

Abstract
The extracellular form of vaccinia virus is referred to as an enveloped virion (EV) because it contains an additional lipoprotein membrane surrounding the infectious mature virion (MV) that must be discarded prior to cell fusion and entry. Most EVs adhere to the surface of the parent cell and mediate spread of the infection to adjacent cells. Here we show that some attached EVs have ruptured envelopes. Rupture was detected by fluorescence microscopy of unfixed and unpermeabilized cells using antibodies to the F13 and L1 proteins, which line the inner side of the EV membrane and the outer side of the MV membrane, respectively. The presence of ruptured EV membranes was confirmed by immunogold transmission electron microscopy. EVs with broken membranes were present on several cell lines examined including one deficient in glycosaminoglycans, which are thought to play a role in breakage of the EV membrane prior to fusion of the MV. No correlation was found between EVs with ruptured membranes and actin tail formation. Studies with several mutant viruses indicated that EV membranes lacking the A34 protein were unbroken. This result was consistent with other properties of A34R deletion mutants including resistance of the EV membrane to polyanions, small plaque formation and low infectivity that can be increased by disruption of the EV membrane by freezing and thawing.
Introduction
Poxviruses are large, enveloped DNA viruses that replicate in the cytoplasm of the host cell (Moss, 2007). Virus assembly begins in specialized factory areas with the formation of a crescent-shaped membrane and progresses to production of the infectious mature virion (MV) (Condit, Moussatche, and Traktman, 2006), which is retained in the cell until lysis or enclosed by a double membrane to form a wrapped virion (Smith, Vanderplasschen, and Law, 2002). The wrapped virion is transported along microtubules to the periphery of the cell, where the outer membrane fuses with the plasma membrane resulting in an extracellular enveloped virion (EV) (Moss and Ward, 2001). Thus, the EV is essentially an MV with an additional membrane. Most EVs remain cell-associated and mediate cell-to-cell spread (Blasco and Moss, 1992), which is enhanced by long cellular protrusions called actin tails (Roper et al., 1998; Sanderson et al., 1998; Wolffe et al., 1997; Wolffe, Weisberg, and Moss, 1998). In addition, some EVs are released into the medium and may contribute to long-range spread (Payne, 1980).
Recent studies indicate that the fusion proteins required for virus entry reside in the MV membrane (Izmailyan et al., 2006; Ojeda, Domi, and Moss, 2006; Ojeda, Senkevich, and Moss, 2006; Senkevich and Moss, 2005; Senkevich et al., 2005; Senkevich, Ward, and Moss, 2004; Townsley, Senkevich, and Moss, 2005a; Townsley, Senkevich, and Moss, 2005b) and that the EV membrane is discarded prior to entry (Carter et al., 2005; Law et al., 2006). It is well known that the EV membrane is fragile and that it is broken or absent in a significant percentage of EVs purified from the medium (Ichihashi, 1996; Roos et al., 1996; Vanderplasschen, Hollinshead, and Smith, 1997; Vanderplasschen and Smith, 1997). During microscopic studies of VACV infected cells, we noted that the outer membrane of some attached EVs also appeared to be broken. Here we document this occurrence and show that EV membrane rupture is not dependent on a specific cell type or formation of actin tails, but is absent or greatly reduced in cells infected with a mutant lacking the A34 EV membrane protein.
Results
EVs with a ruptured outer membrane are present on the surface of infected cells
Six proteins are known to be components of the EV outer membrane. Of these, A56 and B5 are type I integral membrane proteins; A33 and A34 are type II integral membrane proteins; F13 is a peripheral membrane protein; and K2 is associated with A56 as a heterodimer. Except for F13, these proteins have long extracellular domains that are exposed on the surface of the EV. Since F13 resides on the inner aspect of the EV membrane and the cytoplasmic side of the plasma membrane, it should be inaccessible to exogenous antibody. However, when HeLa cells were infected with vF13-HA, a recombinant VACV that has an influenza hemagglutin (HA) epitope tag appended to the C-terminus of F13, staining was detected with an HA MAb. In the experiment depicted in Fig. 1, infected cell monolayers on coverslips were stained directly in the tissue culture wells using primary and secondary MAbs in phosphate buffered saline (PBS) containing 10% fetal bovine serum (FBS) to minimize cell injury. In the top row of Fig. 1, the cells were stained successively with anti-HA and -B5 MAbs to detect F13 and B5, respectively. With the anti-B5 MAb, there was extensive bright punctate staining, presumably representing EV particles, although some B5 detected might have been inserted into the plasma membrane during exocytosis. A subset of the B5-staining particles appeared to react with the anti-HA MAb (Fig. 1, top row). It seemed likely that the exposure of F13 resulted from partial disruption of the outer EV membrane rather than the plasma membrane since there was no intracellular staining. If that interpretation is true, then MV proteins should also be exposed. The latter was confirmed by staining with a MAb to the L1 MV protein, which does not traffic independently of virus particles to the plasma membrane. A subset of the particles that stained with anti-B5 MAb also reacted with anti-L1 MAb (Fig. 1, middle row). Moreover, there was co-staining of many particles with anti-HA and -L1 MAbs (Fig. 1, lower row), indicating that the F13 detected was associated with ruptured EVs. Similar images were obtained when infected cells were fixed with paraformaldehyde but not permeabilized prior to MAb staining (not shown).
Fig. 1.

Detection of EV and MV proteins on the surface of unfixed and unpermeabilized infected cells. HeLa cells were infected with vF13-HA and after 16 h were stained first with mouse anti-HA MAb (top row), mouse anti-L1 MAb (middle row) or rabbit anti-HA polyclonal antibody (bottom row) followed by FITC-conjugated anti-mouse IgG or anti-rabbit IgG antibody. Cells were then stained with rat anti-B5 MAb (top and middle rows), mouse anti-L1 MAb (bottom row) followed by rhodamine red-conjugated anti-rat or anti-mouse IgG antibodies. Stained cells were then analyzed by confocal microscopy. Green, FITC; red, rhodamine red. Bars, 10 μm.
The above experiments were carried out without permeabilization to avoid staining of cytoplasmic F13-HA and L1. For comparison, infected HeLa cells were fixed and treated with digitonin, which selectively permeabilizes the plasma membrane, prior to staining with anti-HA, -L1 or -B5 MAbs. Since the anti-B5 MAb recognizes the luminal domain of B5, digitonin treatment had no effect on staining (Fig. 2). In contrast the patterns of F13 and L1 staining were consistent with their known Golgi membrane and factory localization, respectively (Fig. 2). The absence of such staining in the experiments depicted in Fig. 1, confirmed that F13 and L1 staining had not resulted from inadvertent permeabilization of the plasma membrane and was due to ruptured EV membranes.
Fig. 2.

Detection of EV and MV proteins in fixed and digitonin permeabilized infected cells. HeLa cells infected with vF13-HA for 16 h were fixed with paraformaldehyde and permeabilized with digitonin. Cells were stained with mouse anti-HA MAb (top row), rat anti-B5 MAb (middle row) or rabbit anti-HA polyclonal antibody (bottom row) followed by FITC-conjugated anti-mouse or anti-rat IgG or anti-rabbit IgG antibodies. Cells were then stained with rat anti-B5 MAb (top row) or mouse anti-L1 MAb (middle and bottom rows) followed by rhodamine red-conjugated anti-rat or anti-mouse IgG antibodies. Cells were then analyzed by confocal microscopy. Green, FITC; red, rhodamine red. Bars, 10 μm.
The presence of EVs with ruptured membranes was not specific for HeLa cells, as particulate staining with anti-HA MAb was detected on the surface of hamster BHK-21, monkey BS-C-1, rabbit RK13, mouse L and mouse Sog9 cells that were infected with vF13-HA (Fig. 3). The detection of ruptured EV membranes on Sog9 cells was of particular interest as these cells are deficient in glycosaminoglycans (Banfield et al., 1995), which have been proposed to serve as the agents which disrupt the membranes of EVs on cell contact prior to entry (Law et al., 2006). Rupture of EV membranes on Sog9 cells was confirmed and quantified by electron microscopy in the next section.
Fig. 3.

Detection of ruptured EV particles on the surface of multiple cell lines. BHK, BS-C-1, RK13, L and Sog9 cells were infected with vF13-HA and at 16 h were stained with mouse anti-HA MAb followed by Alexa 488-conjugated anti-mouse IgG antibody. Cells were then fixed and analyzed by confocal microscopy. Bars, 10 μm.
Determination of the integrity of the EV membrane by electron microscopy
The resolution of confocal microscopy was insufficient to characterize the particles with ruptured membranes. Therefore, HeLa cells infected with vF13-HA were stained with anti-HA MAb before processing the samples for transmission electron microscopy. Virions on the cell surface with broken outer membrane were readily detected and these were stained by anti-HA MAb, indicating accessibility of F13 on the inner surface of the EV membrane. EVs with all stages of membrane breakage and shedding were found and there were examples of discarded EV membranes that were resealed in an inside-outside fashion (Fig. 4A-E).
Fig. 4.

Detection of ruptured EV particles on the surface of cells by electron microscopy. HeLa cells were infected with vF13-HA for 16 h. Unfixed and unpermeabilized cells were stained with mouse anti-HA MAb followed by protein A gold. After staining cells were fixed, cryosectioned and examined by transmission electron microscopy.
A similar experiment was carried out on glycosaminoglycan-deficient Sog9 and parental L cells. Based on HA staining, we counted a total of 493 and 472 disrupted EVs on the perimeters of 50 L and Sog9 cells, respectively.
EV particles attached to actin tails have an intact outer membrane
Some EV particles on the cell surface are located at the tips of long protrusions called actin tails, which are necessary for efficient cell-cell virus spread. Since only the MV membrane is fusogenic, we considered the possibility that disruption of the outer membrane occurred on EV particles associated with actin tails in anticipation of cell fusion. To test this hypothesis, unfixed and unpermeabilized cells were incubated with MAbs to B5 or HA prior to actin staining. As expected, anti-B5 MAb decorated virus particles on the tips of many actin tails (Fig. 5, top panel). However, there was no correspondence of F13-HA staining with actin tails (Fig. 5, second panel), unless the EVs were first fixed and permeabilized with Triton X-100 (Fig. 5, third panel). Furthermore, EV with ruptured membranes were present on cells infected with vA36(Y112,132F) a mutant VACV with tyrosine mutations in the A36 protein that prevent actin tail formation, as shown by staining with a MAb to L1 (Fig. 5, bottom panels).
Fig. 5.
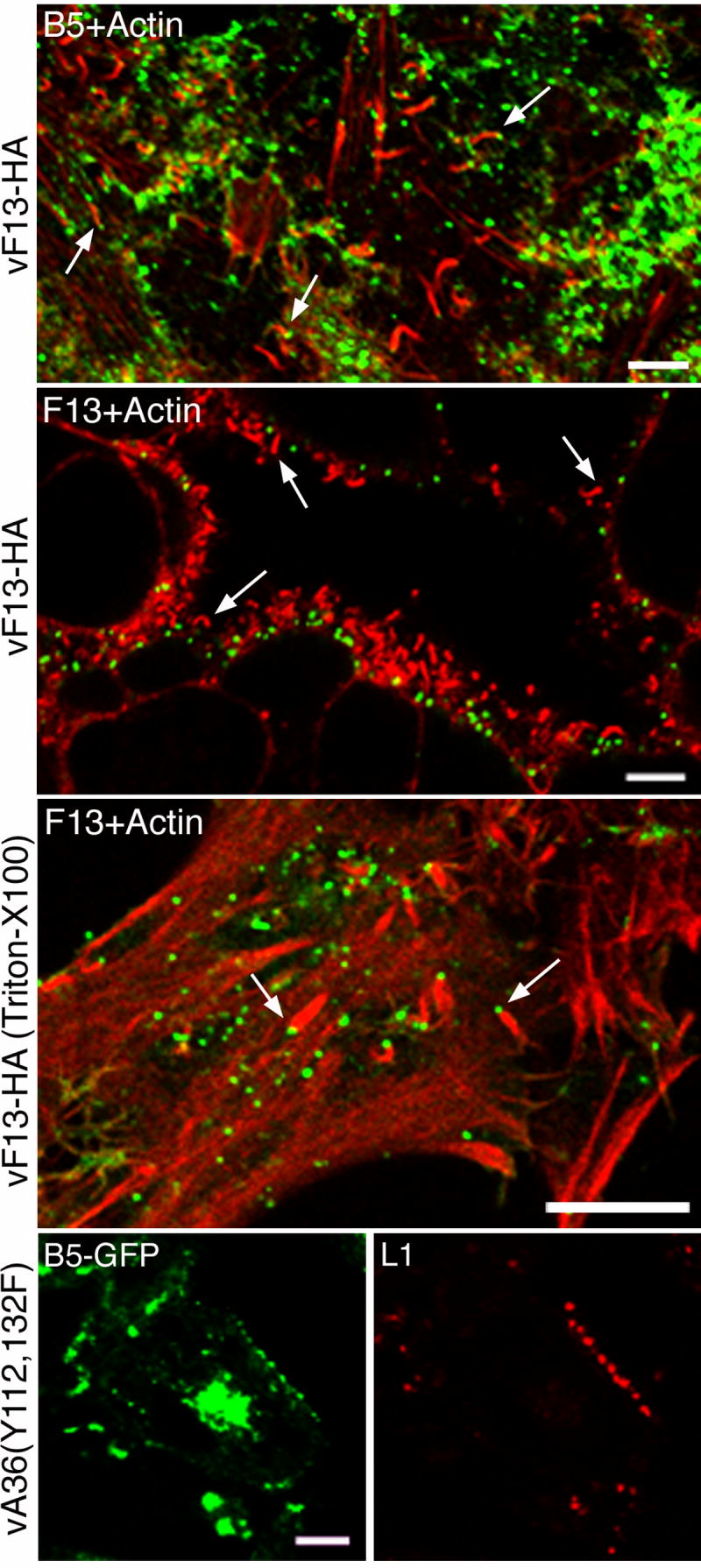
Staining of EV particles and actin tails. HeLa cells were infected with vF13-HA (rows 1-3) or vA36(Y112,132F) (row 4). At 16 h, cells were stained with rat anti-B5 MAb (row 1), mouse anti-HA MAb (rows 2 and 3) and mouse anti-L1 MAb (row 4) followed by Alexa 488-conjugated anti-rat IgG, anti-mouse IgG and Alexa 594-conjugated anti-mouse IgG antibodies. Cells were then fixed, permeabilized with Triton X-100 and stained with Texas red-conjugated phalloidin (rows 1-3). In row 3, cells were first fixed and permeabilized with Triton X-100 before staining. Cells were analyzed by confocal microscopy. Green, Alexa 488; red, Alexa 594 and Texas red. Bars, 10 μm. Arrows point to representative actin tails.
Effect of deleting individual EV proteins on the integrity of the EV membrane
To analyze the role of individual EV proteins in determining the integrity of the EV membrane, cells were infected with vA33Δ, vA34Δ or vA56Δ, with deletions of A33R, A34R or A56R genes, respectively. Unfixed and unpermeabilized cells were stained with MAbs to L1 and B5. The patterns of staining of cells infected with vA33Δ and vA56Δ (Fig. 6) were similar to that of standard VACV (Fig. 1) indicating the presence of EVs with ruptured membranes. In contrast, EV particles on the surface of cells infected with vA34Δ were not stained with anti-L1 MAb suggesting greater stability of the EV membrane lacking A34 (Fig. 6). Few particles staining with anti-L1 MAb were detected on the surface of cells infected with a B5 deletion mutant (data not shown); however the small number of EVs on the cell surface limited our interpretation in this case.
Fig. 6.

Detection of ruptured EVs on the surface of cells infected with VACV mutants. HeLa cells were infected with vA33Δ, vA56Δ or vA34Δ. After 16 h, cells were stained with mouse anti-L1 MAb followed by Alexa 488-conjugated anti-mouse IgG. Cells were then stained with rat anti-B5 MAb followed by Alexa 568 IgG. Cells were then fixed and analyzed by confocal microscopy. Green, Alexa 488; red, Alexa 568 or 594. Bars, 10 μm.
To confirm the result obtained with vA34Δ, we made a new A34R deletion mutant by replacing the A34R gene of vF13-HA with EGFP, allowing use of anti-HA MAb to stain F13. The resulting virus vF13-HA(A34Δ) exhibited a small plaque phenotype similar to that of vA34Δ (data no shown). Cells were infected with vF13-HA(A34Δ) and stained with MAbs to HA, L1 or B5 either with or without fixation and permeabilization. Infected cells were identified by green fluorescence and antibody staining was assessed. In the unfixed, unpermeabilized cells, no staining for F13 or L1 was detected despite punctate staining with MAb to B5 (Fig. 7, rows 1 to 3). However, staining with anti-HA and -L1 MAb was obtained in cells fixed and permeabilized with digitonin (Fig. 7, rows 4 to 6). These data indicate that the EV membrane is stabilized by the absence of A34.
Fig. 7.
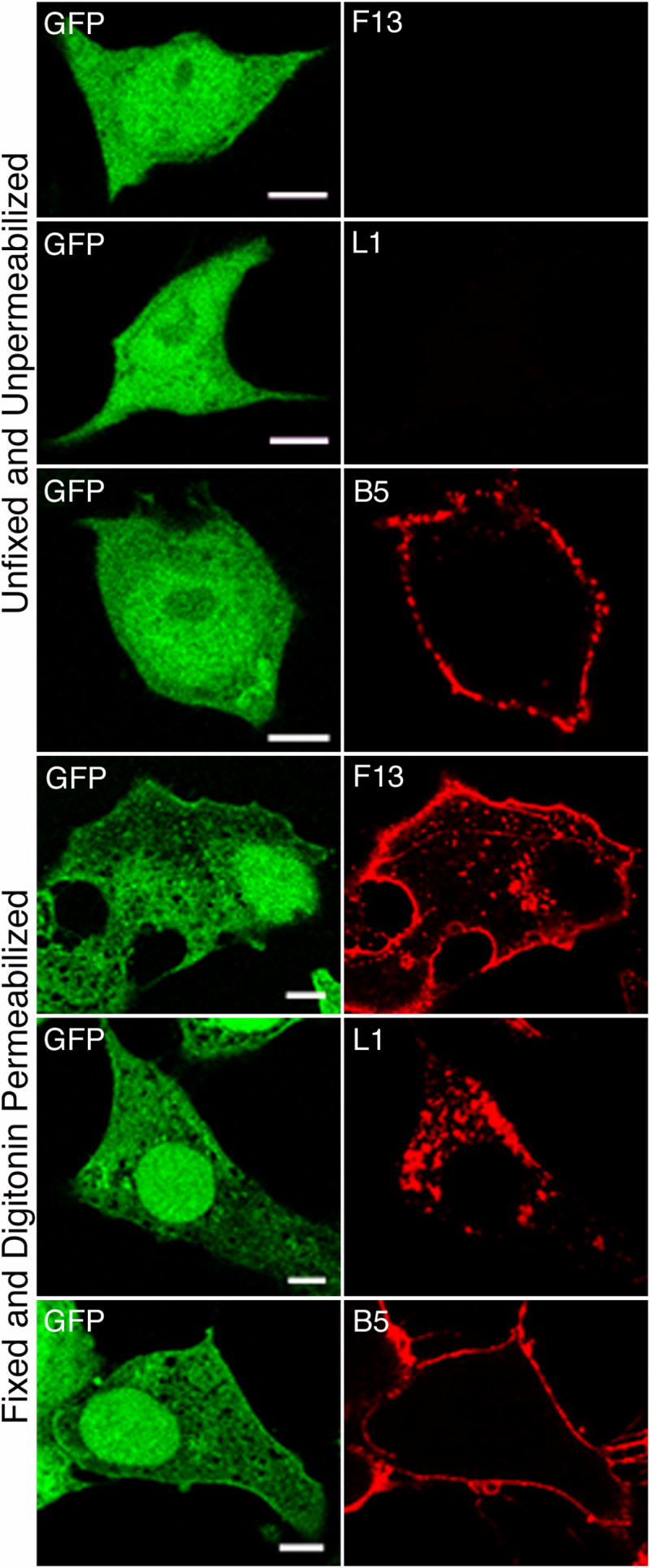
Absence of ruptured EV particles on the surface of cells infected with vaccinia virus with deleted A34R gene. HeLa cells were infected with vF13-HA(A34Δ) and after 16 h were stained with mouse anti-HA MAb (rows 1, 4), mouse anti-L1 MAb (rows 2, 5) and rat anti-B5 MAb (rows 3,6) followed by Alexa 594-conjugated anti-mouse or anti-rat IgG antibodies. Cells in rows 1-3 were unfixed and unpermeabilized; cells in rows 4-6 were fixed with paraformaldehyde and permeabilized with digitonin. Confocal microscopy images are shown. Green, GFP; red, Alexa 594. Bars, 10 μm.
We also examined cells infected with vF13-HA or vF13-HA(A34Δ) by electron microscopy. There were approximately 10-fold more EVs on the surface of cells infected with vF13-HA than vF13-HA(A34Δ), consistent with the greater release of the mutant particles from the cell surface (McIntosh and Smith, 1996). Data compiled from two separate experiments indicated that of 1,379 virus particles on the surface of cells infected with vF13-HA, 81 stained with anti-HA MAb indicating rupture of the outer EV membrane. In contrast, of 780 virus particles on the surface of cells infected with vF13- HA(A34Δ), none stained with anti-HA MAb. Nevertheless, the staining of EVs with antibody to the B5 protein was similar for both viruses. Thus, electron microscopy confirmed the stability of the EV membranes lacking A34.
Discussion
The presence of the fusion protein complex in the MV membrane implies that the outer membrane of the EV form of VACV must be removed prior to virus entry (Moss, 2006). Indeed, stunning pictures of a broken EV membrane shroud above a virion attached to the plasma membrane have recently been published (Law et al., 2006). For technical reasons the latter study and nearly all other studies of EVs have been carried out with particles released into the medium, even though they represent less than 1% of the total made by most VACV strains. The IHD-J strain, an exception that releases large numbers of EVs into the medium, has an amino acid substitution in the A34 protein that is not present in A34 homologs of other orthopoxviruses (Blasco, Sisler, and Moss, 1993). The EV membrane is fragile as even fresh preparations of EVs contain up to 20% with ruptured or missing outer membranes (Ichihashi, 1996; Roos et al., 1996; Vanderplasschen, Hollinshead, and Smith, 1997; Vanderplasschen and Smith, 1997). In addition, the EV membrane can be ruptured by low pH, freezing and thawing or addition of soluble polyanions (Law et al., 2006; Vanderplasschen, Hollinshead, and Smith, 1998). The purpose of the present study was to analyze the state of progeny EV particles on the surface of infected cells, rather than those released into the medium.
Precautions were taken to prevent EV membrane damage due to handling of the cells. Thus, antibodies in PBS containing serum were added directly to the cell monolayers on cover slips in tissue culture wells. When either unfixed or paraformaldehyde fixed but unpermeabilized cells were examined by confocal microscopy, we found some surface staining with MAb to the F13 protein, which is present on the inner aspect of the EV membrane, and to the L1 MV protein. The rupture of the outer membrane of about 6% of the EV particles was determined by immunoelectron microscopy, though this number may be an underestimate since thin sections were examined. We found evidence for ruptured progeny EVs on cells from a variety of sources including Sog9 cells. The latter result was surprising because Law et al (Law et al., 2006) had reported that rupture of the outer membrane of spinoculated EVs did not occur during a 10 to 30 min period on such cells, which are deficient in glycosaminoglycans. This difference could suggest alternative mechanisms of membrane rupture.
We considered possible causes and consequences of EV membrane rupture. Rupture could occur during exocytosis, while on the cell surface but before release into the medium, or after release and reattachment to the cell. The most interesting hypothesis was that membrane rupture of progeny virions was related to the movement of actin tails and that exposure of the MV membrane might facilitate fusion with neighboring cells. However, finding intact EV particles on the tips of actin tails did not support this idea. We also found that tyrosine mutations of the A36 protein, which prevent actin tail formation and reduce plaque size, did not prevent EV membrane disruption. An A33 deletion mutant, which is also defective in actin tail formation, produced EVs with ruptured membranes. Thus, actin tail formation was not correlated with EV membrane rupture. We also considered the possibility that the A56 VACV hemagglutinin protein might help stabilize the EV membrane, as A56 deletion mutants cause extensive cell-cell fusion. Nevertheless, the extent of EV membrane rupture was similar on the surface of cells infected with an A56 deletion mutant and wild type virus. However, we did find that the EV membrane on the surface of cells infected with an A34R deletion mutant was intact. This result was consistent with other properties of the A34R deletion mutant including small plaque formation and low infectivity that can be increased by disruption of the EV membrane by freezing and thawing (McIntosh and Smith, 1996) and resistance of the EV membrane to polyanions (Law et al., 2006).
In conclusion, EVs with ruptured outer membranes are present on the surface of VACV infected cells. Although removal of the envelope is necessary for virus fusion and entry of neighboring cells, the importance of membrane breakage at this stage is unknown. Presumably, the A56 and K2 proteins in the plasma membrane of the parent cell would prevent fuse back and re-entry (Turner and Moyer, 2006; Wagenaar and Moss, 2007). The absence of EVs with broken membranes from cells infected with an A34R deletion mutant provides further evidence that the A34 protein has a role in removing the envelope to allow virus entry.
Materials and Methods
Cells and viruses
BS-C-1, RK13, BHK-21 and HeLa cells were grown and maintained in Eagle’s modified medium supplemented with 10% FBS at 37°C with 5% CO2. L and Sog9 cells were grown in a similar manner except for use of Dulbecco’s modified Eagle’s medium. Recombinant VACV (WR strain) viruses are: vF13-HA expressing HA epitope tagged F13 protein (Husain, Weisberg, and Moss, 2003); vA36(Y112,132F) previously referred to as vB5-GFP(A36(YdF) EGFP tagged B5 and A36 with tyrosine substitutions (Ward, Weisberg, and Moss, 2003); vA34RΔ previously referred to as v41 containing a deletion of the A34R gene (Wolffe et al., 1997); vA33Δ containing a deletion of the A33R gene (Roper et al., 1998); vA56Δ containing a deletion of the A56R gene was kindly provided by Tim Wagenaar; and vB5Δ previously referred to as vSI14 and containing a deletion of the B5R gene (Wolffe, Isaacs, and Moss, 1993). vF13-HA(A34Δ) was constructed in this study by replacing the A34R gene with EGFP.
vF13-HA(A34Δ) was generated by recombination according to the following procedure. Plasmid pGA34-LGR was constructed by a 4-way ligation of polymerase chain reaction products containing the EGFP open reading frame under the VACV A34R gene promoter, two DNA segments of approximately 500 bp representing the left and right sides of the A34R open reading frame and linearized pGEM7 (Promega). HeLa cells were infected with vF13-HA at a multiplicity of 0.1 pfu per cell for 1 h at 37°C and then transfected with plasmid pGA34-LGR and Lipofectamine 2000 (Invitrogen) in Opti-MEM I medium (Invitrogen). After 2 days at 37°C cells were harvested and lysed by three freeze/thaw cycles. Diluted lysate of infected/transfected cells was used to infect BS-C-1 monolayers and tiny green plaques were picked. After several rounds of plaque purification, the viral DNA was screened by polymerase chain reaction for the presence of the inserted DNA.
Viruses were propagated in HeLa cells and titrated by plaque assay on BS-C-1 cells as described (Earl et al., 1998).
Antibodies
Rat anti-B5 MAb 192C was prepared from a hybridoma derived by G. Hiller and mouse anti-L1 MAb 7D11 and anti-A33 MAb from hybridomas provided by A. Schmaljohn. Mouse anti-HA.11 MAb and rabbit polyclonal antibody that recognize the HA epitope and mouse anti-GFP MAb were purchased from Covance (Princeton, N.J.). Fluorescein isothiocynate (FITC)-conjugated anti-mouse, anti-rat and anti-rabbit immunoglobulin antibody, tetramethylrhodamine isothiocyanate (TRITC)-conjugated anti-mouse, anti-rat and rabbit immunoglobulin antibody and rhodamine red-conjugated anti-rat immunoglobulin antibody were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). Alexa 488, 568 or 594-conjugated anti-mouse and anti-rat immunoglobulin antibodies and Texas red-conjugated phalloidin were procured from Molecular Probes Division of Invitrogen.
Confocal Microscopy
Infected cells on cover slips were washed once with PBS and stained with primary antibodies diluted in 10% FBS in PBS for 1 h followed by secondary antibody diluted in 10% FBS-PBS for 30 min at room temperature. Cells were gently washed three times with PBS after incubation with each antibody. Cells were then fixed with 4% paraformaldehyde in PBS at room temperature for 20 min. Alternatively, cells were fixed first and permeabilized for 5 min with digitonin (20 μg/ml) or 0.2% TritonX-100 in PBS on ice or at room temperature, respectively. Cells were then stained with antibodies as above. Cover slips were mounted in 20% glycerol and fluorescence was examined under Leica TCS inverted confocal microscope. Images were analyzed and overlaid using Adobe Photoshop version 7.0.
Transmission electron microscopy
HeLa cells were infected with vF13-HA for 16 h, washed with PBS and incubated with MAb to HA and then with protein A conjugated to gold. The cells were then fixed, cryosectioned and viewed by transmission electron microscopy as previously described (da Fonseca et al., 2000).
Acknowledgments
We thank Norman Cooper for propagating cells and Tim Wagenaar for a vaccinia virus mutant. The study was supported by the Division of Intramural Research, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J Virol. 1995;69:3290–3298. doi: 10.1128/jvi.69.6.3290-3298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Moss B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol. 1992;66:4170–4179. doi: 10.1128/jvi.66.7.4170-4179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco R, Sisler JR, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67:3319–3325. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- Condit RC, Moussatche N, Traktman P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 2006;66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- da Fonseca FG, Wolffe EJ, Weisberg A, Moss B. Effects of deletion or stringent repression of the H3L envelope gene on vaccinia virus replication. J Virol. 2000;74:7518–7528. doi: 10.1128/jvi.74.16.7518-7528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl PL, Cooper N, Wyatt S, Moss B, Carroll MW. Preparation of cell cultures and vaccinia virus stocks. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Vol. 2. John Wiley and Sons; New York: 1998. pp. 16.16.1–16.16.3. [Google Scholar]
- Husain M, Weisberg A, Moss B. Topology of epitope-tagged F13L protein, a major membrane component of extracellular vaccinia virions. Virology. 2003;308:233–242. doi: 10.1016/s0042-6822(03)00063-1. [DOI] [PubMed] [Google Scholar]
- Ichihashi Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology. 1996;217:478–485. doi: 10.1006/viro.1996.0142. [DOI] [PubMed] [Google Scholar]
- Izmailyan RA, Huang CY, Mohammad S, Isaacs SN, Chang W. The envelope G3L protein is essential for entry of vaccinia virus into host cells. J Virol. 2006;80:8402–8410. doi: 10.1128/JVI.00624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Carter GC, Roberts KL, Hollinshead M, Smith GL. Ligand-induced and non-fusogenic dissolution of a viral membrane. Proc Natl Acad Sci USA. 2006;103:5989–5994. doi: 10.1073/pnas.0601025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh AA, Smith GL. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J Virol. 1996;70:272–281. doi: 10.1128/jvi.70.1.272-281.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae: the viruses and their replicaton. In: Knipe DM, editor. Fields Virology. Vol. 2. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2905–2946. [Google Scholar]
- Moss B, Ward BM. High-speed mass transit for poxviruses on microtubules. Nature Cell Biol. 2001;3:E245–E246. doi: 10.1038/ncb1101-e245. [DOI] [PubMed] [Google Scholar]
- Ojeda S, Domi A, Moss B. Vaccinia virus G9 protein is an essential component of the poxvirus entry-fusion complex. J Virol. 2006;80:9822–9830. doi: 10.1128/JVI.00987-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda S, Senkevich TG, Moss B. Entry of vaccinia virus and cell-cell fusion require a highly conserved cysteine-rich membrane protein encoded by the A16L gene. J Virol. 2006;80:51–61. doi: 10.1128/JVI.80.1.51-61.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne LG. Significance of extracellular virus in the in vitro and in vivo dissemination of vaccinia virus. J Gen Virol. 1980;50:89–100. doi: 10.1099/0022-1317-50-1-89. [DOI] [PubMed] [Google Scholar]
- Roos N, Cyrklaff M, Cudmore S, Blasco R, Krijnse-Locker J, Griffiths G. A novel immunogold cryoelectron microscopic approach to investigate the structure of the intracellular and extracellular forms of vaccinia virus. EMBO J. 1996;15:2343–2355. [PMC free article] [PubMed] [Google Scholar]
- Roper R, Wolffe EJ, Weisberg A, Moss B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to- cell spread of vaccinia virus. J Virol. 1998;72:4192–4204. doi: 10.1128/jvi.72.5.4192-4204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson CM, Frischknecht F, Way M, Hollinshead M, Smith GL. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J Gen Virol. 1998;79:1415–1425. doi: 10.1099/0022-1317-79-6-1415. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Moss B. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J Virol. 2005;79:4744–4754. doi: 10.1128/JVI.79.8.4744-4754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. Poxvirus multiprotein entry-fusion complex. Proc Natl Acad Sci USA. 2005;102:18572–18577. doi: 10.1073/pnas.0509239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Ward BM, Moss B. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J Virol. 2004;78:2357–2366. doi: 10.1128/JVI.78.5.2357-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GL, Vanderplasschen A, Law M. The formation and function of extracellular enveloped vaccinia virus. J Gen Virol. 2002;83:2915–2931. doi: 10.1099/0022-1317-83-12-2915. [DOI] [PubMed] [Google Scholar]
- Townsley A, Senkevich TG, Moss B. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is requried for cell entry and cell-cell fusion. J Virol. 2005a;79:10988–10998. doi: 10.1128/JVI.79.17.10988-10998.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley A, Senkevich TG, Moss B. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J Virol. 2005b;79:9458–9469. doi: 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PC, Moyer RW. The cowpox virus fusion regulator proteins SPI-3 and hemagglutinin interact in infected and uninfected cells. Virology. 2006;347:88–99. doi: 10.1016/j.virol.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and spread. J Gen Virol. 1997;78:2041–2048. doi: 10.1099/0022-1317-78-8-2041. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M, Smith GL. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J Gen Virol. 1998;79:877–887. doi: 10.1099/0022-1317-79-4-877. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Smith GL. A novel virus binding assay using confocal microscopy: demonstration that intracellular and extracellular vaccinia virions bind to different cellular receptors. J Virol. 1997;71:4032–4041. doi: 10.1128/jvi.71.5.4032-4041.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar TR, Moss B. Association of vaccinia virus fusion regulatory proteins with the multicomponent entry/fusion complex. J Virol. 2007 doi: 10.1128/JVI.00274-07. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Weisberg AS, Moss B. Mapping and functional analysis of interaction sites within the cytoplasmic domains of the vaccinia virus A33R and A36R envelope proteins. J Virol. 2003;77:4113–4126. doi: 10.1128/JVI.77.7.4113-4126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe EJ, Isaacs SN, Moss B. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J Virol. 1993;67:4732–4741. doi: 10.1128/jvi.67.8.4732-4741.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe EJ, Katz E, Weisberg A, Moss B. The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. J Virol. 1997;71:3904–3915. doi: 10.1128/jvi.71.5.3904-3915.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe EJ, Weisberg AS, Moss B. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology. 1998;244:20–26. doi: 10.1006/viro.1998.9103. [DOI] [PubMed] [Google Scholar]