

Abstract
Somitogenesis is a highly controlled process that results in segmentation of the paraxial mesoderm. Notch pathway activity in the presomitic mesoderm is fundamental for management of synchronized gene expression which is necessary for regulation of somitogenesis. We have isolated an embryonic lethal mutation, SBU2, that causes somite formation defects very similar to Notch pathway mutants. SBU2 mutants generate only 6-7 asymmetrically arranged somites. However, in contrast to Notch pathway mutants, these mutants do not maintain previously formed somite boundaries and by 24 hpf, almost no somite boundaries remain. Other developmental processes disrupted in SBU2 mutants include tail morphogenesis, muscle fiber elongation, pigmentation, circulatory system development and neural differentiation. We demonstrated that these defects are the result of a nonsense mutation within the spt6 gene. spt6 encodes a transcription elongation factor that genetically interacts with the Paf-1 chromatin remodeling complex. SBU2 mutant phenotypes could be rescued by microinjection of spt6 mRNA and spt6 morpholinos phenocopied the mutation. Our real-time PCR analysis revealed that Spt6 is essential for the transcriptional response to activation of the Notch pathway. Analysis of sbu2;mib double mutants indicates that Spt6 deficiency suppresses the neurogenic effects of the mib. Altogether, these results demonstrate that Spt6 is critical for somite formation in zebrafish and suggest that some defects observed in spt6 mutants result from alterations in Notch signaling. However, additional Spt6 mutant phenotypes are likely caused by vital functions of Spt6 in other pathways.
Introduction
Somites establish the segmental outline of the vertebrate embryo. Bilateral pairs of paraxial mesoderm cells flanking the notochord are first organized into epithelial tissue blocks that then separate from more caudal cells in a regulated fashion, giving rise to highly organized transient structures on the dorsal side of the embryo. These structures then differentiate into dermis of the dorsal skin, vertebrae and skeletal muscles of the trunk and tail.
The process of somitogenesis is regulated by synchronized expression of Notch pathway components in the presomitic mesoderm (PSM) (Holley et al., 2000; Jiang et al., 2000; Pourquie, 2001; Pourquie, 2003). These include lunatic fringe in chick and mouse (Forsberg et al., 1998; McGrew et al., 1998), delta homolog deltaC (Jiang et al., 2000) and several Hairy and Enhancer of Split–Related (Her) b-HLH repressor genes her1 and her7 in zebrafish (Henry et al., 2002; Holley et al., 2000; Holley et al., 2002; Oates and Ho, 2002). Inhibiting the Notch pathway in mice and zebrafish severely disrupts posterior somite formation (Conlon et al., 1995; Evrard et al., 1998; Henry et al., 2002; Hrabe de Angelis et al., 1997; Kusumi et al., 1998; Oates and Ho, 2002; Oka et al., 1995; Takke and Campos-Ortega, 1999; van Eeden et al., 1996; Wong et al., 1997; Zhang and Gridley, 1998).
The first Tubingen large scale genetic screen identified five loci, fused somites (fss), beamter (bea), after eight (aei), deadlyseven (des) and white tail/mindbomb (wit/mib), that are required for somite formation in zebrafish (van Eeden et al., 1996). Later, it was shown that four of those genes, bea, aei, des, and wit/mib, encode the respective Notch pathway components: notch ligands deltaC (Julich et al., 2005), deltaD (Holley et al., 2000), notch receptor notch1a (Holley et al., 2002) and mib, an E3 ubiquitin ligase which is required for cellular localization and activity of Delta (Itoh et al., 2003). The absence of posterior somites is a characteristic phenotype observed in all of the Notch pathway mutants. After forming 1 to 9 anterior somites, segmentation of the posterior PSM is interrupted and no new somites are formed (van Eeden et al., 1996).
In a genetic screen to isolate mutations disrupting neural patterning and embryogenesis, we identified an embryonic lethal mutation, SBU2 that has somite defects resembling Notch pathway mutants. In particular, SBU2 mutants only form 6-7 anterior somites. In addition, SBU2 embryos have other phenotypes including defects in muscle fiber differentiation, pigmentation, circulatory system development and neural differentiation. RNA in situ hybridization with mesodermal markers revealed that SBU2 mutants have reduced paraxial mesoderm and little lateral plate mesoderm. However, intermediate mesoderm is largely unaltered suggesting that not all mesodermal subtypes are disrupted by the SBU2 mutation. While early regional neural makers are expressed in SBU2, many markers for differentiated neurons and neural crest are absent.
Using positional cloning, we identified a mutation that produces a premature stop codon within the spt6 gene. The SBU2 defects could be rescued by microinjection of spt6 mRNA into SBU2 mutant embryos. Microinjection of spt6 morpholino oligonucleotides phenocopied the SBU2 phenotype. Together, these results demonstrated that the SBU2 phenotypes are produced by disruption of the spt6 locus. The pandoram313 (pan) mutation was previously shown to disrupt the spt6 locus (Keegan et al., 2002). Since SBU2 is allelic to panm313, we designate our allele panSBU2. The phenotypes of panSBU2 mutants are apparent at an earlier stage and are more severe than the panm313 mutants. This finding demonstrates a broader and earlier requirement for Spt6 in zebrafish embryogenesis then was apparent from characterization of panm313.We present evidence that the phenotypic variations between the two spt6 alleles are the result of genetic background differences.
Spt6 was identified in Saccharomyces cerevisiae as a transcription elongation factor which interacts with histone H3 and H4 (Bortvin and Winston, 1996). Spt6 functions in chromatin disassembly. During transcription, Spt6 allows RNA pol II to pass through the DNA template and re-establishes chromatin structure after RNA pol II passage (Adkins and Tyler, 2006; Hartzog et al., 1998; Kaplan et al., 2003; Saunders et al., 2003; Swanson and Winston, 1992). Hence, Spt6 is required for persistent yet regulated transcription processes in the cell.
In C. elegans, EMB-5, the ortholog of yeast Spt6, genetically interacts with LIN-12 (Notch family transmembrane receptor) (Hubbard et al., 1996). EMB-5 augments the penetrance of LIN-12 related defects on vulval morphology. Furthermore, interactions between the ankyrin repeats of EMB-5 and both LIN-12 and GLP-1 were detected in yeast two-hybrid analysis. These findings suggest that EMB-5/Spt6 might function in modulation chromatin structure upon activation of LIN-12/Notch pathway and regulate transcription of Notch pathway genes (Hubbard et al., 1996).
In spt6 mutants, we found that activation of target genes by ectopic expression of Notch was suppressed. However, analysis of mindbomb;SBU2 double mutants demonstrated that the neurogenic phenotype in mindbomb mutants is suppressed by mutation of Spt6. While the panSBU2 phenotype establishes that Spt6 is broadly required for many aspects of development, we also observe somite defects characteristic of Notch mutants. These studies provide the first evidence for a role for Spt6 in Notch signaling in vertebrates. In addition, we demonstrate a specific developmental requirement for the Spt6 transcription elongation factor in somitogenesis in zebrafish.
Results
Identification of a mutant with disrupted somitogenesis
We performed a haploid ENU mutagenesis screen to identify mutations affecting zebrafish embryogenesis. In SBU2 mutant embryos only 6-7 anterior somites form and further segmentation is severely interrupted (Fig. 1). SBU2 embryos also have a short body length and slightly wide notochord and neural keel which may result from defects in convergence and extension movements. In addition, other defects including tail elongation, anterior neural, circulatory, and pigmentation defects are apparent in SBU2 mutant embryos. After about 20 hrs, many of the previously formed structures start to deteriorate (Fig. 1J).
Figure 1.

Phenotype of SBU2 mutants. (A-J) Lateral views of living WT and SBU2 embryos, anterior to the top and dorsal to the right. (A’-H’) Dorsal views of A-H, anterior to the top. At early somitogenesis, somite boundaries are irregular and indefinite (A’ and B’) in SBU2 embryos. At ~5 somite stage, somites are formed asymmetrically while previously formed somite boundaries start to degenerate (arrows) (C’ and D’). By the 5-somite stage SBU2 mutants have a slightly shorter body length then their wild-type siblings (C and D) and by 13 somite stage, SBU2 mutants are clearly less extended along AP axis than wild type embryos (E and F). While somites continue to form in wild-type embryos, SBU2 mutants have only irregularly formed 6-7 somites and lack posterior somites (arrows) (F’ and H’). By 24-27 hpf, most of the previously formed structures including somite boundaries are degraded or distorted and the embryos degenerate (J). Note: all of the embryos shown in this figure and the following figures are diploids.
The SBU2 somite defects resemble the Notch pathway mutants. Embryos obtained from SBU2 intercrosses do not show any morphological abnormalities during gastrulation (data not shown). During early somitogenesis, SBU2 homozygous embryos can be distinguished from their siblings due to their indistinct and irregularly placed somite boundaries (Fig. 1A-A’ and 1B-B’). At 4-5 somite stage, altered somite boundary formation is evident in SBU2 embryos (Fig. 1D-D’). By the time their heterozygous or wild-type siblings reach the 10-13 somite stage, homozygous SBU2 embryos exhibit only irregularly and asymmetrically formed 6-7 anterior somites (Fig. 1F-F’ and 1H-H’). After that stage, somitogenesis stops in SBU2 mutants. In most SBU2 mutants, previously formed somite boundaries start to degenerate around 4-5 somite stage leaving either incomplete or no somite boundaries by 24 hours post-fertilization (hpf) (Fig. 1J).
Mesodermal gene expression is disrupted in SBU2 mutants
During somitogenesis, the paraxial mesoderm is segmented into highly arranged tissue blocks. Since SBU2 has segmental defects, we examined the effects of the mutation on specification of mesodermal cells. As somites are specifically derived from paraxial mesoderm cells, we first studied the state of differentiated paraxial mesoderm cells in SBU2 embryos. RNA in situ hybridization with paraxial mesodermal markers papc and spt revealed that SBU2 mutants have a reduced expression domain of paraxial mesoderm markers in the A-P dimension (arrowheads, Fig. 2A-D). In addition, the boundaries of the expression domain are less distinct in SBU2 mutants. However, this observation does not explain the somitogenesis defects observed in SBU2, since tri; kny double mutants form very little paraxial mesoderm but somite boundaries still form (Henry et al., 2000).
Figure 2.

SBU2 mutants have mesoderm defects. Whole mount RNA in situ hybridization of mesodermal markers in wild-type and SBU2 mutants. All views are dorsal; anterior to the top. The expression domains of presomitic mesoderm markers papC and spt are reduced in SBU2 mutants (A-D). AP patterning of the somites in SBU2 embryos is also affected; myoD expression at the posterior half of the somites is lost while it is reduced at adaxial cells (E and F), ephB2 at the anterior half of the somites is reduced and more diffused (G and H). gataI expression is diminished in SBU2 mutants (I and J) indicating that lateral plate mesoderm development is highly reduced. There is no apparent difference in pax2a expression between wild type and SBU2 mutants (K and L); SBU2 mutants have no defect in intermediate mesoderm formation. The most anterior and posterior papC and spt expression is marked by arrowheads.
Rostrocaudal polarity of the somites is also disrupted in SBU2 mutants. The segmental expression of myoD in the posterior half of the somites (Weinberg et al., 1996) is lost (Fig. 2E-F) whereas gene expression at anterior half of the somites such as papC (Yamamoto et al., 1998), ephB2 (Durbin et al., 1998) and dld (Holley et al., 2002; Jiang et al., 2000) is reduced and diffuse (Fig. 2A-B, G-H and 3I-L) in SBU2 mutants. Although muscle differentiation is disrupted and myoD expression is completely lost in lateral paraxial mesoderm, adaxial cell specification is less severely disrupted (Fig. 2E-F).
Figure 3.

Somite segmentation and PSM maturation is disrupted in SBU2 mutants. Whole mount RNA in situ hybridization of genes involved in somite segmentation process in wild-type and SBU2 mutants. All views are dorsal; anterior to the top. In wild type embryos, periodic activation of notch signaling provides cycling gene expression of Notch pathway genes such as her1 and her7 (A-D and E-H). dld expression is weak and diffused in presomitic mesoderm as well as anteriormost somites (I-L). tbx24, normally expressed in intermediate and anterior PSM, is also reduced in SBU2 embryos (M-P). Arrows denote stripes of her1, her7 or dld expression.
We also studied differentiation of the other subdivisions of the mesodermal precursors such as lateral plate mesoderm and intermediate mesoderm. An early erythroid lineage marker, gata-1 (Tsai et al., 1989) is not expressed in SBU2 embryos (Fig. 2I, J) suggesting that lateral plate mesoderm in SBU2 embryos is severely disrupted by the 10 somite stage. SBU2 embryos have additional defects in lateral plate mesoderm as evidenced by the failure of heart tube formation (data not shown). However, RNA in situ hybridization with the nephrogenic marker pax2a (Krauss et al., 1991) reveals no difference between wild type and SBU2 embryos (Fig. 2K, L). This suggests that there is no defect in differentiation of intermediate mesoderm. Together these results suggest that the mutation underlying the SBU2 phenotype disrupts somite formation, specification of paraxial and lateral plate mesoderm but intermediate mesoderm is relatively unaffected.
The segmentation of the PSM is regulated by the periodic expression of genes including her1, her7 and deltaC and disruption of these genes causes somitogenesis defects (Gajewski et al., 2003; Henry et al., 2002; Holley et al., 2002; Jiang et al., 2000; Oates and Ho, 2002). To ascertain whether periodic gene expression is altered in SBU2, we assayed expression of these markers (Fig. 3A-L). Distinct stripes of her1 and her7 are observed in wild-type embryos at both the 3 and 10 somite stages. However, in SBU2 mutants, stripes of her1 and her7 are present but exceedingly reduced (arrows, Fig. 3A-H). This shows that expression of molecular clock components is disrupted at early stages of somitogenesis in SBU2 mutants and the loss of molecular clock components correlates with the onset of the segmentation defects. The domain of papc, which is expressed more proximal to somite boundary formation, is spatially disrupted. Thus, it is possible that low levels of periodic gene expression may fail to properly specify expression of downstream genes such as papc that are critical for somite boundary formation (Yamamoto et al., 1998). Expression of another Notch pathway gene deltaD is also reduced but more diffuse in SBU2 mutants (arrows, Fig. 3I-L). Similarly, the expression of tbx24, which is required for PSM maturation (Nikaido et al., 2002) is reduced in SBU2 mutants (Fig 3M-P). These results indicate that somite segmentation as well as PSM maturation is highly disrupted in SBU2 mutants. Muscle differentiation may also be disrupted, as expression of the myogenic regulatory factor MyoD is absent in lateral paraxial mesoderm (Fig. 2F).
Somite boundaries deteriorate in SBU2 mutants
SBU2 mutants have segmentation defects very similar to Notch pathway mutants. To further characterize these defects in SBU2 embryos, we examined the somite structure of 29 somite stage SBU2 mutant embryos using a series of antibodies. Focal adhesion kinase (Fak) is robustly accumulated at somite boundaries during somitogenesis and persists at these boundaries (Henry et al., 2001; Hens and DeSimone, 1995; Polte et al., 1994) (arrows, Fig. 4A, A’). In 29 somite stage SBU2 mutants, Fak fails to accumulate at somite boundaries, even in the anterior of the embryos (Fig. 4B, B’). This observation is unexpected, since anterior somite boundaries formed earlier in development morphologically. Therefore, we conclude that somite boundaries that form are not maintained properly in SBU2 mutants. This result differs from Notch pathway mutant embryos in which irregular posterior somite boundaries eventually form, albeit later in development compared to wild-type embryos (Henry et al., 2005). Thus, the initial somite phenotype of SBU2 and Notch pathway mutant embryos is similar, but the later boundary phenotype of SBU2 is more severe compared to previously characterized Notch pathway mutants.
Figure 4.
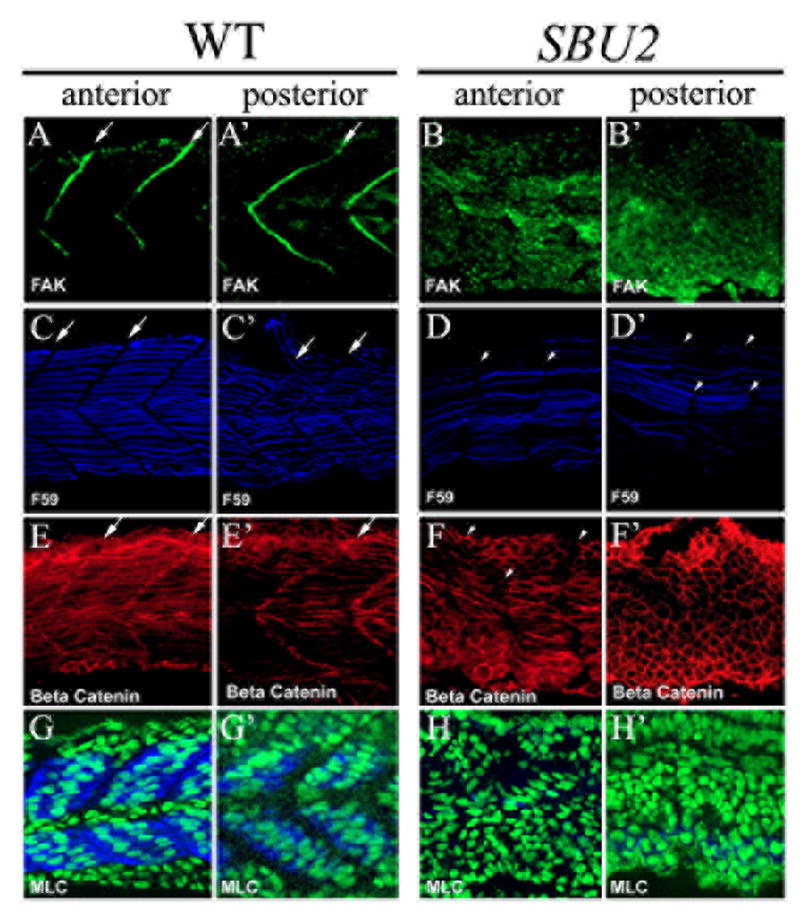
Somite boundary formation and muscle fiber differentiation is disrupted in SBU2 mutants. Confocal sections of anterior (A-H) and posterior (A’-H’) portions of the trunk region 29 somite stage embryos at approximately same medial-lateral and anterior-posterior locations; anterior to the left. Focal adhesion marker phosphorylated Fak is in green; slow muscle marker F59 is in blue; β-catenin which outlines the cell is in red. In wild type embryos, phosphorylated Fak accumulates at myotome borders, revealing regularly formed somite boundaries (A and A’). On the other hand, there is no specific accumulation of phosphorylated FAK in SBU2 mutants (B and B’). The number of the slow muscle fibers is lower both in anterior and posterior (D and D’) compared to wild type muscle fibers (C and C’). In wild-type embryos, fast muscle precursors elongate and attach to the anterior and posterior somite borders (E). In SBU2 mutant embryos, some fast cells elongate in the anterior of the embryos (F). However, in the posterior, few, if any, fast cells elongate (F’). Likewise, the MLC antibody F310 staining (blue) is abundant in wild-type muscle (G and G’) but absent in SBU2 mutant embryos (H and H’), nuclei are labeled with cytoxgreen. Therefore, fast muscle fiber morphogenesis is severely disrupted in SBU2 mutant embryos. Arrows denote somite boundaries. Arrowheads denote improper and degenerated somite boundaries.
It has been hypothesized that migrating slow muscle fibers mediate the belated formation of boundaries in fss-type mutant embryos (Henry et al., 2005; Julich et al., 2005). In SBU2 mutant embryos, boundaries do not form even later in development. We therefore asked if slow muscle fiber morphology is disrupted in SBU2 mutant embryos. Staining with the slow muscle antibody F59 revealed a reduced number of differentiated slow muscle fibers in SBU2 mutants (Fig. 4C, D). Furthermore, the initial segmental defects in these embryos are reflected in slow fiber organization; slow fibers are not as well organized as in wild type embryos. In addition, slow muscle fiber staining becomes fainter and gradually fades away in the posterior (Fig. 4C’, D’). This result is not surprising because small gaps in adaxial (slow muscle precursor) myoD expression (Fig 2F) suggest that there may be fewer slow muscle precursors present in SBU2 embryos which fail to properly differentiate into muscle fibers. The disruption in slow fiber organization at 29 somites in SBU2 is more severe then that of Notch pathway mutants. In both the Notch pathway mutant embryos aei/deltaD and des/Notch1, slow muscle fiber morphology is disrupted early but later recovers (Henry et al., 2005).
Clearly, slow-twitch muscle fiber development is disrupted in SBU2 mutant embryos. Analysis of myoD expression suggests that there might also be a fast muscle defect (Fig. 2F). We utilized an antibody against β-catenin that visualizes all cells to ask if fast muscle morphogenesis is disrupted in SBU2 mutant embryos (Topczewska et al., 2001). In wild-type embryos, the expression and lateral displacement of slow muscle fibers correlates with fast MyHC expression and muscle cell elongation (Blagden et al., 1997; Cortes et al., 2003; Henry and Amacher, 2004). Fast muscle cell elongation is disrupted throughout the axis of SBU2 mutant embryos. Anteriorly, some cells elongate but elongation is inconsistent and disorganized (Fig. 4F). Posteriorly, fast cell elongation is also disrupted (Fig. 4F’). The disruption of fast muscle fiber formation in SBU2 mutant embryos was confirmed by analysis of myosin light chain (MLC) expression. Whereas fibers in wild-type embryos express significant amounts of MLC, protein is undetectable in SBU2 mutant embryos (Figure 4H-H’), Taken together, these data indicate that muscle morphogenesis is more severely disrupted in SBU2 mutant embryos than in Notch pathway mutant embryos.
SBU2 mutants have neural defects
Lateral inhibition mediated by Notch signaling regulates neuron number in both vertebrates and invertebrates. In Drosophila, Notch pathway mutants are neurogenic. In zebrafish, ectopic neurons or neural progenitors are readily apparent in mib (Jiang et al., 1996; Schier et al., 1996). However, the neurogenic defects in des and aei are more subtle (Gray et al., 2001; Holley et al., 2000). To determine whether SBU2 embryos are neurogenic, we performed in-situ hybridization with neural markers. RNA in situ hybridization with midbrain-hindbrain boundary and placode marker, pax2a (Krauss et al., 1991) revealed that placode formation is delayed (arrowheads, Fig. 5D). The overall midbrain-hindbrain boundary and placode expression of pax2a is reduced but the general spatial pattern of pax2a is largely unaffected in SBU2 embryos (Fig. 5B, D). However, the midbrain-hindbrain boundary is not morphologically apparent in living embryos (Fig 1J). This suggests that although these cells initiate proper gene expression at the midbrain-hindbrain, the boundary does not form. Similarly, expression of the hindbrain marker krox20 (Oxtoby and Jowett, 1993) is also delayed in SBU2 embryos. However, as development progresses, krox20 recovers and the overall pattern becomes similar to that of wild type embryos (Fig. 5E-H).
Figure 5.

SBU2 mutants are not neurogenic. Whole mount RNA in situ hybridization of neural markers in wild-type and SBU2 mutants. (A-J) Dorsal views; (A-H) anterior to the top, (I, J) flat mounted, anterior to the right. I’, J’ are lateral views of I and J, anterior to the top right. Morphology of the MHB domain expressing pax2a at early somitogenesis (B) indicates a delay in development of MHB in SBU2 mutants. Later, at 10 somite stage (D) pax2a expression resembles much earlier stage wild type expression domain (A). Likewise, although hindbrain marker krox20 expression is also delayed and relatively reduced (E and F) later in the development, expression pattern appears similar to wild-type embryos (G and H). RNA in situ hybridization using huC probe revealed that SBU2 embryos have reductions in early differentiating neurons (I, I’ and J, J’). Arrows denote MHB and arrowheads denote otic placode.
The neural marker huC, reveals the number of the differentiated neurons in zebrafish embryo during development (Kim et al., 1996). SBU2 embryos express very little, if any, huC (Fig. 5I-I’ and 5J-J’). Similarly, epibranchial neuron marker, phox2a expression is weak or completely absent in SBU2 embryos (data not shown). Together these data indicate that SBU2 embryos generate few differentiated neurons and are not neurogenic. In zebrafish, somitogenesis is more sensitive to disruptions in notch signaling than neurogenesis as evident by the weak neurogenic phenotype in des (Gray et al., 2001). Potential neurogenic effects of SBU2 may be masked by additional requirements for the protein in neurogenesis. Alternatively, SBU2 might not influence Notch-mediated lateral inhibition in neural tissue.
Disruption of spt6 causes the SBU2 phenotype
To identify the gene that is responsible for the SBU2 phenotype, a large genetic mapping panel comprising 6928 meioses was assembled. SBU2 was mapped to a ~5 cM interval in the centromeric region of linkage group 21 between Z7809 and Z10432 Because of the proximity to the centromere, recombination is suppressed in this region. Therefore, the genetic distance corresponds to a larger then average physical distance. Using the sequence information from the Sanger Ensembl genome assembly (www.ensembl.org/Danio_rerio) a series of polymorphic markers was generated to narrow the interval (Fig. 5A). Sequence analysis of the genes at that region revealed a C to T transition at nucleotide 3931 of the coding sequence of the spt6 gene (Fig. 6B, bottom panel). This sequence change was detected in 75/75 mutants analyzed using restriction analysis of a PCR product generated from the locus (see Materials and Methods). 0/95 phenotypically wild-type sibling were homozygous for the sequence change. This mutation creates a premature stop codon in exon 30 (Fig. 6B). The truncated Spt6 protein lacks the C-terminal 415 amino acids which includes a Src homology 2 domain important for recognition and binding to phosphorylated tyrosine (Pawson, 1995) (Fig. 6C).
Figure 6.

SBU2 phenotype is the result of a non-sense mutation on spt6 gene. SBU2 maps to linkage group 21 between Z7809 and Z10432 (A). Sequencing analysis revealed a C to T transition at the 3931st nucleotide of the coding sequence of the spt6 gene (B). Predicted Spt6 protein structure in wild type, panm313 and panSBU2 mutant embryos (C). The premature stop codon in SBU2 is predicted to produce a truncated protein which lacks SH2 domain (C). The defects in homozygous SBU2 embryos can be rescued by injection of spt6 mRNA (G). Microinjection of spt6 morpholino into the progeny from intercrosses of wild-type siblings of SBU2 heterozygotes, phenocopies SBU2 mutants (H). However, in some wild type stocks, the phenotype upon injection of spt6 morpholino is much weaker (I) and resembles pan phenotype (F). (D, E, G, H) are 27-hour-old; (F and I) are 48-hour old embryos. Asterisk denotes stop codon in (B).
To confirm that disruption of the spt6 gene produces the SBU2 phenotype, we assayed the ability of spt6 mRNA to rescue the SBU2 phenotype and determined whether microinjection of morpholinos directed against spt6 phenocopy the SBU2 mutants. spt6 mRNA was injected into embryos from a SBU2 heterozygote intercross. Overexpression of spt6 sense mRNA in SBU2 mutants rescues most of the somitogenesis and tail elongation defects (compare Fig. 6E and 6G). Overexpression of spt6 mRNA doesn’t produce an effect on wild type embryos (data not shown).
Next, we designed a morpholino antisense oligonucleotide to span the boundary between the 30th exon and 30th intron of spt6. spt6 morpholino binding to the spt6 pre-mRNA is predicted to create a missplicing event that deletes exon 30 and causes a frameshift mutation. The protein transcribed from that misspliced spt6 RNA is predicted to closely mimic the Spt6 protein structure in SBU2 mutants. Injection of spt6 morpholino into genotypically wild-type siblings of SBU2 mutants results in embryos that strongly resemble the SBU2 phenotype (compare Fig. 6E and H). Based on linkage, sequence change, rescue and morpholino experiments; we conclude that disruption of spt6 results in the SBU2 phenotype.
An spt6 mutation was previously shown to be the cause of the pandora phenotype (Keegan et al., 2002). The genetic lesion in pan produces a truncated protein of 829 amino acids. We think that both mutations likely produce non-functional proteins. pandora mutants exhibit defects in cardiac differentiation, pigmentation and ear formation. However, stronger defects in these cell types are also observed in SBU2 mutants (data not shown). A complementation test was performed to determine if the two mutations are allelic. Since the two mutations fail to complement, we designate our allele as panSBU2. In some crosses panm313/panSBU2 embryos resemble panm313 mutants and in others panSBU mutants (data not shown). This result suggest that genetic background strongly influence the spt6 mutant phenotype. In addition injection of the spt6 morpholino into some wild-type stocks produces a phenotype (Fig. 6I) more similar to panm313 (Fig. 6F) than panSBU2 (Fig. 6E). Therefore, we conclude that the phenotypic differences between panm313 and panSBU2 most likely stem from genetic background differences and not from difference in properties of the alleles.
Notch signaling is suppressed in SBU2 mutants
Since the somite defects in spt6 mutants resemble Notch pathway mutants, we sought to determine whether Spt6 is required for activation of the Notch pathway. We activated the Notch pathway by microinjection of mRNA corresponding to the intracellular domain of the Notch protein (NICD) into SBU2 embryos and measured transcript levels of Notch target genes at the 10-somite stage (Fig. 7A-C). As expected, activation of Notch pathway induced transcription of Notch pathway target genes, her1, her6, her7 (Fig. 7A-C,). Conversely, homozygous SBU2 mutants have reduced levels of those transcripts (Fig. 7A-C,). This observation is consistent with what was observed in the RNA in situ hybridization analysis of SBU2 (fig 2E-H). However, induction of target genes by the NICD mRNA was suppressed in SBU2 mutant embryos (Fig. 7A-C). It should be noted that disruption of Spt6 function in SBU2 embryos does not have same effect on every Notch target gene. her6 transcript levels are not highly affected by disruption of Spt6 function while her1 and her7, two essential Notch target genes for somite formation, are clearly suppressed. These results demonstrate that Spt6 is required for proper response to Notch activation.
Figure 7.

Spt6 genetically interacts with Notch1a. (A-D) Activation of Notch pathway was monitored by the relative quantification of transcripts of Notch pathway target genes. The intracellular domain of Notch1a (NICD) was injected to 1-4 cell stage embryos obtained from SBU2 incross; cDNA synthesized from total RNA pools from 10 somite embryos were used for real time PCR. Each set of data is normalized to transcript level from the GFP injected wild type siblings. Bars represent means of measurements from three different mRNA pools. Standard error of the mean is also shown. Levels of her1, her6 and her7 transcripts are increased when Notch pathway is activated via NICD injection (third bar in A, B, C). Transcripts of Notch pathway target genes are reduced in SBU2 mutants. (second bar in A, B, C). However, SBU2 mutation impedes activation of Notch pathway upon NICD injection; her1, her6 and her7 transcript levels stay relatively lower with respect to transcript levels in NICD injected wild type siblings (compare third and fourth bar in A, B, C) when Spt6 gene is disrupted in NICD injected embryos.
Genetic interactions between Spt6 mutation and mindbomb
The Notch pathway regulates somitogenesis in zebrafish embryos by controlling the synchronous gene expression at the PSM. Mutation of Notch pathway genes leads to characteristic posterior somite defects (van Eeden et al., 1996) very similar to SBU2 mutants. This suggests that Spt6 might have a function in the Notch pathway in regulation of somite formation. Previous studies also indicate a genetic interaction between the C. elegans homologue of Spt6, EMB-5 and the C. elegans notch homologues LIN-12/GLP-1 (Hubbard et al., 1996). To determine whether mutation of Spt6 enhances the phenotype of Notch pathway mutants in zebrafish, we generated mib;SBU2 double mutants. mindbomb (mib) is an E3 ubiquitin ligase required intracellular localization of the Notch ligand Delta. The overall somite morphology and body shape of the double mutants resembled SBU2 single mutants (Fig. 8A-D). The somite defects caused by disruption of Spt6 is not enhanced by mib mutation.
Figure 8.

mib; SBU2 double mutants reveal an epistatic interaction between Spt6 and Notch pathway. (A-D) Lateral views of 27 hour-old embryos, anterior to the right. (E-H) Dorsal views of huC stained embryos, anterior to the right. All embryos were genotyped following photography. The phenotype of mib; SBU2 mutants (D) resembles SBU2 single mutants (C), rather than mib single mutant embryos (B). However, huC expression profile in mib; SBU2 double mutants (H) is stronger than SBU2 mutants (G) but not as strong as either wild type (E) or mib single mutants (F).
To determine if mutation of spt6 enhances the neurogenic phenotype of mib, we performed RNA in situ hybridization with a huC probe on the double mutants. We observed decreased numbers of huC positive cells in mib;SBU2 double mutants compared to mib single mutants (Fig. 8E-H). The number of huC positive cells in the double mutant was less than in wild-type embryos but more then in SBU2 single mutants (Fig. 8E-H). Disruption of Spt6 does not enhance the neurogenic defects in Mib but instead largely suppresses them. These findings suggest that Spt6 is not required for Notch mediated suppression of neurogenesis.
Discussion
We identified and characterized an embryonic lethal mutation, SBU2, which gives rise to somite abnormalities in zebrafish embryos. These defects are similar to, but more severe than Notch mutant embryos suggesting that additional pathways require SBU2. SBU2 mutants are characterized by severe posterior segmentation and differentiation defects, disruption of gene expression in paraxial and lateral plate mesoderm, but intermediate mesoderm is unaffected. Moreover, although the patterns of many early regional neural markers in the mutant embryos are not severely altered, later, neural differentiation is highly disrupted. We utilized mRNA rescue and morpholino knockdown experiments to demonstrate that the phenotype observed in SBU2 mutant embryos is due to disruption of the spt6 locus. Altogether, our studies indicated that Spt6 has important functions during early patterning and somitogenesis of the zebrafish embryo.
Relationship of SBU2 and pandora
A mutation that disrupts splicing of spt6 and is predicted to give rise to a truncated protein was shown to give rise to pan phenotype (Keegan et al., 2002). Complementation studies between SBU2 and pan confirmed that the two mutations are allelic. In panm313 mutants, however, the truncated Spt6 protein is predicted to be much shorter than the protein produced in SBU2 mutants. The Panm313 protein lacks the S1 RNA binding domain in addition to the SH2 domain. Given the function of Spt6 as a modulator of chromatin structure, the absence of the S1 nucleic acid binding domain in pan is consistent with the loss of function of the Spt6 protein. On the other hand, Spt6 protein produced by panSBU2 which lacks only the SH2 domain might also fail to interact with other proteins in the transcription elongation machinery. Although both of the spt6 alleles likely encode non-functional proteins, the phenotypic differences caused by those mutations are very striking. SBU2 mutants reveal a much earlier and stronger phenotype than pan mutants and die around 2 dpf while pan embryos display defects much later and usually survive more than 7 days. Moreover, microinjection of spt6 morpholino can phenocopy panSBU2 or panm313 depending on the background of the injected embryos (Fig 6). In addition panm313/panSBU2 embryos may resemble panm313 or panSBU. These observations suggest that severity of the defects in spt6 mutants is highly genetic background dependent and variations in levels of maternal Spt6 protein may to some degree impact the extent of the defects observed. Similarly, yeast are sensitive to SPT6 copy number (Clark-Adams and Winston, 1987).
Relationship of Spt6 to the Notch pathway
Spt6 protein has been studied in human, mouse, zebrafish, Drosophila, C. elegans and yeast. It has been shown to interact with many components of the transcriptional machinery. In yeast, Spt6 protein has ATP-independent nucleosome assembly function, controlling transcription of various sets of genes by modulating the assembly state of the nucleosomes during active transcription (Bortvin and Winston, 1996). In addition spt6 mutations can suppress the effects of the mutations affecting the Snf/Swi chromatin remodeling complex in yeast (Malone et al., 1993). Similarly, in Drosophila, it was shown that Spt6 protein is associated only with the active form of RNA pol II and has a role in regulation of transcription (Kaplan et al., 2000). Moreover, human SPT6 antagonistically interacts with histones as well as the transactivator domain of human cytomegalovirus pUL69 to modulate transcription (Winkler et al., 2000). Overall, Spt6 is a part of transcription elongation machinery and regulates the transcription of corresponding genes by modulating the chromatin structure in the cell in wide range of organisms. However, in C.elegans, EMB-5, a homolog of yeast Spt6, was characterized as a regulator of timing of the gut processor division during gastrulation and controller of oogenesis and required for specific synchronous cell division timing of E cells (Nishiwaki et al., 1993).
Many of the Notch pathway components found in invertebrates are conserved in vertebrates as well (reviewed in (Greenwald, 1998). Our work provides further evidence that Spt6 function is also conserved in a vertebrate model and suggests that it interacts with complexes containing the Notch ICD. An interaction between the C. elegans homolog of Spt6, EMB-5 and ankyrin repeats of the LIN-12 and GLP-1, Notch family transmembrane receptors (Hubbard et al., 1996) was identified through yeast 2-hybrid assay. Moreover, when emb-5 function is eliminated from the lin-12 mutant embryos, the penetrance of the lin-12 phenotype is enhanced whereas penetrance of the constitutively active lin-12 phenotype is reduced. In addition, emb-5 is required for the proper germline development regulated by glp-1 (Hubbard et al., 1996). Ectopic activation of glp-1 cannot rescue the phenotype caused by emb-5 mutation (Hubbard et al., 1996) indicating that emb-5 is epistatic to glp-1. These observations also suggest that there is a direct functional interaction between emb-5 and lin-12/glp1.
A recent study also supports the functional interaction between Spt6 and Notch pathway in Drosophila (Tenney et al., 2006). Rad-6/Bre1 histone modification activity is essential for transcription of Notch target genes (Bray et al., 2005). Paf-1 complex is required for this activity (Wood et al., 2003). Knocking-down Rtf-1, a component of the Paf-1 transcription elongation complex, in Nnd-1 notch mutant background enhances the phenotype of the notch mutation. Furthermore, the zebrafish homolog of the yeast Rtf-1 protein was recently shown to be disrupted in a novel zebrafish mutant, white zebra (wze) (T. Akanuma and S. Takada, personal communications). wze also exhibits segmentation defects similar to Notch pathway mutants and panSBU2.
Since genetic studies indicate that Spt6 interacts with Rtf-1 (Costa and Arndt, 2000), there is a strong possibility that Spt6 also functions in Notch signaling along with Paf-1/Bre1 transcription elongation complex. Other studies (reviewed in (Tsukiyama and Wu, 1997)) support the requirement for appropriate chromatin modifying and remodeling functions for proper transcription of particular genes. In this study, we demonstrated that the response to ectopic Notch activation requires Spt6 function. However, it should be noted that disruption of Spt6 function does not have the same effect on every Notch target gene. We observed much stronger effects on her1 and her7 which are required for proper somite formation, then on her6. Likewise, mutation of Spt6 did not enhance the neurogenic phenotype of mib.
In addition to playing a role in Notch signaling, other cellular processes are affected by spt6 mutation since the overall panSBU2 phenotype is much more severe and complex than Notch pathway mutants. Several signaling pathways including FGF and Wnt are required for posterior development. Some of the defects in panSBU may stem from defects in these pathways. Most of the Notch pathway mutants can be raised to adulthood indicating that the somitogenesis defects observed in those embryos improve later in development however in panSBU2 mutants, defects become much worse; even the previously formed structures cannot be maintained, and embryos eventually deteriorate and die. Although spt6 is required for segmentation of the paraxial mesoderm in the zebrafish embryo, it is also required for many other basic cellular functions. While chromatin remodeling factors like Spt6 have broad cellular roles, they also may be required for specific signal transduction pathways and regulate specific developmental processes.
Materials and methods
Fish husbandry and mutagenesis
Adult zebrafish strains and embryos obtained from natural crossings were maintained at 28.5°C. Developmental stages of the embryos were determined according to Kimmel et. al. (Kimmel et al., 1995). For mutagenesis, adult fish were placed in 3.0 mm ENU for 1 hour, once a week for three weeks.
Positional mapping of SBU2
Genomic DNA extractions and PCR conditions were performed as described in (Gates et al., 1999). SSLP primers were designed using the zebrafish SSR program (http://danio.mgh.harvard.edu). PCR products were electrophoresed on 3% MetaPhore (Cambrex, Rockland, ME) agarose gels. New polymorphic marker sequences were:
| scaf1051ssr7 | F: GCCAGTAAATTTTGGCCTTG
R: GACGTGTGAAGCTGCAGAAA |
| baczk17e16ssr1 | F: ACTGTGCTCTGATGCCTCCT
R: TGCAAAAATAAGCAAATAAACAAA |
| baczk17e16ssr3 | F: CATTCCGAAATGACCCAGTT
R: GCCTTTGATTATGTCTGTAAAAGC |
| baczk17e16ssr5 | F: CACAACAACCAACCGCTCTA
R: CGCAGTACTGAAGACGCAAA |
| baczk54m16ssr | F: GCCTCGTCCCTACAAACAAA
R: AACCGCTTGGTATGCTGAAC |
| paczkp6a12ssr | F: CAAACAACAACTGGGCACTTT
R: AGATCCAGATCCGGCTTGA |
| paczkp6a12snpf | F: CACAAAGGAGATGTCATGCTG
R: GCAAGACTTTAACACTGCTACCAGAC |
| scaf1767ssr8 | F: GTGCAGGCCTTGGTGTGTAT
R: AAGGTTACCATAAGCCATTAACAA |
| baczk220h13 | F: CAGGTATTTTCCTACACTAACAACAC
R: AATTGCCAGATGAAACATGC |
| bacfin612ssr | F: ACATCCACACAGCCACTCAT
R: GGGGTGTAATGGTACACTGGA |
Expression constructs, mRNA synthesis and morpholinos
spt6 and myc-NICD expression constructs were kindly provided by Deborah Yelon and Scott Holley and generation of these constructs is previously described (Keegan et al., 2002; Takke et al., 1999).
Capped sense mRNA was synthesized using mMESSAGE mMACHINE Kit (Ambion, Austin, TX). Splice morpholino that targets 30th exon-intron boundary of the spt6 pre-mRNA blocking the proper nuclear processing is designed and synthesized by Gene Tools. The sequence of the morpholino used is 5’-GCCATAGGAACAGCTCACCTCAGTG-3’. Standard control oligo (Gene Tools, Philomath, OR) is used as control. mRNA and morpholino solutions were diluted to desired concentration with 0.2 M KCl supplemented with phenol red. Typically, 100 pg of spt6 mRNA, 500 pg of NICD mRNA and 10 ng of spt6 splice morpholino is injected to one- to two-cell stage embryos.
Whole mount in-situ hybridization and immunohistochemistry
In situ hybridizations were performed according to Thisse et. al. (Thisse et al., 1993). Digoxigenin labeled probes for in situ hybridization were synthesized using T7, T3 or Sp6 RNA polymerase (Roche, Indianapolis, IN). Hybridized probes were detected using NBT/BCIP system (Roche, Indianapolis, IN). Stained embryos were re-fixed in 4% PFA and either stored in 100% methanol or cleared in Benzyl benzoate/benzyl alcohol solution (2:1) and mounted in Canada balsam/methyl salicylate (2.5% v/v) or flat mounted in 70% glycerol. Embryos were viewed with Zeiss Axioplan microscope, digitally photographed with Zeiss Axiocam camera. Images were processed and assembled with Zeiss Axiovision and Adobe Photoshop.
β-catenin, Fak, and F59 staining were performed as previously described (Henry et al 2001; Topczewska et al., 2001; Henry and Amacher, 2004). Embryos were fixed in 4% paraformaldehyde (PFA) for 4 hours at room temperature and incubated in block for 1 hour. Staining was conducted in PBDT (1% BSA, 1% DMSO, 1% Triton X-100 in PBS), embryos were rinsed for 2 hours, then secondary staining proceeded overnight. Antibodies used were: mouse monoclonal anti-slow-twitch myosin (F59) (Devoto, et al. 1996, generous gift of Frank Stockdale) 1:10, rabbit polyclonal anti-pY397FAK (Biosource, Camarillo, CA) 1:50, mouse monoclonal myosin light chain (F310) (University of Iowa Developmental studies hybridoma bank) 1:10 and rabbit polyclonal anti-β-catenin (Abcam, Cambridge, UK) 1:500. Alexa-Fluor 488, 546, and 633 conjugated goat anti-mouse and goat anti-rabbit secondary antibodies (Invitrogen, Carlsbad, CA) 1:200. Images were acquired using a Leica SP2 confocal microscope and a Zeiss ApoTome running on a Zeiss Axio Imager Z1. For confocal images, embryos were mounted in 100% glycerol and visualized using a 40x oil immersion lens. Image quality was optimized using 2-16 line averaging. Images were processed in Adobe Photoshop and collated in Adobe Illustrator.
Genotyping SBU2
All SBU2;mib double mutants and SBU2 embryos younger than 10 somites are genotyped either after they reach to desired stage and imaged or scored after in-situ hybridization. Genomic DNA is extracted from whole embryos using DNeasy tissue kit (Qiagen, Valencia, CA) or followed the protocol previously described. PCR primers used to genotype SBU2 embryos are:
Spt6 pvuII F AATGCTGACTGGTTCTCAGCT
Spt6 R2 GCCGCATAATGAAGATCGAC
Following PCR, products were digested with PvuII which cuts the wild-type allele.
Relative quantitation of gene expression by real-time PCR
Total RNA and genomic DNA from single GFP or NICD mRNA injected embryo obtained from SBU2 incross were extracted by using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol at 10 somite stage. After each embryo is genotyped, three total RNA preps are pooled and SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA) was used to synthesize first strand cDNA from 0.3 ng of total RNA. Real time PCR was carried out using ABI Prism 7900HT Fast Real-Time PCR system (Applied Biosystems, Foster City, CA) in 15 μL of final volume using 2X FastStart SYBR Green Master (Roche, Indianapolis, IN). Primer pairs used to amplify desired transcripts were:
β-actin: GATTCGCTGGAGATGATG/GTCTTTCTGTCCCATACCAA
her1: AGGCGATTCTAGCAAGGACA/CGAGTTATGGGTTTGGATGG
her6: CCTTGGTAGACTCCGAGGAAA/CGCTGAACAAAGAAAACAAGTG
her7: CAACCAACCTAATCAGAGACGA/ TCTGACAGGCAGTCTGATGG
Total RNA amount of each sample is normalized to relative amount of β-actin transcripts in each pool. In each experiment, 3 pools of control and experimental samples were run in duplicates; Ct values of each pool is averaged and relative amounts of gene expression was calculated using relative standard curve prepared for each primer set in each particular real time PCR run.
Acknowledgments
We thank Bernadette Holdener, Peter Gergen, Will Talbot, Keith Gates and Eric Londin for helpful suggestions and comments; Richard Grady, Mihoko Yamamoto and Susanna Li for Fish care; Jack Niemiec for technical assistance. We are also grateful to the many labs that provided reagents and advice, in particular the Yelon laboratory. This work was supported by NIH grant 1RO1HD043998 (HS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adkins MW, Tyler JK. Transcriptional activators are dispensable for transcription in the absence of Spt6-mediated chromatin reassembly of promoter regions. Mol Cell. 2006;21:405–16. doi: 10.1016/j.molcel.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Blagden CS, et al. Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev. 1997;11:2163–75. doi: 10.1101/gad.11.17.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortvin A, Winston F. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996;272:1473–6. doi: 10.1126/science.272.5267.1473. [DOI] [PubMed] [Google Scholar]
- Bray S, et al. Bre1 is required for Notch signaling and histone modification. Dev Cell. 2005;8:279–86. doi: 10.1016/j.devcel.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Clark-Adams CD, Winston F. The SPT6 gene is essential for growth and is required for delta-mediated transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:679–86. doi: 10.1128/mcb.7.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon RA, et al. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–45. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- Cortes F, et al. Cadherin-mediated differential cell adhesion controls slow muscle cell migration in the developing zebrafish myotome. Dev Cell. 2003;5:865–76. doi: 10.1016/s1534-5807(03)00362-9. [DOI] [PubMed] [Google Scholar]
- Costa PJ, Arndt KM. Synthetic lethal interactions suggest a role for the Saccharomyces cerevisiae Rtf1 protein in transcription elongation. Genetics. 2000;156:535–47. doi: 10.1093/genetics/156.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin L, et al. Eph signaling is required for segmentation and differentiation of the somites. Genes Dev. 1998;12:3096–109. doi: 10.1101/gad.12.19.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrard YA, et al. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature. 1998;394:377–81. doi: 10.1038/28632. [DOI] [PubMed] [Google Scholar]
- Forsberg H, et al. Waves of mouse Lunatic fringe expression, in four-hour cycles at two-hour intervals, precede somite boundary formation. Curr Biol. 1998;8:1027–30. doi: 10.1016/s0960-9822(07)00424-1. [DOI] [PubMed] [Google Scholar]
- Gajewski M, et al. Anterior and posterior waves of cyclic her1 gene expression are differentially regulated in the presomitic mesoderm of zebrafish. Development. 2003;130:4269–78. doi: 10.1242/dev.00627. [DOI] [PubMed] [Google Scholar]
- Gates MA, et al. A genetic linkage map for zebrafish: comparative analysis and localization of genes and expressed sequences. Genome Res. 1999;9:334–47. [PubMed] [Google Scholar]
- Gray M, et al. Zebrafish deadly seven functions in neurogenesis. Dev Biol. 2001;237:306–23. doi: 10.1006/dbio.2001.0381. [DOI] [PubMed] [Google Scholar]
- Greenwald I. LIN-12/Notch signaling: lessons from worms and flies. Genes Dev. 1998;12:1751–62. doi: 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- Hartzog GA, et al. Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes Dev. 1998;12:357–69. doi: 10.1101/gad.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CA, Amacher SL. Zebrafish slow muscle cell migration induces a wave of fast muscle morphogenesis. Dev Cell. 2004;7:917–23. doi: 10.1016/j.devcel.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Henry CA, et al. Roles for zebrafish focal adhesion kinase in notochord and somite morphogenesis. Dev Biol. 2001;240:474–87. doi: 10.1006/dbio.2001.0467. [DOI] [PubMed] [Google Scholar]
- Henry CA, et al. Somites in zebrafish doubly mutant for knypek and trilobite form without internal mesenchymal cells or compaction. Curr Biol. 2000;10:1063–6. doi: 10.1016/s0960-9822(00)00677-1. [DOI] [PubMed] [Google Scholar]
- Henry CA, et al. Interactions between muscle fibers and segment boundaries in zebrafish. Dev Biol. 2005;287:346–60. doi: 10.1016/j.ydbio.2005.08.049. [DOI] [PubMed] [Google Scholar]
- Henry CA, et al. Two linked hairy/Enhancer of split-related zebrafish genes, her1 and her7, function together to refine alternating somite boundaries. Development. 2002;129:3693–704. doi: 10.1242/dev.129.15.3693. [DOI] [PubMed] [Google Scholar]
- Hens MD, DeSimone DW. Molecular analysis and developmental expression of the focal adhesion kinase pp125FAK in Xenopus laevis. Dev Biol. 1995;170:274–88. doi: 10.1006/dbio.1995.1214. [DOI] [PubMed] [Google Scholar]
- Holley SA, et al. Control of her1 expression during zebrafish somitogenesis by a delta-dependent oscillator and an independent wave-front activity. Genes Dev. 2000;14:1678–90. [PMC free article] [PubMed] [Google Scholar]
- Holley SA, et al. her1 and the notch pathway function within the oscillator mechanism that regulates zebrafish somitogenesis. Development. 2002;129:1175–83. doi: 10.1242/dev.129.5.1175. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis M, et al. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997;386:717–21. doi: 10.1038/386717a0. [DOI] [PubMed] [Google Scholar]
- Hubbard EJ, et al. Evidence for physical and functional association between EMB-5 and LIN-12 in Caenorhabditis elegans. Science. 1996;273:112–5. doi: 10.1126/science.273.5271.112. [DOI] [PubMed] [Google Scholar]
- Itoh M, et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev Cell. 2003;4:67–82. doi: 10.1016/s1534-5807(02)00409-4. [DOI] [PubMed] [Google Scholar]
- Jiang YJ, et al. Notch signalling and the synchronization of the somite segmentation clock. Nature. 2000;408:475–9. doi: 10.1038/35044091. [DOI] [PubMed] [Google Scholar]
- Jiang YJ, et al. Mutations affecting neurogenesis and brain morphology in the zebrafish, Danio rerio. Development. 1996;123:205–16. doi: 10.1242/dev.123.1.205. [DOI] [PubMed] [Google Scholar]
- Julich D, et al. beamter/deltaC and the role of Notch ligands in the zebrafish somite segmentation, hindbrain neurogenesis and hypochord differentiation. Dev Biol. 2005;286:391–404. doi: 10.1016/j.ydbio.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, et al. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003;301:1096–9. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]
- Kaplan CD, et al. Spt5 and spt6 are associated with active transcription and have characteristics of general elongation factors in D. melanogaster. Genes Dev. 2000;14:2623–34. doi: 10.1101/gad.831900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan BR, et al. The elongation factors Pandora/Spt6 and Foggy/Spt5 promote transcription in the zebrafish embryo. Development. 2002;129:1623–32. doi: 10.1242/dev.129.7.1623. [DOI] [PubMed] [Google Scholar]
- Kim CH, et al. Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci Lett. 1996;216:109–12. doi: 10.1016/0304-3940(96)13021-4. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, et al. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- Krauss S, et al. Expression of the zebrafish paired box gene pax[zf-b] during early neurogenesis. Development. 1991;113:1193–206. doi: 10.1242/dev.113.4.1193. [DOI] [PubMed] [Google Scholar]
- Kusumi K, et al. The mouse pudgy mutation disrupts Delta homologue Dll3 and initiation of early somite boundaries. Nat Genet. 1998;19:274–8. doi: 10.1038/961. [DOI] [PubMed] [Google Scholar]
- Malone EA, et al. Molecular and genetic characterization of SPT4, a gene important for transcription initiation in Saccharomyces cerevisiae. Mol Gen Genet. 1993;237:449–59. doi: 10.1007/BF00279450. [DOI] [PubMed] [Google Scholar]
- McGrew MJ, et al. The lunatic fringe gene is a target of the molecular clock linked to somite segmentation in avian embryos. Curr Biol. 1998;8:979–82. doi: 10.1016/s0960-9822(98)70401-4. [DOI] [PubMed] [Google Scholar]
- Nikaido M, et al. Tbx24, encoding a T-box protein, is mutated in the zebrafish somite-segmentation mutant fused somites. Nat Genet. 2002;31:195–9. doi: 10.1038/ng899. [DOI] [PubMed] [Google Scholar]
- Nishiwaki K, et al. emb-5, a gene required for the correct timing of gut precursor cell division during gastrulation in Caenorhabditis elegans, encodes a protein similar to the yeast nuclear protein SPT6. Mol Gen Genet. 1993;239:313–22. doi: 10.1007/BF00276929. [DOI] [PubMed] [Google Scholar]
- Oates AC, Ho RK. Hairy/E(spl)-related (Her) genes are central components of the segmentation oscillator and display redundancy with the Delta/Notch signaling pathway in the formation of anterior segmental boundaries in the zebrafish. Development. 2002;129:2929–46. doi: 10.1242/dev.129.12.2929. [DOI] [PubMed] [Google Scholar]
- Oka C, et al. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- Oxtoby E, Jowett T. Cloning of the zebrafish krox-20 gene (krx-20) and its expression during hindbrain development. Nucleic Acids Res. 1993;21:1087–95. doi: 10.1093/nar/21.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–80. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Polte TR, et al. Focal adhesion kinase is abundant in developing blood vessels and elevation of its phosphotyrosine content in vascular smooth muscle cells is a rapid response to angiotensin II. J Cell Biochem. 1994;55:106–19. doi: 10.1002/jcb.240550113. [DOI] [PubMed] [Google Scholar]
- Pourquie O. Vertebrate somitogenesis. Annu Rev Cell Dev Biol. 2001;17:311–50. doi: 10.1146/annurev.cellbio.17.1.311. [DOI] [PubMed] [Google Scholar]
- Pourquie O. The segmentation clock: converting embryonic time into spatial pattern. Science. 2003;301:328–30. doi: 10.1126/science.1085887. [DOI] [PubMed] [Google Scholar]
- Saunders A, et al. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094–6. doi: 10.1126/science.1085712. [DOI] [PubMed] [Google Scholar]
- Schier AF, et al. Mutations affecting the development of the embryonic zebrafish brain. Development. 1996;123:165–78. doi: 10.1242/dev.123.1.165. [DOI] [PubMed] [Google Scholar]
- Sirotkin HI, et al. bozozok and squint act in parallel to specify dorsal mesoderm and anterior neuroectoderm in zebrafish. Development. 2000;127:2583–92. doi: 10.1242/dev.127.12.2583. [DOI] [PubMed] [Google Scholar]
- Swanson MS, Winston F. SPT4, SPT5 and SPT6 interactions: effects on transcription and viability in Saccharomyces cerevisiae. Genetics. 1992;132:325–36. doi: 10.1093/genetics/132.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takke C, Campos-Ortega JA. her1, a zebrafish pair-rule like gene, acts downstream of notch signalling to control somite development. Development. 1999;126:3005–14. doi: 10.1242/dev.126.13.3005. [DOI] [PubMed] [Google Scholar]
- Takke C, et al. her4, a zebrafish homologue of the Drosophila neurogenic gene E(spl), is a target of NOTCH signalling. Development. 1999;126:1811–21. doi: 10.1242/dev.126.9.1811. [DOI] [PubMed] [Google Scholar]
- Tenney K, et al. Drosophila Rtf1 functions in histone methylation, gene expression, and Notch signaling. Proc Natl Acad Sci U S A. 2006;103:11970–4. doi: 10.1073/pnas.0603620103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, et al. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development. 1993;119:1203–15. doi: 10.1242/dev.119.4.1203. [DOI] [PubMed] [Google Scholar]
- Topczewska JM, et al. The winged helix transcription factor Foxc1a is essential for somitogenesis in zebrafish. Genes Dev. 2001;15:2483–93. doi: 10.1101/gad.907401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SF, et al. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature. 1989;339:446–51. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, Wu C. Chromatin remodeling and transcription. Curr Opin Genet Dev. 1997;7:182–91. doi: 10.1016/s0959-437x(97)80127-x. [DOI] [PubMed] [Google Scholar]
- van Eeden FJ, et al. Mutations affecting somite formation and patterning in the zebrafish, Danio rerio. Development. 1996;123:153–64. doi: 10.1242/dev.123.1.153. [DOI] [PubMed] [Google Scholar]
- Weinberg ES, et al. Developmental regulation of zebrafish MyoD in wild-type, no tail and spadetail embryos. Development. 1996;122:271–80. doi: 10.1242/dev.122.1.271. [DOI] [PubMed] [Google Scholar]
- Winkler M, et al. Functional interaction between pleiotropic transactivator pUL69 of human cytomegalovirus and the human homolog of yeast chromatin regulatory protein SPT6. J Virol. 2000;74:8053–64. doi: 10.1128/jvi.74.17.8053-8064.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, et al. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–92. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Wood A, et al. The Paf1 complex is essential for histone monoubiquitination by the Rad6-Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem. 2003;278:34739–42. doi: 10.1074/jbc.C300269200. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, et al. Zebrafish paraxial protocadherin is a downstream target of spadetail involved in morphogenesis of gastrula mesoderm. Development. 1998;125:3389–97. doi: 10.1242/dev.125.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Gridley T. Defects in somite formation in lunatic fringe-deficient mice. Nature. 1998;394:374–7. doi: 10.1038/28625. [DOI] [PubMed] [Google Scholar]