

Abstract
Background
Intimal hyperplasia remains the principal lesion in the development of restenosis after vessel wall injury. Cell signaling in vascular smooth muscle cells remains a potential molecular target to modulate the development of intimal hyperplasia. The aim of this study is to define a baseline pattern of histological changes and kinase activation in a murine model.
Methods
The murine femoral wire injury model was employed in which a micro wire was passed through a branch of the femoral artery and used to denude the common femoral artery. Pluronic gel was used to apply MAPK inhibitors (PD98059, SB230580 and SP600125) on the exterior of the vessels. Specimens were perfusion-fixed and sections were stained for morphometry using an ImagePro system. Additional specimens of femoral artery were also harvested and snap frozen for western blotting and zymography to allow for the study of kinase and protease activation. Contralateral vessels were used as controls
Results
The injured femoral arteries developed intimal hyperplasia, which is maximal at 28 days and does not change substantially between day 28 and day 56. Sham operated vessels did not produce such a response. Cell apoptosis peaked within 3 days and cell proliferation peaked at 7 days after injury. There is a time dependent increase in kinase activity immediately after injury. MEK1/2 activation peaks at 20 mins after injury and is followed by a peak in ERK1/2 activation at 45 mins. The stress kinases p38MAPK and JNK peak between 10 and 20 mins. Activation of akt is later at 45mins and 120 mins and activation of p70S6K was biphasic. There was a time dependent increase in uPA/PAI-1 expression and activity after injury. Local application of MAPK inhibitors (PD98059, SB230580 and SP600125) within a pluronic gel reduced respective MAPK activity, decreased cell proliferation and enhanced cell apoptosis, increased PAI-1 and decreased uPA expression and activity; at 14 days there was a decrease in intimal hyperplasia.
Conclusions
These data demonstrate that femoral wire injury in the mouse induces a consistent model of intimal hyperplasia and that it is associated with a time dependent increase in signaling kinase activity. Interruption of these pathways will interrupt uPA/ PAI-1 pathway and decrease intimal hyperplasia development. Accurate characterization of cell signaling is a necessary step in the development of molecular therapeutics
INTRODUCTION
The introduction and widespread use of endoluminal therapies (angioplasty and intravascular stenting) and the subsequent reports of high restenosis rates have increased awareness of the significance of vessel remodeling in the current interventional environment (1). As a result, there has been an increased stimulus to study the biology and pathophysiology of a vessel’s response to injury. Intimal hyperplasia is the universal response of vessels to injury and involves abnormal migration and proliferation of vascular smooth muscle cells with the associated deposition of extracellular connective tissue matrix that is then followed by remodeling of this new tissue (1). A protease in remodeling is urokinase (uPA) and Plasminogen Activator -1 (PAI-1). The biology of a vessel’s response to injury is complex and has many biological tangents that testify to the therapeutic dilemma it poses to control. Cell signaling in vascular smooth muscle cells remains a potential molecular target to modulate the development of intimal hyperplasia (2). The aim of this study is to characterize the cell kinetics and the early cell signaling in the murine femoral artery model, to examine their relationship to uPA/PAI-1 expression and activity and to determine if interruption of MAPK activity will effect lesion progression and uPA/PAI-1 pathway.
METHODS
Experimental Design
6 week old male FVB mice underwent femoral wire injury model and were harvested at various time points over a 56 day period. Specimens were perfusion-fixed and sections were stained for morphometry using an ImagePro system. Additional specimens of femoral artery were also harvested and snap frozen for western blotting and zymography to allow for the study of kinase and protease activation. Additional vessels were also coated in pluronic gel with and without MAPK kinase inhibitors (PD98059 – ERK inhibitor; SB230580 – p38MAPK inhibitor; and SP600125 - JNK inhibitor) and harvested at various time points to determine the impact of local inhibitor therapy. Contralateral vessels were used as controls. Animal care and procedures were conducted at the University of Rochester Medical Center in accordance with state and federal laws and under protocols approved by the University of Rochester Animal Care and Use Committee. Animal care complied with the “Guide for the Care and Use of Laboratory Animals” issued by the Institute of Laboratory Animal Resources.
Femoral wire injury
Endoluminal injury to the common femoral artery was produced by 3 passages of a 0.25-mm-diameter angioplasty guidewire (Advanced Cardiovascular Systems) (3). While being viewed under a surgical microscope, a groin incision was made and the femoral artery is dissected free and temporarily clamped at the level of the inguinal ligament while an arteriotomy was made on the profunda branch. The guidewire is then inserted, the clamp was removed, and the wire was advanced to the level of the aortic bifurcation and pulled back. After removal of the wire, the arteriotomy site was ligated. The skin incision was closed in one layer with a 6-0 interrupted vicryl simple sutures. Control sham-operated arteries underwent dissection, temporary clamping, arteriotomy, and ligature, without passage of the wire. Non-operated normal femoral arteries were used as additional controls. Based on the data derived from the control experiments, additional mice underwent femoral wire injury after being randomly allocated to receive a pluronic gel with or without (PD98059 [25μM], SB230580 [25μM] and SP600125 [25μM],). The vessels were harvested for western blotting to detect peak kinase activity and peak plasminogen activator expression and activity and final morphometry. Pluronic gel (1ml of 30% F127 Pluronic gel; Sigma, St Louis, MO) was applied to the external surface of the femoral artery at the end of the procedure as previously described (4, 5). The pluronic gel remains in a liquid form at 4°C and rapidly forms a gel on exposure to body temperature. It is dissipated within 4 hrs.
Morphometry
Following perfusion fixation, each vessel was bisected and central segments from each of these halves were embedded in paraffin and cross sections were cut (10, 100μm apart). Standard morphometric measurements were performed on histological cross sections stained with Hematoxylin/Eosin using SPOT camera system. The luminal, intimal and medial areas of each vessel are determined. Following determination of dimensions of each vessel, a ratio of the intimal and medial areas was calculated.
Determination of in vivo proliferation and apoptosis
Labeling of proliferating VSMC in specimens is performed using the thymidine analogue 5-bromo-2í deoxyuridine (BrdU; Boehringer-Mannheim Corp., Indianapolis, IN; administered by Intraperitoneal injection 24 hours prior to sacrifice, 60 μg/g body weight). The number of BrdU-stained VSMC nuclei are counted and the BrdU labeling index (% of unlabelled cells) is calculated on 10 slides (100μm apart). Labeling of apoptotic VSMC on adjacent sections from each vessel specimens are evaluated using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL; Boehringer-Mannheim Corp.) after processing the tissue according to the manufacturer’s guidelines on 10 slides (100μm apart). Stained VSMC nuclei are counted and the TUNEL labeling index (% of unlabelled cells) is calculated.
En Face SMC Migration Assay
Femoral arteries were perfusion-fixed as described above, and the vessels were opened longitudinally and pinned down onto on agar plate with the luminal surface facing upward. Arteries were stained with hematoxylin for 10 seconds and incubated in Scott’s blue for 2 minutes. Migrated VSMCs were identified as the hematoxylin-stained cells on the luminal surface of the endothelium-denuded area. The length of the femoral artery was visualized under a microscope at x100 magnification, and the total number of VSMC above and below the internal elastic lamina are counted and results expressed as % cells on the luminal surface of the artery.
Immunoblotting
Following wire injury femoral arteries were rapidly retrieved, cleaned once in ice-cold dPBS and then paced in ice-cold kinase buffer (200μl; 25mM Hepes, pH7.5, 10% glycerol, 5mM EDTA, 5mM EGTA, 150mM NaCl, 100mM benzamidine, 0.1% 2-mercaptoethanol, 1% Triton X-100, 1mM pepstatin A, 2mg/ml leupeptin and 20 kallikrein inhibitor units/ml aprotinin). Protein determinations were performed using a bicinchonic acid assay (Biorad Laboratories, Richmond, CA) with bovine serum albumen as the standard. Equal amounts of protein lysates (20μg) were loaded onto a 10% polyacrylamide gel separated by electrophoresis and then transferred to a nitrocellulose membrane (Biorad) (6). The membrane was blocked in 5% non-fat milk. The blot was then immunostained with antibodies against either phosphorylated or unphosphorylated target protein. Protein bands were visualized using an anti-mouse IgG horseradish peroxidase-conjugated antibody followed by enhanced chemiluminescence (ECL, Amersham Bioscience, Piscataway, NJ) using the manufacturer’s protocols. Band intensity was measured using Gel-Imaging software (Kodak, Rochester, NY). All experiments were performed at least three times.
Plasminogen Zymography
Casein Zymography is performed as described previously (7) with minor modifications. Equal amounts of proteins are mixed with SDS sample buffer without reducing agent and subjected to 12.5% SDS-PAGE containing casein (0.25 mg/ml) and plasminogen (20μg/ml). After electrophoresis, the gels are washed twice in 2.5% Triton X-100 for 30 min each at room temperature to remove SDS and then incubated for 18 hrs at 37°C in buffer containing 100mM Glycine, pH8.0. The proteins in the gel are stained with 0.2%Coomassie Brilliant Blue in 30% Methanol: 10% Acetic acid solution for 2-3 hrs and then destained in 30% Methanol: 10% acetic acid.
Reverse Plasminogen Zymography
Reverse Plasminogen Zymography is performed as described previously (7) with minor modifications. Equal amounts of proteins are mixed with SDS sample buffer without reducing agent and subjected to 12.5% SDS-PAGE containing casein (0.25 mg/ml), plasminogen (20μg/ml) and u-PA (0.03U/ml). After electrophoresis, the gels are washed twice in 2.5% Triton X-100 for 30 min each at room temperature to remove SDS and then incubated for 18 hrs at 37°C in buffer containing 100mM Glycine, pH8.0. The proteins in the gel are stained with 0.2%Coomassie Brilliant Blue in 30% Methanol: 10% Acetic acid solution for 2-3 hrs and then destained in 30% Methanol: 10% acetic acid.
Data and Statistical Analysis
Activated kinases are normalized to the total kinase. All data are presented as the mean ± standard error of the mean (s.e.m.) of six observations and statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant. Non-significant p-values were expressed as p=ns.
RESULTS
Lesion development
The injured femoral arteries developed intimal hyperplasia, which was maximal at 28 days and did not change substantially between day 28 and day 56 (Fig. 1). Sham operated on vessels did not produce such a response (Fig. 1). Using BrdU as a marker of cell proliferation, we identified a peak in VSMC proliferation at 7 days after injury (Fig. 2). Using TUNEL staining as a marker for cell apoptosis, we identified a peak in VSMC apoptosis within 1-3 day after injury and a second smaller peak at 28 days after injury (Fig. 2). We did not see significant cell proliferation and the rate of apoptosis was <5% at 3 days in the sham-operated animals. Femoral arteries were opened enface and stained with a nuclear stain and the number of VSMC on the intimal surface were counted in the central area of the artery. There was a time dependent VSMC migration in the vessels peaking at day 7 (Fig. 3). Endothelium was intact and no migration was observable in the sham-operated animals.
Figure 1.
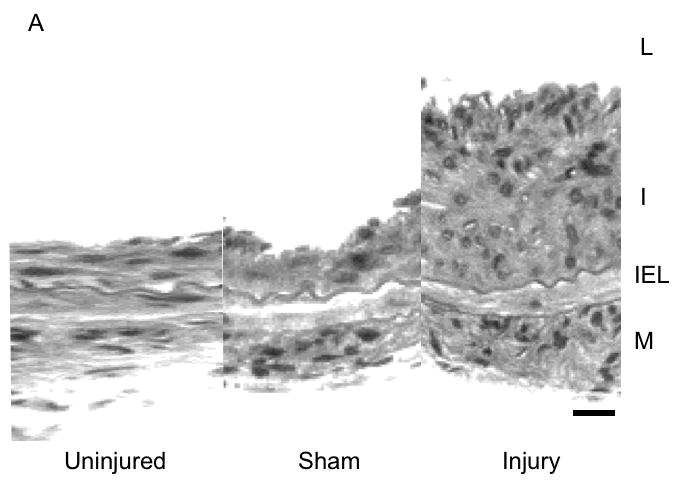
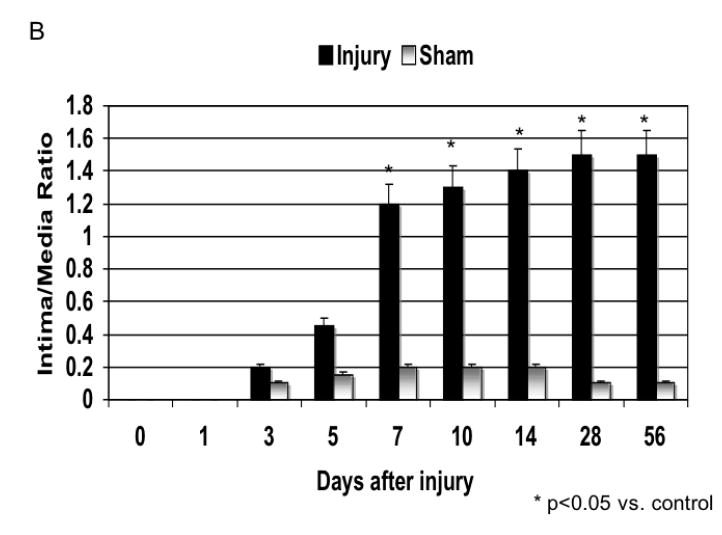
Development of intimal hyperplasia over the time course of the study in wire injured femoral arteries. (A) Representative cross-sectional photomicrograph showing an uninjured, sham and injured vessel at day 56 vessel wall (L, lumen; I, intima; M, media; IEL, internal elastic lamina). (B) Quantification of the development of intimal hyperplasia over time in the injured and sham operated animals. Values are the mean± s.e.m of the intima:media area ratio (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant. Non-significant p-values were expressed as p=ns
Figure 2.
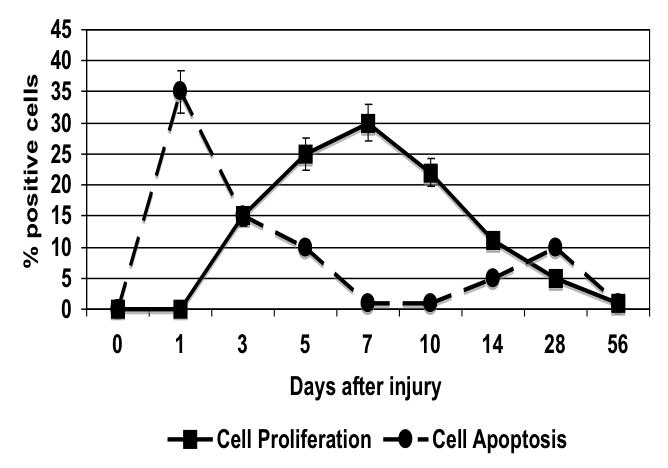
The time course of cell proliferation as determined by BrdU staining and cell apoptosis as determined by TUNEL in wire injured femoral vessels. Values are the mean± s.e.m (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Figure 3.
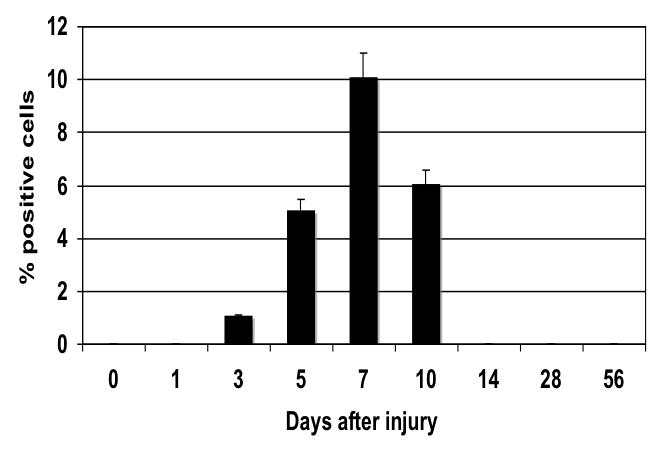
The time course of cell migration onto the luminal surface as determined by enface microscopy in wire injured femoral vessels. Values are the mean±s.e.m. (n=6 per group) and statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Kinase activation
There was no significant change in total kinase concentrations relative to α-actin expression. There was a time dependent increase in phosphorylated kinase expression relative to the total kinase expression present immediately after injury (Fig. 4A). MEK1/2 activation peaked at 20 mins after injury (data not shown) and is followed by a peak in ERK1/2 activation at 45 mins, which was sustained (Fig. 4B). The stress kinases p38MAPK and JNK peaked between 10 and 20 mins (Fig. 4B). Activation of akt occured later at 45 and 120 mins and activation of p70S6 Kinase was biphasic occurring at 20 and 60 mins after injury (Fig. 4C and 4D).
Figure 4.
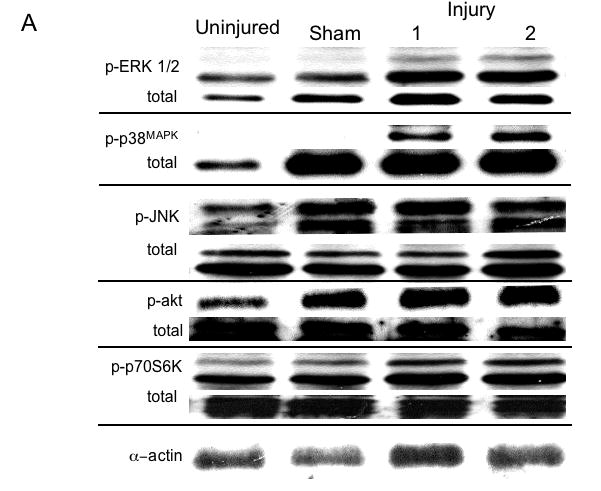
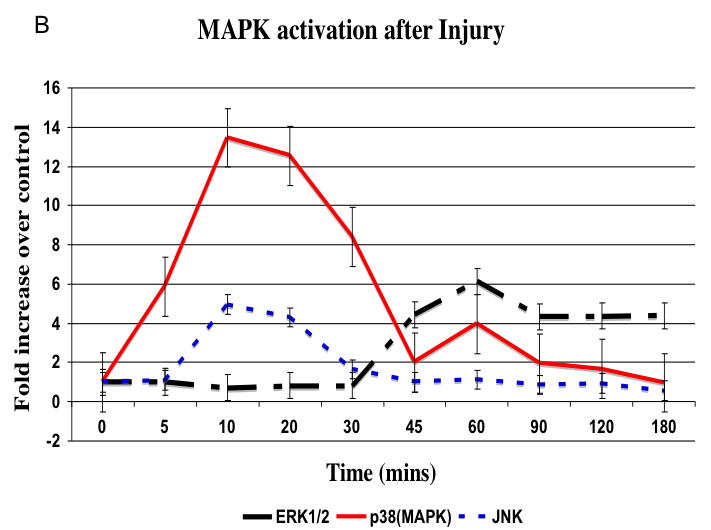
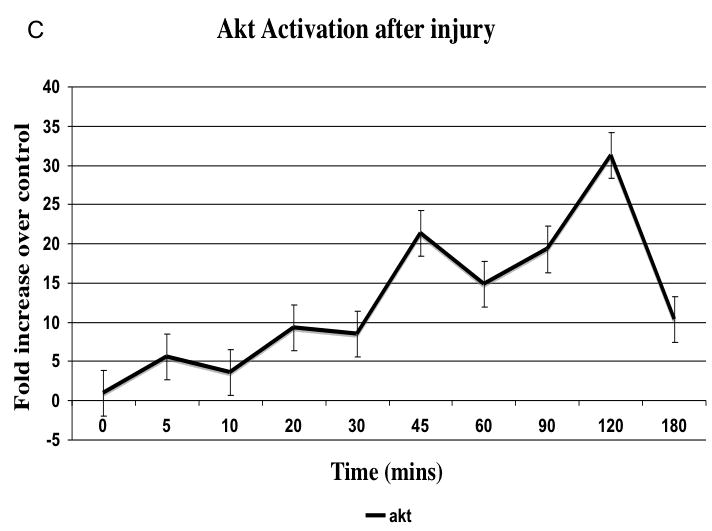
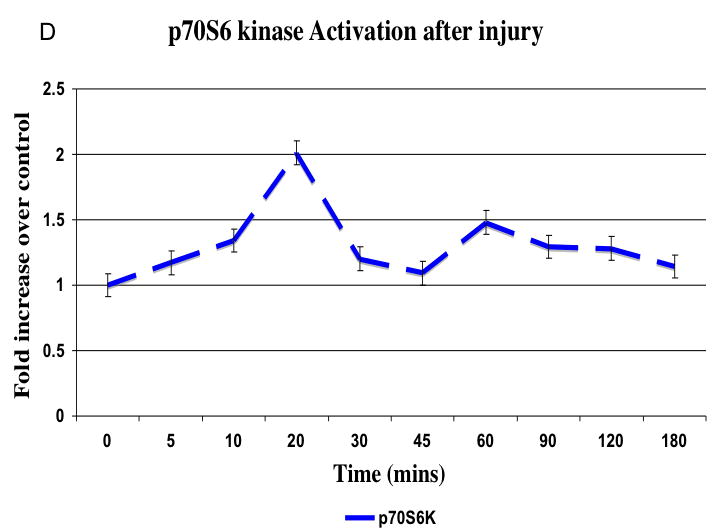
Representative western blots from uninjured, sham and injured femoral arteries at the maximal intensity of activation for each kinase are shown in (A). The time course of activated ERK1/2, p38MAPK and JNK (B) and activated akt (C) and p70S6 kinase (D) in the wire injured femoral vessels over time Activated kinases are normalized to the total kinase. All data are presented as the mean ± s.e.m. (n=6 per group) and statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Plasminogen Activation
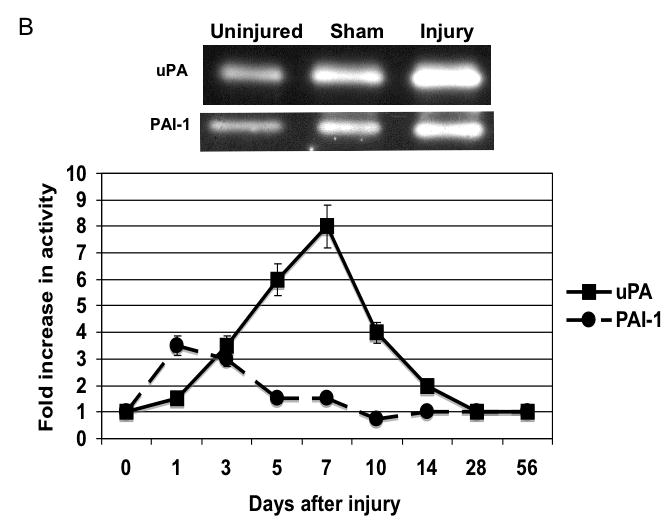
There were time dependent alterations in uPA/PAI-1 activity as determined by plasminogen zymography and reverse plasminogen zymography. uPA activity peaked at 7 days while PAI-1 activity was decreased early from 0 to 3 days and returned to baseline thereafter. There were time dependent changes in expression of uPA and PAI-1 by western blotting after injury (Fig. 5A). PAI -1 expression was increased early from 0 to 3 days and returned to baseline thereafter. In contrast, uPA protein showed a steady increase to rose at day 7 and peaked at day 28 (Fig. 5B).
Figure 5.
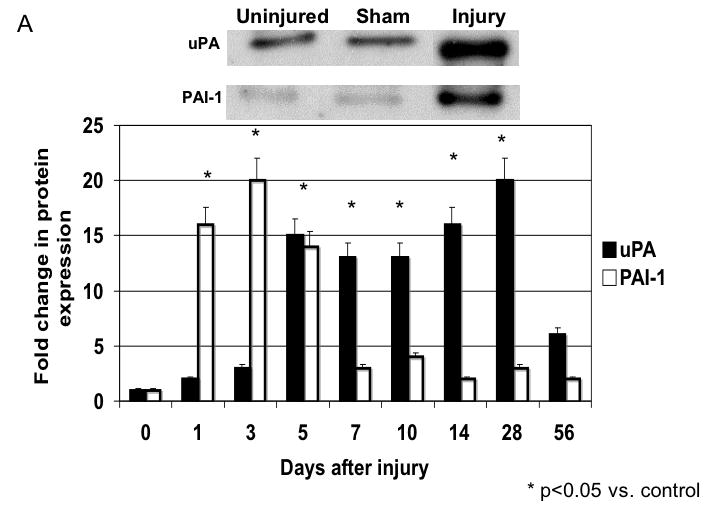
A The expression of uPA and PAI-1 in the wire inured femoral vessels over time. Top panel shows a representative western blots for uPA and PAI-1. Bottom panel shows the quantification of the data for the uninjured, sham and injured vessels. Values are the Mean± s.e.m. fold increase from baseline expression
B The activity of uPA and PAI-1 as determined by plasminogen and reverse plasminogen zymography in lysates from wire injured femoral vessels. Top panel shows a representative zymograms for uPA and PAI-1 for the uninjured, sham and injured vessels. Bottom panel shows the quantification of the data. Values are the mean± s.e.m. fold increase from baseline expression (n=6 per group).
Local inhibition of MAPK
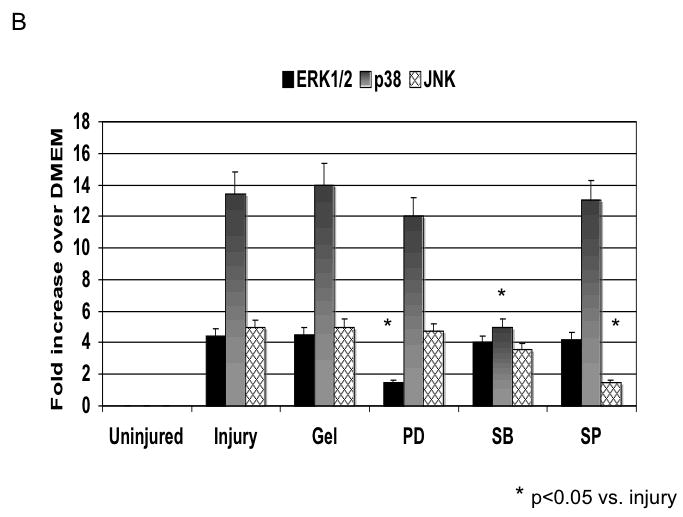
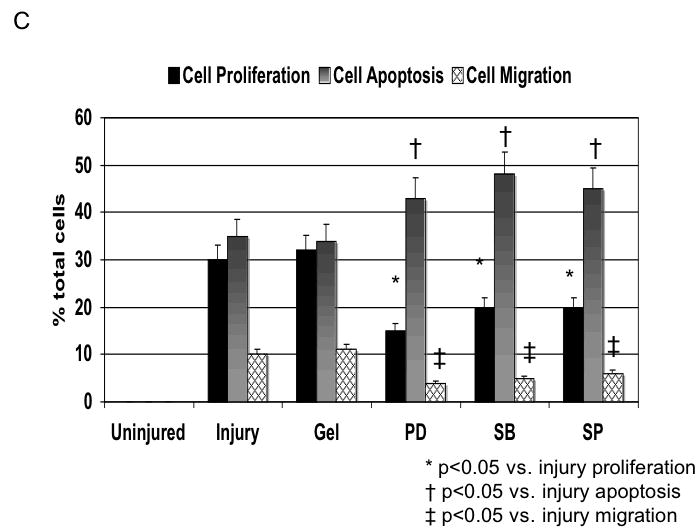
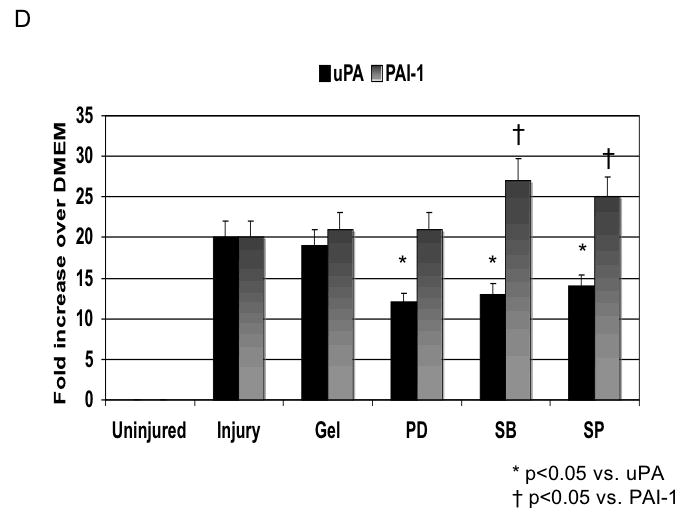
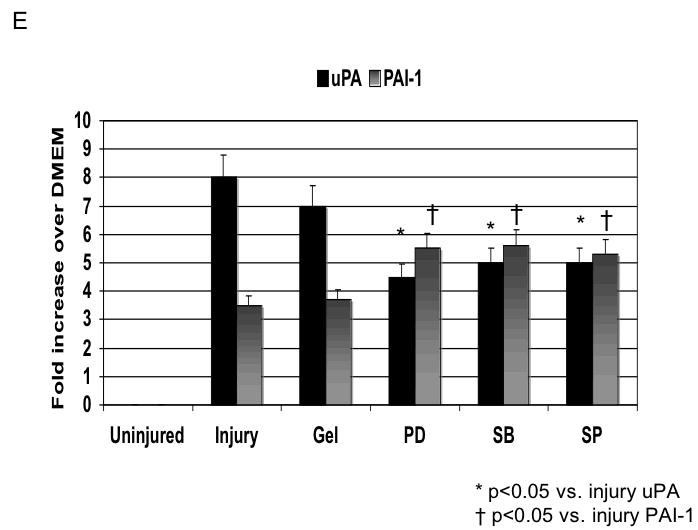
Application of pluronic gel to the femoral artery resulted in a small increase in intimal medial area due to the greater dissection but this was not significant (Fig, 6A). Application of PD98059, SB230580 and SP600125 in pluronic gel, each resulted in a decrease in intimal hyperplasia and a decrease in the peak activation of the relevant MAPK (Fig, 6A and 6B). These changes in MAPK were associated with an increase in maximal cell apoptosis and a decrease in maximal cell proliferation (Fig, 6C). When we examined PAI-1 expression, we found it was increased and this was associated with an increase in its activity. In contrast, the peak expression (Fig, 6D) and activity of uPA was markedly decreased (Fig, 6E).
Figure 6.
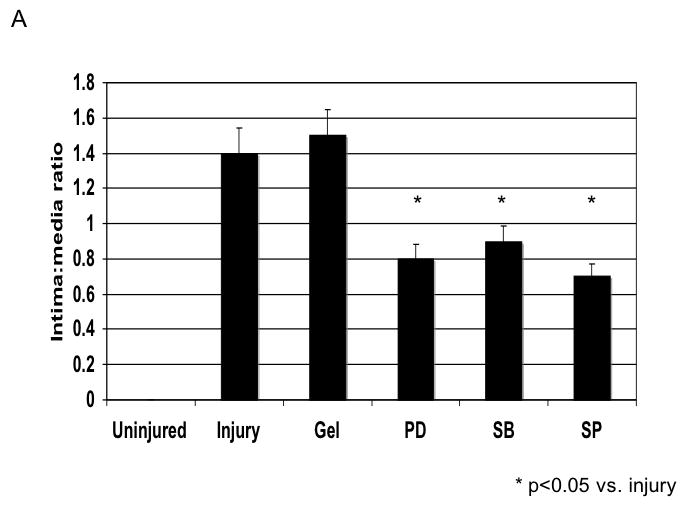
A A comparison of the effect of pluronic gel with and without PD98059 (PD), SB230580 (SB) and SP600125 (SP) on intimal hyperplasia development at 14 days after wire injury. Values are the Mean± s.e.m of the intima:media area ratio (n=6 per group).. Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant. Non-significant p-values were expressed as p=ns
B The effect of pluronic gel with and without PD98059 (PD), SB230580 (SB) and SP600125 (SP) on maximal kinase activation for ERK1/2, p38MAPK and JNK. All data are presented as the mean ± s.e.m. (n=6 per group). and statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
C Changes in cell proliferation, cell apoptosis and cell migration in wire injured femoral vessels in the presence of pluronic gel with and without PD98059 (PD), SB230580 (SB) and SP600125 (SP). Values are the mean± s.e.m. (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
D /E Alterations in the expression (D) and activity (E) of uPA and PAI-1 in wire injured femoral vessels in the presence of pluronic gel with and without PD98059 (PD), SB230580 (SB) and SP600125 (SP). Values are the mean± s.e.m. (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
DISCUSSION
Response to injury
The development of intimal hyperplasia after arterial injury is a universal response and is a wound healing response (1) (Fig. 7). Cell death and cell apoptosis occur within the wall with the first several days. The surface is covered with organizing clot and an acute inflammatory reaction is promoted. Medial VSMC and adventitial fibroblast proliferation occurs which is subsequently followed by VSMC migration from the media into the intimal where a neointima begins. These newly migrated VSMC proliferate and lay down a matrix rich in proteogylcans, which allows for the accelerated increase in the volume of the neointima. Re-endothelization moderates this process. Interruption of cell signaling can either prevent or accelerate VSMC apoptosis and the ability of VSMC to proliferate or migrate. The present data shows that early increases in cell signaling occur and their interruption will alter the development of cell proliferation and migration and thus reduce intimal hyperplasia development.
Figure 7.
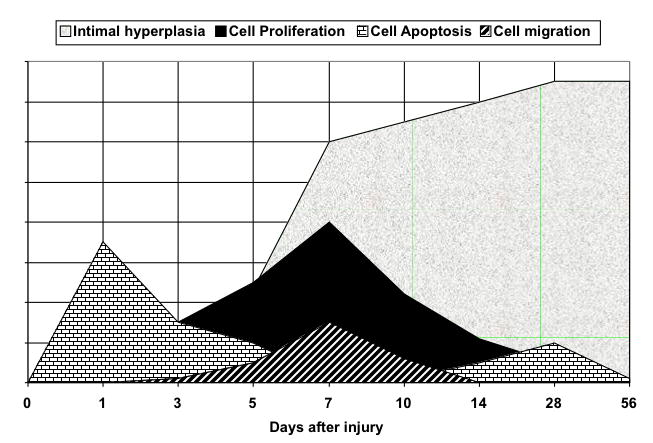
Events leading to the development of arterial intimal hyperplasia. There are two phases of cell apoptosis and a sustained period of cell proliferation followed by cell migration. The neointima increases rapidly with the cell proliferation and migration and by the accumulation of extracellular matrix around the newly arrived cells.
Cell kinetics
We and others have only detected a single peak in VSMC proliferation at 7 days in the murine model (8). The intimal hyperplasia development was consistent and peaked at 28 days and was maintained out to 56 days. In the rat model, VSMC proliferation within the media, which is normally less than 1%, increases to over 20% within 48 hours after angioplasty (9-11) and returns to baseline to have a second peak in the neointima at 7 to 10 days after injury. There have been concerns that in the rat model, involution of the intimal hyperplasia occurs beyond 28 days window. In both models, endothelial cell recoverage was achieved within 28 days. We did not perform extensive staining for inflammatory infiltrates in this series of experiments because others have shown that one-hour after injury in the mouse model, the denuded surface was covered with platelets and leukocytes, predominantly neutrophils (3). This was associated with the accumulation of P-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1. Expression of these adhesion molecules was not seen in the underlying medial VSMC. At 24 hours, few neutrophils remained on the denuded surface. At 1 week, macrophages and platelets were present in the vessel wall, partially covered by regenerated endothelium. A similar pattern of inflammatory infiltration and surface activation has been reported in the rat and rabbit angioplasty models (1). The degree of intimal hyperplasia that develops in a vessel is dependent on the length and depth of the injury (12-14). In addition, distension of VSMC without significant endothelial cell injury has been shown to result in VSMC l proliferation. The length of the injury influences the duration of the re-endothelialization process. Re-endothelialization occurs from the margins of the denuded area and possibly from the endothelial cells of the vasa vasorum. The longer there is an incomplete endothelial cell covering, the greater time the VSMC are without the modulating influence of the endothelial cells, and the longer the replication phases of the VSMC will be (15, 16). We noted near complete endothelialization with 28 days and only one peak in VSMC proliferation. We also noted an immediate increase in the number of apoptotic cells in the media after passage of the wire suggesting significant mural injury. This level of injury fits with the aforementioned theory as we did see a robust intimal hyperplastic response in the vessels. A similar early loss of cells has been documented in the immediate aftermath of angioplasty in the rat carotid artery; apoptosis can be identified at 1-2 hours and appears to disappear by 4 hours (17). No apoptosis can be identified in the wall at 3 days after injury. Application of MAPK kinase inhibitors increased the apotosis rates in the treated vessels. A second peak in apoptosis is noted in the wall by 7 days after injury with up to 50% of the cells showing signs of apoptosis. By day 14, the number of apoptotic cells is again markedly decreased. Cell migration forms an important part of the injury response in the injured artery. We did see a peak in VSMC migration into the intima, which would be consistent with the observations of others (18). When human restenotic lesions have been examined, the lesions are mature and have been found to have low proliferative and apoptotic indices; migration has never been determined in these lesions; furthermore they have a higher composition of extracellular matrix compared to cell content (19). Thus the precise correlation to the animal model remains elusive and one can only presume that the cell kinetics described is in fact occurring.
Cell signaling
Smooth muscle cell proliferation is a key event in neointimal formation after balloon angioplasty. Mitogen-activated protein (MAP) kinases play a pivotal role in transmitting transmembrane signals required for cell proliferation. Within 30 minutes of balloon catheter injury to rat carotid artery, a marked increase in extracellular signal-regulated kinase-1/2 (ERK1/2) activity has been observed. This activity remained elevated for 12 hours but had decreased to control levels by day 1. PD98059, a selective inhibitor of mitogen-activated protein kinase / ERK kinase-1 (MEK1), decreased ERK1/2 activity in injured arteries and also reduced the medial cell replication (20). In the murine model, we also saw a sustained increase in ERK1/2 activity beginning at 45 mins after injury. Application of the ERK1/2 inhibitor also decreased ERK1/2 activation, cell proliferation and intimal hyperplasia development. Both stress kinases showed early peaks in activation that preceded the peak in ERK1/2 activation. This pattern of early and rapid stress kinase activation has been seen after balloon angioplasty in the rat carotid: JNK1 activities in the vessel wall increased rapidly, reaching a high level in 5 minutes and maintained for 1 hour (21), while a similar peak in p38MAPK activation has also being reported to coincide with the JNK peak (22). Inhibition of either p38MAPK or JNK also reduced intimal hyperplasia. These data suggest that pulse therapy against MAPK may be beneficial and at least in several experimental studies against ERK1/2 and p38MAPK, this has worked (20, 22, 23). There has been no cell signaling work reported from early human arterial lesions.
One method of pulsed and sustained therapy to and injured area is the drug eluting stent. Rapamycin eluting stents are used clinically, Rapamycin targets mTOR, a kinase, above p70S6 kinase. VSMC that exhibit a distinct embryonic-like growth phenotype, different from traditional VSMCs, are major contributors to intimal thickening. Growth of these VSMCs is dependent on constitutive Akt and mTOR/p70S6 kinase signaling and is actively inhibited through the timed acquisition of the endogenously produced growth suppressor PTEN (24). The phosphoinositide 3-kinase [PI(3)K]-akt pathway is a key signaling pathway for both apoptotic pathways and replication of mammalian cells. After balloon catheter injury of rat carotid arteries, akt, a downstream target of PI(3)K, was phosphorylated at 30 and 60 minutes after injury and to a lesser degree after 6 hours and 1 and 2 days but not after 7 days. Wortmannin a PI(3)K inhibitor reduced the levels of phosphorylated akt. This reduction in akt phosphorylation was associated with a significant reduction in VSMC replication quantified between 24 to 48 hours compared with controls (25). Injury to the arterial wall also stimulated the activity of p70S6 kinase from 30 minutes to 12 hours, suggesting an alternative pathway in mitogenic signaling of early cell replication (20). In the mouse model, rapamycin inhibits the development of intimal hyperplasia through a p27-independent mechanism and this effect was mediated through decreased VSMC proliferation and enhanced VSMC apoptosis (26). We did see an early robust and sustained increase in activation of akt in our model, which would be consistent with data in rat and as akt is an intermediate signaling molecule in the apoptosis pathway would be consistent with the morphological findings with one day of apoptosis of the VSMC.
Plasminogen activation
Elevated uPA and decreased PAI-1 levels are predictors for angiographic coronary restenosis (27). We saw a steady rise in uPA protein, which stabilized at 14 days and a peak in uPA activity at 7 days in this model. Both responses were blunted by the presence of MAPK inhibitors. After injury to the rat carotid artery, there is a time-dependent enhanced expression of uPA and tPA and increased plasminogen activator activity on smooth muscle cells within the wall compared to the uninjured state (28). Tranexamic acid, a plasmin inhibitor, will inhibit smooth muscle cell migration after carotid balloon injury (29). If uPA is placed in a pluronic gel around an injured carotid, there is a significant increase in the earlier development of intimal hyperplasia due to enhanced migration and proliferation of smooth muscle cells. (30). Infusion of tPA after arterial injury appears to decrease medial cell proliferation and suppress intimal hyperplasia (31). This may be associated with reduction in mural thrombus burden and earlier recovery of endothelial cell coverage. We saw a rapid increase in PAI-1 protein early with a slow rise in PAI-1 activity over the first 3 days of injury. Both responses were blunted by the presence of MAPK inhibitors. This rise in PAI-1 protein was closely associated with the period of cellular apoptosis. Both uPA and PAI-1 expression is regulated in VSMC by bFGF and PDGF both of which have been shown to play a role in the development of intimal hyperplasia after balloon injury. Increased expression of PAI-1 will promote proliferation when VSMCs are exposed to tumor necrosis factor. (32). However, plasmin-mediated apoptosis of VSMC can occur by plasminogen activation by either t-PA or u-PA and can be impaired by PAI-1 (33, 34). In the rat model, expression of PAI-1, the endogenous plasminogen activator inhibitor, is increased 7-fold three hours after arterial balloon injury and returns to baseline by 2 days; it then increases again from day 4 to 7 (35, 36). Elevated PAI-1 protein expression (induced by gene therapy) in the rat carotid enhances thrombosis and endothelial cell regeneration while inhibiting intimal thickening (37). Overexpression of uPA or a deficiency of PAI-1 in transgenic mice has been shown to promote neointimal formation in response to transmural electrical injury. A lack of uPA leads to a reduction in neointimal hyperplasia. (38). In human coronary arteries with either pure fibrointimal proliferation or developed atherosclerotic plaques, the degree of staining showed PAI-1 expression to be greater than uPA (39). A similar pattern was seen in two normal coronary arteries. There were no significant differences in the extent of staining in any vascular compartment between atherosclerotic arteries and those with only fibrointimal proliferation.. Dual labeling studies demonstrated co-localization of all four PA system components in endothelial cells, smooth muscle cells, and macrophages, with a predominance of PAI-1. In normal human saphenous veins, expression of uPA and PAI-1 is associated with the endothelium, as well as with intimal and medial VSMC (40). In stenosing saphenous vein grafts, increasing protein expression for uPA, but not PAI-1, is seen in neointimal VSMC and in migrating SMC (40). Thus, the murine model appears to mirror the findings for the UPA protease system in human vessels well.
Conclusion
Femoral wire injury in the mouse induces a consistent model of intimal hyperplasia and that it is associated with a time dependent increase in signaling kinase activity. The characteristics mirror the commonly used rat carotid injury model. Interruption of these pathways will interrupt uPA/ PAI-1 pathway and decrease intimal hyperplasia development. Accurate characterization of cell signaling is a necessary step in the development of molecular therapeutics and a murine model allows for transgenic approaches to be employed to study the pathobiology of restenosis
Acknowledgments
This research was supported by grants for Mark G. Davies, MD, PhD, from the American College of Surgeons Junior Faculty Award and from the Mentored Clinical Scientist Development Award, sponsored by the NIH-NHLBI/Lifeline Foundation (K08 HL67746) and for Suzanne Nicholl received grant support from the AHA through a Post Doctoral Fellowship (0225696T).
Supported by: U.S. Public Health Service HL67746 American Heart Association (NY) 0225696T
Footnotes
Presented at the Annual Meeting of the Association of Academic Surgery, 2nd Academic Surgical Congress, Phoenix, AZ (February 9-12, 2007).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies MG, Hagen P-O. Pathobiology of Intimal Hyperplasia. Br J Surg. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 2.Berk BC. Vascular smooth muscle growth: autocrine growth mechanisms. Physiol Rev. 2001;81(3):999–1030. doi: 10.1152/physrev.2001.81.3.999. [DOI] [PubMed] [Google Scholar]
- 3.Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- 4.Simons M, Edelman ER, Dekeyser J-L, Langer R, Rosenberg RD. Antisense c-myb oligonucleotides inhibit intimal arterial smooth muscle cell accumulation in vivo. Nature. 1992;359:67–70. doi: 10.1038/359067a0. [DOI] [PubMed] [Google Scholar]
- 5.Fulton GJ, Davies MG, Koch WJ, Dalen H, Svendsen E, Hagen P-O. Antisense oligonucleotide to proto-oncogene c-myb inhibits the formation of intimal hyperplasia in experimental vein grafts. J Vasc Surg. 1997;25:453–463. doi: 10.1016/s0741-5214(97)70255-6. [DOI] [PubMed] [Google Scholar]
- 6.Tanski WJ, Fegley AJ, Roztocil E, Davies MG. Domain dependent actions of urokinase on smooth muscle cell responses. J Vasc Surg. 2004;39:214–22. doi: 10.1016/s0741-5214(03)01031-0. [DOI] [PubMed] [Google Scholar]
- 7.Lijnen HR, VanHoef B, Lupu F, Moon L, Carmeliet P, Collen D. Function of the plasminogen/plasmin and MMP systems after vascular injury in mice with targeted inactivation of fibrinolytic genes. Arterioscler Thromb Vasc Biol. 1998;18(7):1035–45. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- 8.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32(11):2097–104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 9.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I: Smooth muscle cell growth in the absence of endothelium. Lab Invest. 1983;49:327–333. [PubMed] [Google Scholar]
- 10.Clowes AW, Schwartz SM. Significance of quiescent smooth muscle cell migration in the injured rat carotid artery. Circ Res. 1985;56:139–145. doi: 10.1161/01.res.56.1.139. [DOI] [PubMed] [Google Scholar]
- 11.Hanke H, Strohschneider T, Oberhoff M, Betz E, Karsch KR. Time course of smooth muscle cell proliferation in the intima and media of arteries following experimental angioplasty. Circ Res. 1990;67:651–659. doi: 10.1161/01.res.67.3.651. [DOI] [PubMed] [Google Scholar]
- 12.Sarembock IJ, LaVeau PJ, Sigal SL, Timms I, Sussman J, Haudenschild C, et al. Influence of inflation pressure and balloon size on the development of intimal hyperplasia after balloon angioplasty. A study in the atherosclerotic rabbit. Circulation. 1989;80:1029–1040. doi: 10.1161/01.cir.80.4.1029. [DOI] [PubMed] [Google Scholar]
- 13.VanErven L, Post MJ, Velema E, Borst C. In the normal rabbit femoral artery increasing arterial wall injury does not lead to increased intimal hyperplasia. J Vasc Res. 1994;31:153–162. doi: 10.1159/000159041. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto H, Nozaki S, Misumi K, Kurose M, Sohara H, Amitani S, et al. Smooth muscle cell proliferation in the arterial intima after stretch injury - relationship between and severity of stretching and intimal hyperplasia in the New Zealand White rabbits. Jikken Dobutsu. 1996;45:89–93. doi: 10.1538/expanim.45.89. [DOI] [PubMed] [Google Scholar]
- 15.Clowes AW, Reidy MA, Clowes MM. Mechanisms of stenosis after arterial injury. Lab Invest. 1983;49:208–215. [PubMed] [Google Scholar]
- 16.Clowes AW, Clowes MM, Fingerle J, Reidy MA. Regulation of smooth muscle cell growth in injured artery. J Cardiovasc Pharmacol. 1989;14(Suppl 6):S12–S15. [PubMed] [Google Scholar]
- 17.Perlman H, Maillard L, Krasinski K, Walsh K. Evidence for the rapid onset of apoptosis in medial smooth muscle cells after balloon injury. Circulation. 1997;95:981–7. doi: 10.1161/01.cir.95.4.981. [DOI] [PubMed] [Google Scholar]
- 18.Bendeck MP, Irvin C, Reidy MA. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ Res. 1996;78:38–43. doi: 10.1161/01.res.78.1.38. [DOI] [PubMed] [Google Scholar]
- 19.Farb A, Kolodgie FD, Hwang JY, Burke AP, Tefera K, Weber DK, et al. Extracellular matrix changes in stented human coronary arteries. Circulation. 2004;110(8):940–7. doi: 10.1161/01.CIR.0000139337.56084.30. [DOI] [PubMed] [Google Scholar]
- 20.Koyama H, Olson NE, Dastvan F, Reidy MA. Cell replication in the arterial wall - activation of signaling pathway following in vivo injury. Circ Res. 1998;82:713–721. doi: 10.1161/01.res.82.6.713. [DOI] [PubMed] [Google Scholar]
- 21.Hu Y, Cheng L, Hochleitner BW, Xu Q. Activation of mitogen-activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol. 1997;17(11):2808–16. doi: 10.1161/01.atv.17.11.2808. [DOI] [PubMed] [Google Scholar]
- 22.Davies MG, Mason DP, Tran PK, Daum G, Clowes AW. Inhibition of intimal hyperplasia by the p38MAPK inhibitor SB203580. FASEB J. 2000;14:A450. abstract. [Google Scholar]
- 23.Ohashi N, Matsumors A, Furukawa Y, Ono K, Okada M, Iwasaki A, et al. Role of p38 mitogen-activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:2521–2526. doi: 10.1161/01.atv.20.12.2521. [DOI] [PubMed] [Google Scholar]
- 24.Mourani P, Garl P, Wenzlau J, Carpenter T, Stenmark K, Weiser-Evans M. Unique, highly proliferative growth phenotype expressed by embryonic and neointimal smooth muscle cells is driven by constitutive Akt, mTOR, and p70S6K signaling and is actively repressed by PTEN. Circulation. 2004;109(10):1299–306. doi: 10.1161/01.CIR.0000118462.22970.BE. [DOI] [PubMed] [Google Scholar]
- 25.Shigematsu K, Koyama H, Olson EN, Cho A, Reidy MA. Phosphatidylinositol 3-Kinase signaling is important for smooth muscle cell replication after arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:2373–2378. doi: 10.1161/01.atv.20.11.2373. [DOI] [PubMed] [Google Scholar]
- 26.Roque M, Reis E, Cordon-Cardo C, Taubman M, Fallon J, Fuster V, et al. Effect of p27 deficiency and rapamycin on intimal hyperplasia: in vivo and in vitro studies using a p27 knockout mouse model. Lab Invest. 2001;81(6):895–903. doi: 10.1038/labinvest.3780298. [DOI] [PubMed] [Google Scholar]
- 27.Strauss BH, Lau HK, Bowman KA, Sparkes J, Chisholm RJ, Garvey MB, et al. Plasma urokinase antigen and PAI-1 antigen levels predict angiographic coronary restenosis. Circulation. 1999;100(15):1616–22. doi: 10.1161/01.cir.100.15.1616. [DOI] [PubMed] [Google Scholar]
- 28.Clowes AW, Reidy MA, Clowes MM, Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid. Circ Res. 1990;67:61–67. doi: 10.1161/01.res.67.1.61. [DOI] [PubMed] [Google Scholar]
- 29.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 30.Plekhanova OS, Parfenova EV, Bibilashvily RS, Stepanova VV, Erne P, Bobik A, et al. Urokinase plasminogen activator enhances neointima growth and reduces lumen size in carotid arteries. J Hypertens. 2000;18(8):10065–9. doi: 10.1097/00004872-200018080-00011. [DOI] [PubMed] [Google Scholar]
- 31.Kanamasa K, Otani N, Ishida N, Inoue Y, Ikeda A, Morii H, et al. Suppression of cell proliferation by tPA during the early phase after balloon injury minimizes intimal hyperplasia in hypercholesterolemic rabbits. J Cardiovasc Pharmacol. 2001;37(2):155–62. doi: 10.1097/00005344-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Budd RC, Kelm RJJ, Sobel BE, Schneider DJ. Augmentation of proliferation of vascular smooth muscle cells by plasminogen activator inhibitor type 1. Arterioscler Thromb Vasc Biol. 2006;26(8):1777–83. doi: 10.1161/01.ATV.0000227514.50065.2a. [DOI] [PubMed] [Google Scholar]
- 33.Rossignol P, Angles-Cano E, Lijnen HR. Plasminogen activator inhibitor-1 impairs plasminogen activation-mediated vascular smooth muscle cell apoptosis. Thromb Haemost. 2006;96(5):665–70. doi: 10.1160/TH06-06-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossignol P, Luttun A, Martin-Ventura JL, Lupu F, Carmeliet P, Collen D, et al. Plasminogen activation: a mediator of vascular smooth muscle cell apoptosis in atherosclerotic plaques. J Thromb Haemost. 2006;4(3):664–70. doi: 10.1111/j.1538-7836.2005.01765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasentaub D, Forough R, Clowes AW. Plasminogen activator inhibitor type I and tissue inhibitor of metalloproteinases-2 increase after arterial injury in rats. Circ Res. 1997;80:490–496. doi: 10.1161/01.res.80.4.490. [DOI] [PubMed] [Google Scholar]
- 36.Hamdan AD, Quist WC, Gagne JB, Feener EP. ACE inhibition suppresses PAI-1 expression in the neointima of balloon injured rat aorta. Circulation. 1996;93:1073–1078. doi: 10.1161/01.cir.93.6.1073. [DOI] [PubMed] [Google Scholar]
- 37.Hasentaub D, Lea H, Clowes AW. Local plasminogen activator inhibitor Type 1 overexpression in rat carotid artery enhances thrombosis and endothelial regeneration while inhibiting intimal thickening. Arterioscler Thromb Vasc Biol. 2000;20:853–859. doi: 10.1161/01.atv.20.3.853. [DOI] [PubMed] [Google Scholar]
- 38.Shen GX. Vascular cell-derived fibrinolytic regulators and atherothrombotic vascular disorders. Inter J Mol Med. 1998;1(2):399–408. [PubMed] [Google Scholar]
- 39.Raghunath PN, Tomaszewski JE, Brady ST, Caron RJ, Okada SS, Barnathan ES. Plasminogen activator system in human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1432–1443. doi: 10.1161/01.atv.15.9.1432. [DOI] [PubMed] [Google Scholar]
- 40.Salame MY, Samani NJ, Masood I, deBono DP. Expression of the plasminogen activator system in the human vascular wall. Atherosclerosis. 2000;152(1):19–28. doi: 10.1016/s0021-9150(99)00441-4. [DOI] [PubMed] [Google Scholar]