells. We first examined the relative expression of claudin-1, -3, -4, -5, -7, and -18 in freshly isolated alveolar type II pneumocytes from control-fed and alcohol-fed rats. Perhaps somewhat surprisingly and in contrast to what was observed in the whole lung, chronic alcohol ingestion appeared to have no significant effects on the expression of claudin-1, claudin-3 or claudin-7 (Figure 2). In contrast, the trend toward increased expression of claudin-5 in the lungs of alcohol-fed rats was associated with increased (P=0.02) expression of claudin-5 in freshly isolated type II pneumocytes. However, as shown in Figure 2, claudin-5 protein was barely detectable in type II pneumocytes from control-fed rats, but this low-level expression did increase more than 2-fold in alcohol-fed rats (although as shown in the representative immunoreactive band below the summary data, claudin-5 protein even in the cells from alcohol-fed rats was not abundant). In each case, the data shown are the mean ± SEM of at least six rats. Note that claudin-18 protein, which is relatively more specific for alveolar epithelial cells, was detectable in these type II pneumocytes. In parallel to the absence of any meaningful effects of alcohol on claudin expression in type II pneumocytes shown in Figure 2 (with the exception of an increase in claudin-5), alcohol ingestion had no effect on claudin-1, -3, -5, or -7 mRNA expression as determined by real-time PCR (not shown). Overall, the alcohol-induced changes in the pattern of claudin protein expression observed in the whole lung (as shown in Figure 1) did not appear to be explained by changes in claudin protein expression in the type II pneumocytes.
Figure 2.

The relative protein expression, as determined by western immunoblot analysis, of claudin-1, -3, -4, -5, -7, and -18 in alveolar epithelial type II pneumocytes freshly isolated from rats fed either the Lieber-DeCarli liquid diet with 36% of the total calories as maltin-dextrin (control) or an isocaloric Lieber-DeCarli liquid diet with 36% of the total calories as ethanol (alcohol). For each specific claudin the relative amount of protein was quantified by determining the densitometry of the bands on the western immunoblots, and then normalized to the densitometry of the GAPDH band from the same sample. Each claudin in the alcohol-fed group was then expressed as a percentage of change compared to the lung tissue from control-fed rats. Claudin-5 protein was barely detectable in type II pneumocytes from control-fed rats, but this low-level expression did increase more than 2-fold in alcohol-fed rats (although as shown in the representative immunoreactive band below the summary data, claudin-5 protein even in the cells from alcohol-fed rats was not abundant). In contrast to the whole lung (Figure 1), claudin-18 protein expression was detected in type II pneumocytes. Each value represents the mean ± SEM of at least six rats. There were no significant differences (P≥ 0.05) between the cells from control-fed and alcohol-fed rats for any of the claudins analyzed. Representative immunoreactive bands for the respective claudins are shown below the summary data.
Effects of alcohol ingestion on claudin expression in alveolar epithelial monolayers
As type I pneumocytes actually account for ~95% of the alveolar surface and therefore most of the alveolar epithelial barrier, we proposed that chronic alcohol ingestion might alter claudin expression in these cells, particularly as there were no apparent effects on claudin protein expression in the type II pneumocytes, with the exception of claudin-5 (Figure 2). Ideally, we would have performed western blot analyses for claudin protein expression in freshly isolated type I pneumocytes. Although it has been shown to be technically possible to isolate type I pneumocytes (Dobbs et al., 1998), this method is not widely available. Therefore, we employed a standard technique that we and others have used in which freshly isolated type I pneumocytes are cultured for 6-7 days, by which time they form monolayers that consist primarily of flat cells that resemble type I pneumocytes and that express type I pneumocyte-specific markers such as aquaporins (Dobbs et al., 1988; Borok et al., 1998b). Using this approach, we identified that the alcohol-induced changes in claudin protein expression observed in the whole lungs (Figure 1) were strikingly similar to changes seen in these alveolar epithelial monolayers. Specifically, and as shown in Figure 3, alveolar epithelial monolayers derived from alcohol-fed rats had significantly (P<0.05) decreased expression of claudin-1, -7, and -18 proteins, and there was a trend toward decreased expression of claudin-3 protein (P=0.07). In contrast, alcohol had no effect on claudin-4 protein expression. Finally, alveolar epithelial monolayers derived from alcohol-fed rats had significantly (P<0.05) increased expression of claudin-5 protein. In each case the data shown are the mean ± SEM for alveolar epithelial monolayers derived from at least six rats. In contradistinction to our findings in the whole lungs, the effects of alcohol on the relative mRNA expression of claudin-1, -3, and -7 correlated to the protein expression in the alveolar epithelial monolayers. Specifically, and as shown in Figure 4, alveolar epithelial monolayers derived from alcohol-fed rats had a significant (P<0.05) decrease in claudin-1 and claudin-7 gene expression compared to monolayers from control-fed rats, and claudin-3 gene expression bordered on being significantly decreased (P=0.05). In contrast, claudin-5 gene expression was the same in monolayers from control-fed and alcohol-fed rats. These differences in mRNA expression and protein content for claudin-5 could be due to post-translation mechanisms, but this was not explored in this study. Overall, at the level of the alveolar epithelial monolayer where the majority of the physical barrier is comprised of type I pneumocytes, our data suggest that chronic alcohol ingestion altered both the gene and protein expression for several claudins. As these claudins are the key components of the tight junctions within the alveolar epithelial barrier, these changes can plausibly be linked to alcohol-induced permeability in the lung.
Figure 3.
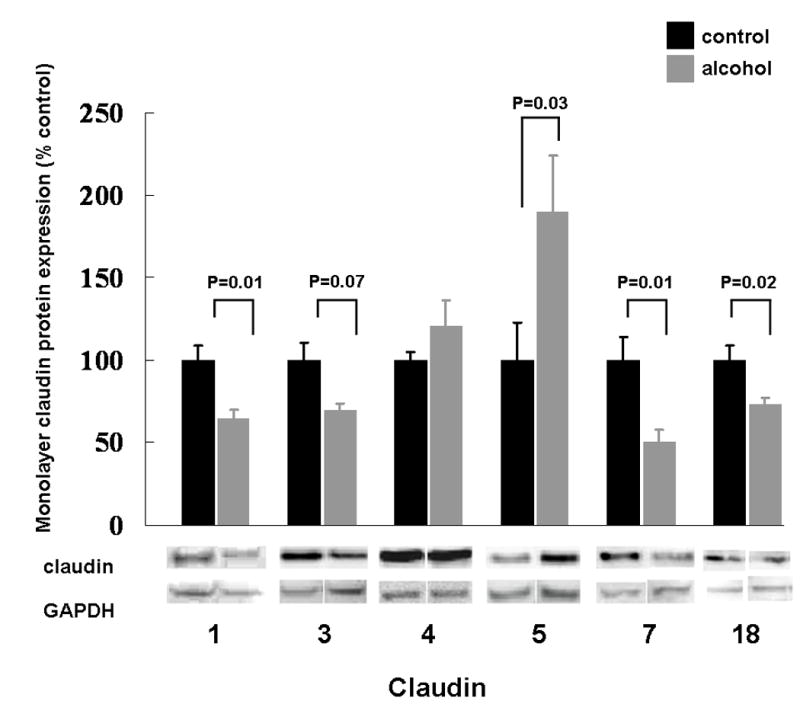
The relative protein expression, as determined by western immunoblot analysis, of claudin-1, -3, -4, -5, -7, and -18 in alveolar epithelial monolayers (7 days in culture) derived from type II pneumocytes isolated from rats fed either the Lieber-DeCarli liquid diet with 36% of the total calories as maltin-dextrin (control) or an isocaloric Lieber-DeCarli liquid diet with 36% of the total calories as ethanol (alcohol). The alveolar epithelial monolayers include cells that have predominantly a type II phenotype and cells that have predominantly a type I phenotype. For each specific claudin the relative amount of protein was quantified by determining the densitometry of the bands on the western immunoblots, and then normalized to the densitometry of the GAPDH band from the same sample. Each claudin in the alcohol-fed group was then expressed as a percentage of change compared to the lung tissue from control-fed rats. Each value represents the mean ± SEM of at least six rats, and the P values that were < 0.10 for comparisons between the control-fed and alcohol-fed rats are shown. Representative immunoreactive bands for the respective claudins are shown below the summary data.
Effects of alcohol ingestion on claudin-7 localization by immunocytochemistry in alveolar epithelial monolayers
Previously, we have reported that chronic alcohol ingestion increases alveolar epithelial permeability in vivo as well as in alveolar epithelial monolayers in vitro (Guidot et al., 2000). Those previous findings, coupled with the findings in this study, suggested that alcohol ingestion alters tight junction assembly and/or function in the alveolar epithelium. Therefore, we extended the current study and examined the relative cellular localization of claudin-7 protein in alveolar epithelial monolayers derived from control-fed and alcohol-fed rats. Claudin-7 was chosen for immunocytochemistry study because its expression is relatively specific for alveolar epithelial type I pneumocytes (as is claudin-1), and because it was significantly inhibited by chronic alcohol ingestion. As shown in Figure 5, Claudin-7 was localized primarily within the plasma membranes in the alveolar epithelial cell monolayers from control-fed rats (left panel). In contrast, claudin-7 protein was irregularly distributed within the plasma membranes of alveolar epithelial monolayers from alcohol-fed rats (right panel). In parallel, monolayers from alcohol-fed rats had multiple foci of intracellular accumulations of claudin-7 (see right panel and magnified area in insert). Overall, these images suggest that at least qualitatively, alcohol impaired cell membrane localization of claudin-7 protein.
Figure 5.
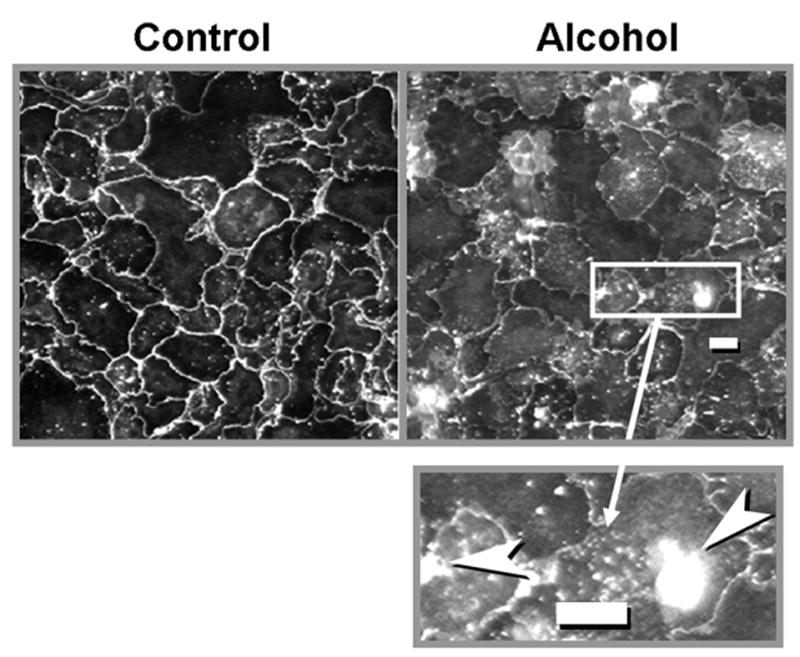
Immunocytochemical localization of claudin-7 protein (see text for details), in alveolar epithelial monolayers from control-fed rats (left panel) and alcohol-fed rats (right panel). As is evident in these representative images, claudin-7 protein is localized primarily to the cell membranes in the control monolayers on the left. In contrast, alcohol monolayers showed patchy localization with many areas where little or no claudin-7 could be visualized in cell membranes. The inset in the lower right is a magnified image of part of the right panel, and illustrates the areas of intracellular accumulation of claudin-7 that were seen in the monolayers from alcohol-fed rats. The size bar in the right panel and in the inset represents 10 microns.
Discussion
Chronic alcohol abuse significantly increases the risk of developing ARDS in response to acute stresses such as sepsis or trauma (Moss et al., 1996; Moss et al., 2003; Moss and Burnham, 2003). A cardinal feature of ARDS is increased alveolar epithelial permeability leading to alveolar flooding with fluid and proteins that impair normal gas exchange and cause respiratory failure (Ware and Matthay, 2000; Ware and Matthay, 2001). We had previously shown in this same experimental rat model that chronic alcohol ingestion alone, even in the absence of any acute stresses such as sepsis, significantly increases alveolar epithelial permeability (Guidot et al., 2000). Although we have also described multiple defects in alveolar epithelial function mediated by chronic alcohol ingestion (Brown et al., 2001b; Brown et al., 2001a; Guidot and Brown, 2000; Joshi et al., 2006), to date we had not identified a discrete mechanism by which alcohol disrupts the alveolar epithelial barrier. As tight junctions mediate the cell-cell interactions that regulate paracellular permeability, we speculated that alcohol targets these complexes.
This study provides new evidence that alcohol-induced alveolar epithelial barrier dysfunction is associated with changes in claudin protein expression and localization within the type I pneumocyte. Specifically, we determined that chronic alcohol ingestion decreased both gene and protein expression of claudin-1 and claudin-7, two critical components of type I pneumocyte tight junctions, in alveolar epithelial monolayers derived from these rats. In addition, there was a strong trend toward decreased expression of claudin-3 in alveolar epithelial monolayers derived from alcohol-fed rats. In parallel, alcohol increased the protein expression of claudin-5, which we have shown in other systems to be associated with leakier epithelial barriers (Wang et al., 2003). These findings are consistent with a previous report in which claudin-1 and claudin-3 decreased solute permeability, and claudin-5 increased permeability, in NIH/3T3 fibroblasts and in IB3.1 human airway cells (Coyne et al., 2003). Finally, alcohol decreased the expression of claudin-18 protein, another claudin with relatively increased expression in epithelial cells. However, as with claudin-1 and claudin-7, claudin-18 was decreased only within the alveolar epithelial monolayers. The immunocytochemistry analyses of claudin-7 suggest that alcohol impairs claudin localization within the cell membrane of the alveolar epithelium. Although it remains unknown at present just how alcohol-induced changes in claudin protein expression and localization could be affecting alveolar epithelial barrier function, our findings are consistent with other recent reports showing reduced claudin-7 expression from breast and esophageal carcinoma that correlated with higher tumor grade and metastases (Sauer et al., 2005; Usami et al., 2006). Finally, the general pattern of altered claudin expression in the alveolar epithelial monolayers (Fig. 3) was mirrored in the whole lung (Fig. 1). As claudin-1 and claudin-7 in particular are relatively specific for type I pneumocytes (Daugherty et al., 2004), this argues that the observed effects in alveolar epithelial monolayers in vitro are relevant to alcohol-induced alveolar epithelial barrier dysfunction in vivo. Taken together, these findings suggest that chronic alcohol ingestion impairs the expression and formation of tight junctions in the alveolar epithelium, and that these effects (at least for the claudins examined in this study) are likely specific to the type I pneumocyte.
Although these findings are consistent with the observed effects of chronic alcohol ingestion on alveolar epithelial permeability, they were initially somewhat surprising. Specifically, with the exception of an increase in claudin-5 expression (which as noted has been associated with increased epithelial permeability), the expression of other claudin proteins in the type II pneumocyte, particularly claudin-1 and claudin-7, were not affected by chronic alcohol ingestion. This is in contrast to myriad alcohol-induced defects in type II pneumocyte function we have identified previously including surfactant synthesis and secretion, signaling receptor expression, and glutathione homeostasis and response to inflammatory stimuli (Bechara et al., 2003; Brown et al., 2001b; Guidot and Brown, 2000; Holguin et al., 1998; Joshi et al., 2006). Further, type II pneumocytes are the progenitor cells for type I pneumocytes in vivo and the alveolar epithelial monolayers in our studies were formed by type II pneumocytes that assume a type I pneumocyte-like phenotype after several days in culture. There are many previous reports demonstrating that type II pneumocytes lose many of their characteristic features such as surfactant protein expression and lamellar bodies and acquire the appearance and characteristics of type I pneumocytes, including aquaporin-5 (AQP-5) and RTI40 expression (Dobbs et al., 1985; Dobbs et al., 1988; Borok et al., 1998b; Borok et al., 1998a; Campbell et al., 1999; Danto et al., 1995; Wang et al., 2003). In this study, the alcohol-induced changes in claudin gene and protein expression we identified appear to be manifested primarily in the type I pneumocyte, but not in the type II pneumocytes from which they develop. This suggests that chronic alcohol ingestion somehow impairs the programming of claudin expression as type II pneumocytes transdifferentiate into type I pneumocytes. Further, these effects persist even when the type II pneumocytes are isolated and cultured in the absence of alcohol. Therefore, it is not likely to be a direct effect of alcohol per se, but rather it is likely mediated by a consequence of chronic alcohol ingestion that persists long after the alcohol is removed from the system. Although we do not yet know how alcohol mediates these effects, one distinct possibility is oxidant stress. We showed previously that type II pneumocytes isolated from alcohol-fed rats have profound glutathione depletion and oxidant stress that does not resolve even after 7-8 days in culture in the absence of alcohol (Guidot and Brown, 2000). It is plausible that as the type I pneumocyte phenotype develops and their tight junctions require a different combination of claudins, particularly claudin-1 and claudin-7, the alcohol-induced chronic oxidant stress interferes with this programmed change. Whether or not this could be mediated by direct oxidative damage to claudin proteins, or by another mechanism such as the actions of transforming growth factor β1 (Bechara et al., 2004; Bechara et al., 2005), is at present unknown.
These alcohol-mediated changes in claudin expression, and the associated increase in paracellular permeability in the alveolar epithelium, must be reconciled with the fact that alcohol abuse alone does not cause pulmonary edema. Specifically, there is no clinical evidence that alcohol abuse by itself can cause lung injury in humans, and the alcoholic rat lung is not more prone to edema in the absence of an acute inflammatory stress such as endotoxin (Holguin et al., 1998). We have shown previously that in contrast to the disruptions in the paracellular barrier, chronic alcohol ingestion actually augments the fluid transport capacity of the alveolar epithelium, in part by increasing epithelial sodium channel density and function on the apical surface (Guidot et al., 2000). Therefore, it appears that the paracellular leak into the alveolar space is compensated for by increased net vectorial transport of salt and water back out of the airspace into the pulmonary interstitial compartment. However, during acute stresses such as sepsis, the paracellular leak increases under the influence of inflammatory mediators such as transforming growth factor β1 (Bechara et al., 2004; Bechara et al., 2005), and overwhelms the compensatory fluid transport that limits edema formation in the otherwise healthy alcoholic.
Although future studies will be necessary to examine the precise mechanisms by which alcohol abuse perturbs the alveolar epithelial barrier, this study is important because it is a first step in identifying the molecular basis by which alcohol abuse impairs alveolar epithelial paracellular permeability, and could have ramifications for our understanding of alcohol-induced alterations of other biological barriers such as the gut epithelium. Further, as the complexities of the tight junctions that mediate tissue barriers continue to be elucidated, there will be exciting opportunities to identify how alcohol abuse interferes with their assembly and function and thereby renders even otherwise healthy individuals susceptible to acute lung injury.
Acknowledgments
This work was funded by a Merit Review from the Department of Veterans Affairs, the National Institute on Alcohol Abuse and Alcoholism (P50 AA013757, T32 AA013528) and the National Heart Lung and Blood Institute (R01 HL083120). The authors wish to thank Robert Raynor and Kylene Werner for their technical expertise. None of the authors have any commercial affiliations or conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bechara RI, Brown LAS, Eaton DC, Roman J, Guidot DM. Chronic ethanol ingestion increases expression of the angiotensin II type 2 (AT2) receptor and enhances tumor necrosis factor-α- and angiotensin II-induced cytotoxicity via AT2 signaling in rat alveolar epithelial cells. Alcohol Clin Exp Res. 2003;27(6):1006–1014. doi: 10.1097/01.ALC.0000071932.56932.53. [DOI] [PubMed] [Google Scholar]
- Bechara RI, Brown LAS, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170:188–194. doi: 10.1164/rccm.200304-478OC. [DOI] [PubMed] [Google Scholar]
- Bechara RI, Pelaez A, Palacio A, Joshi PC, Hart CM, Brown LA, Raynor R, Guidot DM. Angiotensin II mediates glutathione depletion, transforming growth factor beta1 expression, and epithelial barrier dysfunction in the alcoholic rat lung. Am J Physiol Lung Cell Mol Physiol. 2005;289:L363–L370. doi: 10.1152/ajplung.00141.2005. [DOI] [PubMed] [Google Scholar]
- Borok Z, Danto SI, Lubman RL, Cao Y, Williams MC, Crandall ED. Modulation of t1alpha expression with alveolar epithelial cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol. 1998a;272:L155–L164. doi: 10.1152/ajplung.1998.275.1.L155. [DOI] [PubMed] [Google Scholar]
- Borok Z, Lubman RL, Danto SI, Zhang XL, Zabski SM, King LS, Lee DM, Agre P, Crandall ED. Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin 5. Am J Respir Cell Mol Biol. 1998b;18:554–561. doi: 10.1165/ajrcmb.18.4.2838. [DOI] [PubMed] [Google Scholar]
- Brown LAS, Harris FL, Bechara R, Guidot DM. Effect of chronic ethanol ingestion on alveolar type II cell glutathione and inflammatory mediator-induced apoptosis. Alcoholism: Clinical and Experimental Research. 2001a;25(7):1078–1085. [PubMed] [Google Scholar]
- Brown LAS, Harris FL, Guidot DM. Chronic ethanol ingestion potentiates TNF- mediated oxidative stress and apoptosis in rat alveolar type II cells. Am J Physiol Lung Cell Mol Physiol. 2001b;281:L377–L386. doi: 10.1152/ajplung.2001.281.2.L377. [DOI] [PubMed] [Google Scholar]
- Campbell L, Hollins AJ, Al-Eid A, Newman GR, von Ruhland C, Gumbleton M. Caveolin-1 expression and caveolae biogenesis during cell transdifferentiation in lung alveolar epithelial primary cultures. Biochem Biophys Res Commun. 1999;262:744–751. doi: 10.1006/bbrc.1999.1280. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166–L1178. doi: 10.1152/ajplung.00182.2003. [DOI] [PubMed] [Google Scholar]
- Danto SI, Shannon JM, Borok Z, Zabski SM, Crandall ED. Reversible transdifferentiation of alveolar epithelial cells. Am J Respir Cell Mol Biol. 1995;12:497–502. doi: 10.1165/ajrcmb.12.5.7742013. [DOI] [PubMed] [Google Scholar]
- Daugherty BL, Mateescu M, Patel AS, Wade K, Kimura S, Gonzales LW, Guttentag S, Ballard PL, Koval M. Developmental regulation of claudin localization by fetal alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1266–L1273. doi: 10.1152/ajplung.00423.2003. [DOI] [PubMed] [Google Scholar]
- Dobbs LG, Gonzalez R, Matthay MA, Carter EP, Allen L, Verkman AS. Highly water-permeable type I alveolar epithelial cells confer high water permeability between the airspace and vasculature in rat lung. Proc Natl Acad Sci USA. 1998;95:2991–2996. doi: 10.1073/pnas.95.6.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs LG, Williams MC, Brandt AE. Changes in biochemical characteristics and pattern of lectin binding of alveolar type II cells with time in culture. Biochim Biophys Acta. 1985;846:155–166. doi: 10.1016/0167-4889(85)90121-1. [DOI] [PubMed] [Google Scholar]
- Dobbs LG, Williams MC, Gonzalez R. Monoclonal antibodies specific to apical surfaces of alveolar type I cells bind to surfaces of cultured, but not freshly isolated, type II cells. Biochim Biophys Acta. 1988;970:146–156. doi: 10.1016/0167-4889(88)90173-5. [DOI] [PubMed] [Google Scholar]
- Frutos-Vivar F, Ferguson ND, Esteban A. Epidemiology of acute lung injury and acute respiratory disease syndrome. Semin Resp Crit Care Med. 2006;27:327–336. doi: 10.1055/s-2006-948287. [DOI] [PubMed] [Google Scholar]
- Guidot DM, Brown LAS. Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed rats. Alcohol Clin Exp Res. 2000;24(7):1070–1076. [PubMed] [Google Scholar]
- Guidot DM, Modelska K, Lois M, Jain L, Moss IM, Pittet J-F, Brown LAS. Ethanol ingestion via glutathione depletion impairs alveolar epithelial barrier function in rats. Am J Physiol Lung Cell Mol Physiol. 2000;279:L127–L135. doi: 10.1152/ajplung.2000.279.1.L127. [DOI] [PubMed] [Google Scholar]
- Holguin F, Moss IM, Brown LS, Guidot DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest. 1998;101:761–768. doi: 10.1172/JCI1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Mitchell PO, Fernainy K, Roman J, Eaton DC, Guidot DM. GM-CSF receptor expression and signaling is decreased in lungs of ethanol-fed rats. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1150–L1158. doi: 10.1152/ajplung.00150.2006. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Ritzenthaler JD, Roman J, Fernandez AL, Eaton DC, Brown LA, Guidot DM. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J Immunol. 2005;175:6837–6845. doi: 10.4049/jimmunol.175.10.6837. [DOI] [PubMed] [Google Scholar]
- Levitt JE, Matthay MA. Treatment of acute lung injury: historical perspective and potential future therapies. Semin Resp Crit Care Med. 2006;27:426–438. doi: 10.1055/s-2006-948296. [DOI] [PubMed] [Google Scholar]
- Licker M, de Perrot M, Spiliopoulos A, Robert J, Diaper J, Chevalley C, Tschopp J-M. Risk factors for acute lung injury after thoracic surgery for lung cancer. Anesth Analg. 2003;97:1558–1565. doi: 10.1213/01.ANE.0000087799.85495.8A. [DOI] [PubMed] [Google Scholar]
- Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of Acute Respiratory Distress Syndrome in adults. JAMA. 1996;275(1):50–54. [PubMed] [Google Scholar]
- Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med. 2003;31(4 Suppl):S207–S212. doi: 10.1097/01.CCM.0000057845.77458.25. [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, Cotsonis GA. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003;31(3):869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- Pelaez A, Bechara RI, Joshi PC, Brown LAS, Guidot DM. Granulocyte/macrophage colony-stimulating factor treatment improves alveolar epithelial barrier function in alcoholic rat lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L106–L111. doi: 10.1152/ajplung.00148.2003. [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Sauer T, Pederson MK, Ebeltoft K, Naess O. Reduced expression of claudin-7 in fine needle aspirates from breast carcinomas correlate with grading and metastatic disease. Cytopathology. 2005;16:193–198. doi: 10.1111/j.1365-2303.2005.00257.x. [DOI] [PubMed] [Google Scholar]
- Usami Y, Chiba H, Nakayama F, Ueda J, Matsuda Y, Sawada N, Komori T, Ito A, Yokozaki H. Reduced expression of claudin-7 correlates with invasion and metastasis in squamous cell carcinoma of the esophagus. Hum Path. 2006;37:569–577. doi: 10.1016/j.humpath.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Velasquez A, Bechara R, Lewis JF, Malloy J, McCaig L, Brown LAS, Guidot DM. Glutathione replacement preserves the functional surfactant phospholipid pool size and decreases sepsis-mediated lung dysfunction in ethanol-fed rats. Alcohol Clin Exp Res. 2002;26(8):1245–1251. doi: 10.1097/01.ALC.0000024269.05402.97. [DOI] [PubMed] [Google Scholar]
- Wang F, Daugherty B, Keise LL, Wei Z, Foley JP, Savani RC, Koval M. Heterogeneity of claudin expression by alveolar epithelial cells. Am J Respir Cell Mol Biol. 2003;29:62–70. doi: 10.1165/rcmb.2002-0180OC. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163(6):1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]