

Abstract
Cyt c (cytochrome c) has been traditionally envisioned as rapidly diffusing in two dimensions at the surface of the mitochondrial inner membrane when not engaged in redox reactions with physiological partners. However, the discovery of the extended lipid anchorage (insertion of an acyl chain of a bilayer phospholipid into the protein interior) suggests that this may not be exclusively the case. The physical and structural factors underlying the conformational changes that occur upon interaction of ferrous cyt c with phospholipid membrane models have been investigated by monitoring the extent of the spin state change that result from this interaction. Once transiently linked by electrostatic forces between basic side chains and phosphate groups, the acyl chain entry may occur between two parallel hydrophobic polypeptide stretches that are surrounded by positively charged residues. Alteration of these charges, as in the case of non-trimethylated (TML72K) yeast cyt c and Arg91Nle horse cyt c (where Nle is norleucine), led to a decline in the binding affinity for the phospholipid liposomes. The electrostatic association was sensitive to ionic strength, polyanions and pH, whereas the hydrophobic interactions were enhanced by conformational changes that contributed to the loosening of the tertiary structure of cyt c. In addition to proposing a mechanistic model for the extended lipid anchorage of cyt c, we consider what, if any, might be the physiological relevance of the phenomenon.
Keywords: cytochrome c, extended lipid anchorage, hydrophobic interaction, membrane binding, molten globule state, spin state change
Abbreviations: CL, cardiolipin; cyt c, cytochrome c; DOPS, DL-threo-3,4-dihydroxyphenylserine; IMS, intermembrane space; Nle, norleucine; PC, L-α-phosphatidylcholine; PE, L-α-phosphatidylethanolamine; PG, phosphatidylglycerol
INTRODUCTION
In the mitochondrial IMS (intermembrane space), cyt c (cytochrome c) is involved in respiration by transferring electrons from cyt c reductase (bc1 complex) to cyt c oxidase [1]. However, when released in the cytosol, cyt c is instrumental in the early stages of apoptosis. Its recruitment in the apoptosome complex facilitates activation of several caspases that ultimately lead to cell death [2]. The exact mechanism of cyt c release from the mitochondrion is not understood; nevertheless, it is known to be a controlled process and not a result of mitochondrial degradation.
The basic assumption that cyt c binds to the mitochondrial inner membrane through electrostatic attraction to the phospholipid head groups has been challenged from time to time and, most recently, by findings that demonstrate the presence of hydrophobic interactions between cyt c and phospholipid acyl chains that extend outwards from the lipid bilayer [3]. Although most of the cyt c can be dissociated from digitonin-permeabilized mitochondria at physiological ionic strength (150 mM), a small portion (10%) remains membrane-bound (membrane-bound cyt c) [4]. As a result, it has been postulated that there are two pools of cyt c in the IMS: (i) a loosely bound pool and (ii) a pool that binds more strongly to the inner membrane and interacts with the complexes in the electron transport chain [5]. The loosely bound cyt c is sensitive to changes of an electrostatic nature such as ionic strength, surface charge density and pH, while membrane-bound cyt c is not sensitive to electrostatic alterations [6,7]. Membrane-bound cyt c has been suggested to participate in hydrophobic interactions with membrane phospholipids and has been implicated with caspase activation in the early stages of apoptosis [8].
The nature of the reaction between cyt c and the mitochondrial membranes and especially its signature component, CL [cardiolipin; diphosphatidylglycerol; 1,3-bis(sn-3′-phosphatidyl)-sn-glycerol], has been the subject of considerable scrutiny for more than three decades, since the early survey by Nicholls [9]. Indeed, the body of work is worthy of extensive review, but will be only briefly summarized here.
At low to moderate ionic strength with non-binding ions, a stable complex between cyt c and membrane models is formed that will withstand ultracentrifugation [10] and gel permeation chromatography [11] or centrifugal concentration [12]. However, at high ionic strength or with binding buffers, that complex is in dynamic equilibrium with free cyt c [12–15], characteristic of electrostatic binding via specific cationic groupings. Some authors have reported that, as with the permeabilized mitochondria described above [4], a fraction can remain tightly bound to anionic liposomes [16]. The binding surface has been identified as including multiple lysine residues, and although different authors proposed different binding surfaces [11,16–19], the most frequently identified ones are residues 72, 73, 86 and 87.
Many studies also demonstrated that tertiary structural changes take place in cyt c upon binding [7,13,20–23], but α-helical content seems unaffected by interaction with DOPS (DL-threo-3,4-dihydroxyphenylserine) vesicles [7] or PC (L-α-phosphatidylcholine)/PE (L-α-phosphatidylethanolamine)/CL liposomes [15]. This is seen in some cases to alter the haem iron co-ordination state [10,22,24] and elevate [25] or lower [11] redox potential – although it is not known whether these two phenomena are co-ordinated. Some authors suggested that there is an accompanying partial penetration of the membrane [10,14,16,21], while others went as far as to suggest complete penetration, which comes as a result of the ability of cyt c to induce non-bilayer structures in certain types of liposomes (specifically those containing CL) [26]. Residues on the opposite face of the protein from the binding surface are completely exposed to solvent [18] and may cross-link liposomes via a second binding site [17]. Additionally, the protein remains susceptible to protease digestion [7]. The partial penetration of the membrane is proposed to be accompanied by a hydrophobic interaction [10,11,14,25], and a concrete proposal that the hydrophobic surface stretch of cyt c (residues 81–85) inserts into the membrane has been advanced [10] for the ferric protein to explain the disruption of Met-80 co-ordination to the haem iron.
The alternative, that the lipid inserts into the protein, as in ferrous cyt c, should also be considered, since monomeric lipids induce the same spectral changes [27]. In addition, the protein–membrane interaction is also influenced by acyl chain length and degree of unsaturation [12,24,27], suggesting that such a binding is more likely to result from lipid insertion into protein. The nature of the phospholipid head group governs the mode of binding to the protein as well as its strength – various spectroscopic techniques suggest that the conformational change induced in ferricytochrome c depends on whether PG (phosphatidylglycerol), PS or phosphatidylinositol (all anionic) is employed [21] and that while both DCP (dicetylphosphate) and PC/PE/CL micelles affect the structure of ferrous cyt c, only DCP causes detectable change in the ferric protein [15]. This observation also suggests that there may be differences in the mode of binding to physiological CL between the two redox states. In summary, we know that, upon interaction with phospholipid liposomes as well as with simple amphipathic molecules [27], cyt c undergoes conformational changes that are consistent with the loosening of its tertiary structure, alterations in the haem environment and loss of the Met-80-iron ligation. In ferrous cyt c, the spectral changes involve coalescing of the α and β bands in the absorbance spectrum. Although these changes might easily be assumed to represent oxidation of cyt c, and indeed had been in the past [28], recent work demonstrating their reversible nature implies that oxidation is not involved [3,27,24].
Stewart et al. [27] first hypothesized that fatty acid acyl chain penetration would perturb the interior of the cyt c, causing a displacement of the Met-80 side chain from co-ordination with the iron, hence the spin state transition. In following up this finding, Tuominen et al. [3] provided evidence for a direct interaction between the hydrophobic interior of cyt c and the phospholipid acyl chains, in what they have called the extended lipid anchorage of cyt c. Fluorescence spectroscopy of Zn cyt c and dibrominated PG demonstrated a direct contact between the Zn2+-haem and the brominated acyl chain. The entry point for acyl chain insertion is not known; however, suggestions for binding sites involving surface openings of hydrophobic channels that lead to the Met-80 side group have been made. Such regions may involve Lys-72, Lys-73, Lys-39 [27] and Asn-52 [29].
In the present study, we have investigated some of the properties of the electrostatic and hydrophobic interactions between cyt c and unilamellar phospholipid liposomes (PC/PE/CL) that serve as membrane models for the mitochondrial inner membrane, under baseline conditions that perhaps more closely model the physiological reality than those generally used. We have studied the timeframe in which the binding process occurs and thus more accurately investigated binding affinity. We have evaluated the effects that factors such as ionic strength, pH, liposome size and content, nucleotides and various metabolites have on the interaction. We have also investigated the liposome interactions of cytochromes from different species, as well as cyt c variants prepared using semisynthesis and mutagenesis, in order to identify residues that might be important to anchorage. As a result, we are able to present a model of the process by which cyt c may be impaled by one of the acyl chains of membrane CL.
EXPERIMENTAL
Materials
Cyt c (oxidized form) from horse, chicken, rabbit, tuna, pigeon, cow and the yeasts Candida krusei and Pichia membranaefaciens were purchased from Sigma–Aldrich (Oakville, ON, Canada). CL from bovine heart, PC and PE from hen's-egg yolk (in powder forms) were also purchased from Sigma–Aldrich.
Saccharomyces cerevisiae iso-1-cytochrome c mutants
The S. cerevisiae strain containing the C102T mutation was constructed as previously described [30]. The replacement of Cys-102 was done in order to prevent dimerization and, since C102T exhibits structural and functional characteristics identical with those of S. cerevisiae iso-1-cyt c [31], it is denoted as the ‘wild-type’ in the present study. Cyt c variants in which Lys-73 was replaced with hydrophobic amino acids by site-directed mutagenesis (K73V, K73M, K73L and K73I) were prepared as previously described [32,33]. S. cerevisiae iso-1-cyt c lacking the trimethylation of Lys-72 (TML72K) was expressed in Escherichia coli by co-expression of the genes coding for cyt c (CYC1) and yeast cyt c haem lyase (CYC3) [34].
Production of Arg91Nle cyt c (where Nle is norleucine) through semisynthesis
The haem-containing fragment of cyt c was generated by CNBr digestion [35]. The cyt c fragment (1–65) containing the haem group covalently bound to Cys-14 and Cys-17 was isolated by separating the digestion products on a Sephadex-50 column equilibrated with 7% (v/v) formic acid. The synthetic non-haem cyt c peptide (66–104) containing the Nle-91 substitution was synthesized by the late Professor Ian Clark-Lewis using solid-phase methods as described previously [36]. The haem fragment of native horse cyt c and the synthetic fragment containing the Nle-91 substitution were dissolved at a 1:1 molar ratio in 50 mM potassium phosphate buffer (pH 7.0). The haem-containing cyt c fragments were reduced by addition of excess sodium dithionite, and further exposure to oxygen was avoided by sealing the solution in a 1 ml airtight glass syringe. The fragment mixture was incubated in the dark, at room temperature (20 °C), for 24 h. Semisynthetic Arg91Nle cyt c was separated from the non-reacted fragments by gel-exclusion chromatography in formic acid (7%).
Preparation of cyt c solutions
Isolation of cyt c from S. cerevisiae and E. coli was performed as previously described [34,37] and purification of the protein was performed using an SP-Trisacryl M resin. Ferricytochrome c dissolved in 50 mM potassium phosphate buffer (pH 7.0) was reduced by excess sodium dithionite and was immediately eluted with 20 mM potassium phosphate buffer (pH 7.0) through a Sephadex-G25 column in order to remove inorganic reaction products. The concentration of reduced cyt c was determined by using a molar absorption coefficient (ϵ) of 106.1 mM−1·cm−1 at 410 nm [3]. Ferrocytochrome c was diluted appropriately with degassed 20 mM phosphate buffer (pH 7.0) and was used immediately in the binding assays.
Preparation of liposomes
PC, PE and CL dissolved in 99.9% chloroform and 99.9% methanol (2:1, v/v) were combined and the solvent was removed using rotary evaporation at room temperature. The lipid film was rehydrated with degassed 20 mM phosphate buffer (pH 7.0) to a total phospholipid concentration of 2 mM. Multilamellar phospholipid vesicles were produced by shaking the flask containing the lipid solution in a shaker–incubator (250 rev./min, 30 min and 30 °C). Small unilamellar vesicles were prepared using a Mini-Extruder (Avanti Polar Lipids, Alabaster, AL, U.S.A.) with the lipid dispersion being subjected to 19 passes through a polycarbonate membrane with 100 nm pore size. For the experiments summarized in Figure 1, membranes with pore sizes of 50, 100, 400 and 800 nm were employed to generate liposomes of varied radius of curvature.
Figure 1. Time-dependence of the interaction between cyt c and phospholipid liposomes.

Horse cyt c (6 μM) and PC/PE/CL (5:4:3) liposomes (160 μM total phospholipid content) in 20 mM potassium phosphate (pH 7.0) were incubated at 37 °C. The liposomes with diameter ranging from 50 to 800 nm [50 nm (□), 100 nm (●), 400 nm (○) and 800 nm (■)] were prepared by extrusion through polycarbonate membranes with the respective pore sizes.
Binding assays
Samples containing 6 μM ferrocytochrome c and various amounts of phospholipid liposomes were incubated for up to 4 h at 37 °C in 2 ml quartz cuvettes covered with flat lids and the absorbance of cyt c was measured at different time points. The change in the absorption spectrum of cyt c resulting from the addition of liposomes was monitored by measuring the decrease in absorbance at 550 nm as the α and β bands coalesced. The addition of liposomes into the cyt c solution was expected to slightly increase attenuance and light scattering, which in turn would cause an upward shift of the entire spectrum. In order to deduce changes in A550 caused by the spin state change only, an isosbestic point close to A550 was chosen as a reference point. The absorbance value at 526.5 nm does not change with the transition of the haem iron from low to high spin, therefore changes in the A550–A526.5 difference would represent only spectral changes caused by the spin state change and would correct for the baseline shift from the liposome addition. For example, if there were no changes in the A550 due to spin state change, the A550–A526.5 difference would remain the same, even during an increase in absorbance from increases in attenuance. In the event of spin state shift however, the A550–A526.5 is expected to decrease as saturation is approached and α and β bands coalesce. Changes in A550–A526.5 of the control cyt c solution containing no liposomes resulted from oxidation of cyt c and were, therefore, subtracted from the A550–A526.5 value of all samples. The degree of spectral change at a given point in time was then determined using eqn (1):
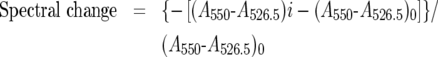 |
(1) |
where (A550–A526.5)i represents the absorbance difference in the presence of a particular concentration of liposomes (i) and (A550–A526.5)0 represents the absorbance difference of the control, in the absence of the liposomes. The progress of the reaction was monitored until the spectral change reached a constant value. The maximum spectral change at various concentrations of phospholipids was then plotted against the concentration of CL added with the liposome solution. The amount of CL required to achieve half-saturation ([S]0.5) was determined by analysing the data by non-linear regression using the GraphPad Prism software (version 4) from GraphPad Software (San Diego, CA, U.S.A.) with one of the following equations:
Hyperbolic single site binding:
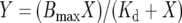 |
(2) |
where X is the concentration of CL in the assay mixture and Y is the spectral change. Bmax is the maximum spectral change and Kd is the concentration of CL required for 1/2 Bmax.
Sigmoidal dose–response with variable slope:
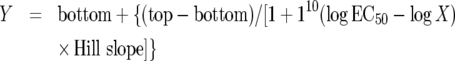 |
(3) |
where X is the concentration of CL, Y is the spectral change that starts at the bottom and goes to the top with a sigmoid shape. EC50 is the concentration of CL at (top−bottom)/2 and the Hill slope represents the co-operativity involved in the binding process.
RESULTS AND DISCUSSION
The change that is the observational focus of the present study is a transition from low- to high-spin ferrous haem spectrum, and we monitored the change at 550 nm, where the relative change is greatest. We accounted for the spectral shift caused by the addition of liposomes by subtracting the absorbance value at A526.5 from all samples (see the Experimental section for a detailed explanation) and, as expected, observed a decline in A550–A526.5 with increasing liposome concentrations.
Time-dependence of cyt c–phospholipid liposome interaction
Previous studies investigating the interaction of cyt c with phospholipid liposomes that results in the spectroscopic transition from low- to high-spin ferrous haem have sometimes relied on the assumption that the tight binding of protein to the lipid membranes is a rapid equilibrium process [3,15,24]. However, in moving to incubation conditions intended to more closely simulate the mitochondrial environment, we found that the association required some time to reach completion (Figure 1). In addition, we noted that there was a relationship between radius of curvature (mean diameter) and binding affinity (as measured by degree of spectral change); a decrease in the radius of curvature as in the case of liposomes with a diameter of 800 nm led to a significant decrease in the degree of spectral change in cyt c (Figure 1).
Although the electrostatic bonding between basic cyt c and acidic phospholipids will be in rapid equilibrium at low ionic strength [15,29,38], the lipid anchorage of cyt c to the liposomes may be viewed as the rate-limiting step of the overall strong binding of cyt c to the phospholipid membranes. This may be because the thermodynamic barrier associated with the passage of an acyl chain through the polar surface of the bilayer is likely to be very high. The mechanism that we are proposing for this strong attachment involves initial electrostatic interactions between cyt c and the membrane, which result in conformational changes in both protein and lipid that make acyl chain insertion in cyt c thermodynamically plausible, by reducing this barrier. This proposal is supported by early work showing that cyt c does affect the bilayer structure of membrane models containing CL [26]. Additionally, we hypothesize that high lipid curvature contributes to the decrease in this thermodynamic barrier by the resulting increase in the conformational strain of CL acyl chains in the bilayer.
Physical factors affecting interaction of cyt c with the phospholipid membrane models
In order to elucidate the mechanism of insertion, we investigated the hydrophobic and the electrostatic components of the association of cyt c with the liposomes by altering conditions such as phospholipid content, ionic strength and pH. Previous studies have emphasized the importance of acidic phospholipids in the binding of liposomes to cyt c [15]. When the total phospholipid concentration was maintained the same and CL was replaced by PC, we found that strength as well as the extent of interaction between cyt c and the lipid membranes was diminished. [S]0.5 for the interaction with PC/PE (2:1, mol/mol) was determined to be 150.5±21.7 μM (total phospholipid content) and the maximum spectral change was found to be 0.78±0.08 (Figure 2). The weaker binding and the inability to saturate the spectral change indicative of extended lipid anchorage imply that electrostatic interactions (including H-bonding) are important not only to initiating the strong binding, but also to sustaining it.
Figure 2. Effect of CL on binding of cyt c to phospholipid vesicles.

Binding of horse cyt c (6 μM) to PC/PE/CL (5:4:3) (□) and PC/PE (2/1) (■) liposomes in 20 mM potassium phosphate (pH 7.0) at 37 °C. The [S]0.5 for cyt c binding to liposomes containing CL was 40 μM (total phospholipid content), while that for cyt c binding to liposomes containing no CL was 150 μM (total phospholipid content).
All studies that have considered the issue [11–15,19,23] have shown that increasing ionic strength decreases the association of cyt c with the liposomes. However, in our system, we found that what was significantly affected was the rate at which the overall interaction reached equilibrium. We studied the effect of the ionic strength on the interaction of cyt c with PC/PE/CL liposomes by monitoring the spectral change to cyt c in potassium phosphate buffer (pH 7.0) of two different concentrations, 20 and 100 mM (Figure 3A). Spectral change at 20 mM occurred much more rapidly than at 100 mM, but appeared to reach a plateau at a similar level. This observation is consistent with the notion that electrostatic equilibria, which will vary with ionic strength, are important for the association of cyt c with membranes, and with our conclusion that these are a necessary prelude to the formation of the ‘extended lipid anchorage’.
Figure 3. Effect of ionic strength and pH on binding of cyt c to phospholipid liposomes.

(A) The time required for completion of interaction between horse cyt c (6 μM) and PC/PE/CL (5:4:3) (40 μM CL content) in 20 mM (■) and 100 mM (□) potassium phosphate buffer (pH 7.0) at 37 °C. (B) Interaction of horse cyt c (6 μM) with PC/PE/CL (5:4:3) (40 μM) in potassium phosphate buffer at various pH values at 37 °C.
Additionally, we incubated cyt c with PC/PE/CL liposomes in 20 mM potassium phosphate buffer at pH values 5.5, 6.0, 6.5, 7.0 and 7.5. The spectral properties of free cyt c do not vary over this pH range, so the change in this case shows that the affinity increased as pH fell, modestly but significantly. Figure 3(B) suggests that, not only are the electrostatics important to the first step of a two-step binding model, but that electrostatic interaction as well as extended lipid anchorage contributed to the continued strong binding.
Binding of cyt c from different species to liposomes and determination of [S]0.5
In order to compare the binding strengths of various cytochromes c to the liposomes, [S]0.5 was determined by non-linear regression using eqns (2) and (3) for horse and S. cerevisiae iso-1-cyt c respectively. The [S]0.5 for horse ferrocytochrome c was found to be 10.0±1.9 μM, while that for S. cerevisiae iso-1 C102T was found to be 22.0±1.8 μM (Figure 4). We examined the binding of various vertebrate and fungal cyts c to the liposomes at a single CL concentration because of the limited quantities of some of the rarer cytochromes, rather than by titration to determine [S]0.5. Using a CL concentration, in the assays involving chicken, rabbit, cow, tuna or pigeon cyt c, of 10 μM ([S]0.5 for horse cyt c) and for C. krusei and P. membranaefaciens cyt c of 20 μM ([S]0.5 for yeast cyt c), we were able to compare the binding strength of the novel cytochromes with these reference points. We found that the binding affinities for avian and mammalian cyt c were the same as for horse cyt c, although lower for tuna (Table 1). However, the same conclusion could not be drawn for fungal cyt c (Table 1).
Figure 4. Binding assay for cyt c and phospholipid liposomes.

Horse (■) or yeast (□) cyt c (6 μM) was titrated with PC/PE/CL (5:4:3) liposomes in 20 mM potassium phosphate buffer (pH 7.0) at 37 °C. [S]0.5 (concentration of CL required for half saturation) for horse cyt c binding was determined to be 10.0±1.9 μM, while that for yeast cyt c was found to be 22.1±1.8 μM.
Table 1. [S]0.5 values determined for the binding of vertebrate and fungal cyt c to PC/PE/CL (5:4:3) liposomes.
| Cyt c | [S]0.5 (μM) |
|---|---|
| Vertebrates | |
| Horse | 10±1.9 |
| Rabbit, cow, chicken and pigeon | ∼10 |
| Tuna | >10 |
| Fungi | |
| S. cerevisiae iso-1 (C102T) | 22±1.8 |
| P. membranaefaciens | >22 |
| C. krusei | <22 |
| Semisynthetic horse analogue | |
| Arg91Nle | 56.0±12.0 |
The binding affinity of horse cyt c for the phospholipid liposomes is significantly greater than that of yeast cyt c and the value was more consistent in vertebrates than in fungi, but in all cases the apparent affinity was sufficiently strong as to indicate the possibility of physiological relevance to the interaction. The tertiary structures of the various cytochromes c are very similar, so differences in amino acid sequence are expected to account for their different affinities for the liposomes. There are small differences in the arrangement of surface lysine residues for those cytochromes that differ in binding strength, but it is difficult to explain the difference in the affinity for the phospholipid liposomes on the basis of initial electrostatic interaction. It should also be noted that the yeast protein binding curve exhibits apparent sigmoidicity (Figure 4). This phenomenon was also noted for the horse protein when association was monitored by observing the transition to high-spin ferrous haem induced by PC/PE/CL liposomes using MCD (magnetic CD) spectroscopy [15], when complexing the horse protein to PC/PE liposomes (Figure 2), and when 3 mM ATP was present (results not shown). In all these cases, in fact, [S]0.5 is substantially greater than for horse ferrocytochrome under our standard conditions. Sigmoidicity is generally considered an indicator of co-operativity, and with a high density (25%) of CL head groups in the membrane and multiple potential binding groups on the protein, co-operativity seems likely. At low [CL], however, molecular crowding by cyt c of the surface might be operative, forcing alternative electrostatic binding configurations on protein–protein interactions that would be anti-co-operative. Such phenomena have been noted, at least at low ionic strength, by other workers [14].
Interaction of yeast cyt c variants with phospholipid liposomes
S. cerevisiae iso-1-cyt c varying at positions 25 (P25R), 58 (L58R), 72 (TML72K and TML72R), 73 (K73V, K73M, K73L and K73I) and 79 (K79R) were incubated with PC/PE/CL liposomes. The CL concentration in the assay mixture was maintained constant at 20 μM ([S]0.5 for the wild-type C102T). We found that the binding strength of P25R and K79R mutants with the liposomes was approximately the same as that of C102T. TML72R could not be studied in this system due to autoxidation on a timescale of minutes. L58R and TML72K mutants showed weaker binding to liposomes, while all position-73 mutants displayed a higher affinity for the liposomes than did the wild-type. Those yeast cyt c variants whose interaction with the liposomes was significantly different from that of the wild-type were titrated with PC/PE/CL liposomes and the [S]0.5 values were determined. Since all position-73 mutants displayed the same affinity for the liposomes, [S]0.5 was only determined for K73V. We found that the [S]0.5 was 50.0±5.2 μM for TML72K and 11.5±5.0 μM for K73V (Table 2).
Table 2. [S]0.5 values determined for the binding of S. cerevisiae iso-1-cyt c variants to PC/PE/CL (5:4:3) liposomes.
| Mutant cyt c | [S]0.5 (μM) |
|---|---|
| C102T | 22.0±1.8 |
| P25R | ∼22 |
| L58R | >22 |
| K73V | 11.5±5.0 |
| K73M, K73L and K73I | <22 |
| K79R | ∼22 |
| TML72K | 50.0±5.2 |
In yeast TML72K, the [S]0.5 is more than doubled, although this is a very subtle change to the side chain. Trimethylation of lysine in cyt c is a post-translational modification event that takes place in fungi and higher plants. Its functional significance is not known, but it has been speculated that it increases the affinity of yeast cyt c for the mitochondrion [39] and it is considered to be a factor in the absence of apoptotic activity in yeast cyt c [40,41]. Our observation is of a substantial decrease in affinity in the absence of trimethylation. Since the effect of this modification is at a surface charge, the decrease may be attributable to the electrostatic component of the cyt c–CL interaction. The methyl groups create a quaternary amino group and that results in a permanent charge on Lys-72. Removal of this permanent charge in TML72K may be the reason for the decrease in the affinity for the phospholipid liposomes, since the proximity of other charged residues on the protein surface will cause a decrease in the pK of an unmodified amino group, resulting in only partial charge at physiological pH. It has been suggested that the pK of Lys-72 in cyt c may be 7.8 [42]. Another effect of trimethylation is the loss of the hydrogen-bonding potential. H bonds formed by lysine N-H are, unlike electrostatic effects, highly directional. Their formation with oxygen atoms of either CL head groups or neighbouring protein residues could conceivably distort the initial interaction complex and reduce overall affinity.
Four of the mutants examined incorporated arginine residues, either as replacements for other surface charge (TML72R and K79R) or as substitutions for aliphatic hydrocarbon side chains (P25R and L58R). Surprisingly, the replacement of the tertiary ammonium group of TML-72 by a guanidino group proved to be structurally destabilizing, causing rapid autoxidation of the protein and rendering [S]0.5 determination impossible. The substitution of arginine in K79R or for P25R (a variable surface residue that is lysine throughout the Animal Domain), both located on the front surface of the protein, did not alter the [S]0.5, indicating that no competing binding sites for bisphosphoglycerol head groups had been created. Residue 58 is also very evolutionary variable and is rarely leucine, and is located at the neck of the ‘bottom loop’ of the cytochrome, in a three-residue stretch of antiparallel β-sheet [43], from which it extends downwards. This loop makes a flattened surface that has been suggested to form a CL-binding site [20] and incorporates Asn-52 that has been suggested as one possible site of entry for the acyl chain anchor [27,29]. If this were the case, then introduction of additional positive charge on this surface might be expected to increase the strength of anchorage; in fact it is weakened ([S]0.5 is increased), which argues against this possibility.
Lys-73 is another surface residue that is part of the domain that interacts with the redox partners; therefore we utilized yeast cyt c variants in which lysine at position 73 was replaced by non-polar amino acids (methionine, leucine, isoleucine and valine) and observed that all the substitutions increased the affinity of cyt c for the liposomes. The [S]0.5 for K73V was found to be 11.5±5.0 μM (Table 2), which is close to that of horse cyt c (10.0±1.9 μM). It initially appears paradoxical that eliminating a positive charge, which is implicated in the electrostatic binding of cyt c to membranes and believed to be a necessary preliminary to acyl chain insertion, should actually enhance the latter. Again, two effects may be in operation here. The side chain of Lys-73 is part of the ‘left side’ face of cyt c that also includes Lys-72, Lys-86 and Lys-87 and which is proposed to be the binding surface for anionic phospholipid-containing bilayers [17,18] that is preliminary to formation of the extended lipid anchorage, and may correspond to the C-site described by Rytomaa et al. [29]. However, it is also an element of the front surface of the protein, where Lys-13, Lys-73, Lys-79 and Lys-86 ring the exposed haem edge and constitute the binding site for redox enzymes. Loss of the amino group at position 73 might tip the balance between alternative binding sites in favour of the left-side face. In addition, it has been shown that non-polar substitutions at position 73 in iso-1 yeast cyt c increase the stability of the denatured state, yet they seem to have no effect on the native state of cyt c [32,33]. Other studies have shown that binding of cyt c to non-esterified fatty acids, CL bilayers, SDS micelles and DOPS liposomes, causes partial unfolding of the protein where the closed packed configuration is not maintained [44–46]. This conformation, however, resembles more of a molten globule state rather than the denatured state, since the secondary structure is maintained. Thus it could be that replacement of Lys-73 with non-polar amino acids increases the affinity of cyt c for liposomes by reducing the thermodynamic barrier to transfer of an acyl chain from the lipid bilayer to the protein interior as a consequence of an easier partial denaturation process. This supposition is strongly supported by the observation (results not shown) that the lipid binding equilibrium is achieved very much more rapidly with K73V than with the wild-type yeast protein.
The role of Arg-91 in the binding of cyt c to liposomes and the effect of ATP
Invariant Arg-91 is one of the residues that comprise the ATP-binding site in cyt c [47,48]. This residue's α-carbon atom lies on the inner face of the terminal α-helix of the protein and the bulk of the side chain fills space in the protein interior with the charged guanidino group just emergent on the left face [49]. The replacement with the linear aliphatic Nle thus duplicated the space-filling role of the side chain while eliminating charge. We found that interaction of horse heart cyt c with PC/PE/CL liposomes was significantly affected by this substitution: [S]0.5 value for Arg91Nle was found to be 56.0±12.0 μM, approx. 5 times greater than the value for the wild-type (Table 3). However, the time required for the reaction to reach equilibrium was extremely short compared with horse and yeast cytochromes as maximum spectral change was achieved within 15 min of incubation time. The implication is that the loss of the guanidino group eliminates one component of the anionic binding site and a stabilizing element of the CL anchorage complex. But it does not, as we are proposing for changes to Lys-73, destabilize the protein interior and make ligand displacement more favourable. This assertion is supported by observations that the pK of the alkaline isomerization, in which Met-80 is displaced by a lysine residue, monitored by the disappearance of the 695 nm charge transfer band, is unchanged in Arg91Nle cyt c [50].
Table 3. Effect of 3 mM ATP on [S]0.5 values for binding of cyt c to phospholipid liposomes.
| [S]0.5 (μM) | |||
|---|---|---|---|
| Cyt c | ATP… | None | 3 mM |
| Horse | 10.0±1.9 | 34.0±7.5 | |
| Arg91Nle | 56.0±12 | 6.9±1.6 | |
| C102T | 22.0±1.8 | 33.8±3.1 | |
The present results suggest that Arg-91 is also found in the liposome-binding site as defined by Tuominen et al. [3]. This view was supported by the observation that 3 mM ATP significantly decreased the affinities of horse and yeast cyt c for the liposomes. The [S]0.5 of both horse and yeast cyt c increased to 34.0±7.5 and 33.8±3.1 μM respectively (Table 3). It seems likely that ATP competes with the CL head groups for a binding site in cyt c. However, since 3 mM ATP (a typical intracellular concentration and the Kd for this binding site [51]) was necessary to achieve this modest decrease in affinity, competition is weak, giving us an idea of the strength of the electrostatic interactions between CL-containing membranes and cyt c. This is possibly due to the fact that the density of CL head groups (25%) in the artificial membrane bilayer, if, as expected, evenly spaced, would allow multiple interactions with a single cyt c molecule, various surfaces of which contain multiple basic groups.
Although ATP might be expected to have little effect on the association of horse Arg91Nle with liposomes, it was surprising to note that the affinity increased (Table 3). There is evidence that when the Arg-91-binding site in Arg91Nle cyt c is compromised, ATP occupies an alternative binding configuration destabilizing the tertiary structure that would lead to a more facile penetration by an acyl chain and increased affinity of Arg91Nle cyt c for the phospholipid liposomes. This evidence was obtained by experiments to determine the autoxidation rate in the presence and absence of ATP. The reaction requires penetration of the protein interior and ligand displacement by dissolved O2, so rate will be correlated with structural stability. Addition of ATP to cyt c in 20 mM potassium phosphate buffer (pH 7.0) decreased the rate of oxidation of reduced horse and yeast cyt c while increasing that for Arg91Nle cyt c (results not shown), demonstrating that ATP not only does bind to the semisynthetic analogue, but significantly destabilizes rather than stabilizes the interior, indicating an alternative binding mode.
Conclusion
The results obtained in the present study, and by others, lead us to propose the following mechanistic model for the extended lipid anchorage of cyt c to CL-containing membrane models (Figure 5). It is quite clear from the results that the functionally required, and experimentally well established, electrostatic association of cyt c with the membrane surface is a necessary prerequisite step for the insertion of an acyl chain into the protein interior. The effects of pH, ionic strength, polyanions and the modification of specific positively charged lysine residues of the protein surface provide evidence for this suggestion. The binding surface must be located on the surface such that penetration by the acyl chain (optimally 18:2) may allow direct contact between that chain and the sixth co-ordinating position at the left side of the haem. The grouping indicated by our results and by others [17,18] meets that requirement, is essentially planar and comprises Lys-72, Lys-73, Lys-86, Lys-87 and Arg-91 (Figure 6A). This positively charged patch of basic residues may complement the negatively charged membrane surface. While the electrostatic interaction may be characterized by on and off rates in the range of ms−1, the acyl chain insertion marked by the low- to high-spin transition is a slow process when the ionic composition of the medium is 20 mM Pi, even at 37 °C. Not surprisingly, the thermodynamic barrier may be very high, since such a transfer requires that the chain pivots through the highly polar interface. However, the conformational changes, widely noted, that result from the electrostatic interactions may assist to reduce the barrier from insurmountable to achievable. Such conformational changes may include an increase in the negative curvature of the membrane surface, in addition to a loosening of the tertiary structure of cyt c [7,13,20–23].
Figure 5. Extended lipid anchorage.

A block diagram of the suggested topography of the extended lipid anchorage of cyt c on phospholipid membrane surfaces and the amino acid residues comprising the binding surfaces in cyt c.
Figure 6. Hydrophobic crevice.

(A) The cleft that may accommodate one of the CL acyl chains in cyt c consists of non-polar residues comprising polypeptide strands 67–71 and 82–85 (ochre) with positively charged residues Arg-91 and Lys-72 (blue) at either extremity. (B) Space-filling model representing the two hydrophobic stretches. The protein structure was generated with the VMD molecular modelling program (Protein Data Bank accession code 1AKK).
An essential aspect of the transfer mechanism is that there should be a route of entry to the protein for the pivoting acyl chain. Clearly such a route to full insertion cannot cross any stretch of polypeptide backbone, but must lie in a crevice (possibly made of hydrophobic residues) between them, as well as being the right length. An ideal candidate for such a route identifies itself: one of the acyl chains of a CL molecule anchored at its pivotal bisphosphoglycerol head group to the amino group of Lys-72 (trimethylated in yeast cytochromes) would be capable of slipping between the non-polar polypeptide strands 67–71 and 82–85 without encountering much polarity (Figure 6A). This crevice is the apparent binding site of ATP in ferrocytochrome c and was suggested as an entry point for acyl chains by Stewart et al. [27]. The representation in Figure 6(B) shows a slight gap between the hydrophobic linings on either side of the crevice, to permit the viewer sight of the interior. In fact, these surfaces are in van der Waals contact; however, they may not contribute too much to the thermodynamic barrier of the acyl chain insertion and they also may be altered by conformational change upon electrostatic binding.
Once it has been established, the lipid anchorage of the phospholipids is remarkably stable. For horse ferrocytochrome c (6 μM), a concentration of slightly more than 10 μM CL is required to achieve half-saturation of the transition. That an acyl chain should be so stable outside the lipid bilayer reflects both the hydrophobicity of the haem crevice and perhaps the strain on the bilayer of the conical CL structure presumably relieved by the flipping out of the acyl chain.
The hydrophobic cleft for acyl chain insertion in cyt c that we have proposed is surrounded by lysine residues, which comprise part of the interaction domain with the redox partners. Therefore it would be of interest to investigate the electron transfer rate of cyt c bound to CL-containing phospholipid liposomes into which are incorporated redox partners, and whether any change might be related to alterations in cyt c orientation or modulated redox potential (which remains controversial [11,25]) in the lipid-anchored state. Additional experiments involving cyt c variants or modified CL-containing liposomes would provide further insight on the mechanism of the acyl chain insertion in the protein interior.
Is there likely to be any physiological relevance to this form of binding? First it is important to re-emphasize that our observations were made with the ferrous form of the protein. Lipid anchorage has not been proved for ferricytochrome c, although many of the observations of other workers noted above could be so interpreted.
In normal respiration, cyt c cycles very rapidly between redox states and is assumed to diffuse rapidly between reductase and oxidase. The cyt c-binding site on oxidase and reductase is not co-planar with the membrane surface, but partly perpendicular to it, so it is conceivable that the extended lipid anchorage may orient cyt c more correctly than normal electrostatic binding. But inspection of the crystal structure of the reductase suggests that a productive complex with cyt c could not be achieved if it were membrane-anchored, and so association/dissociation would be required for a normal redox cycle. Perhaps as a consequence, the evidence available currently [4] suggests that only a small fraction of the total cyt c may be tightly bound under normal circumstances. We must thus consider the alternative possibility that the extended lipid anchorage is a means of disabling cyt c by sequestration under extraordinary circumstances for regulatory or other purposes. In addition, it may be possible that this form of cyt c is involved in the apoptotic triggering process, presenting an abnormal conformation of the protein that might facilitate its exit from the mitochondrial compartment in which is normally locked, or else enhance its presentation to the apoptosome for active complex formation. In this context, it is of interest to note recent work that shows that cellular ATP concentrations (1–5 mM) that significantly inhibit cyt c–CL association also inhibit caspase activity of the apoptosome [52] and that the lysine residues of the membrane-binding surface are also associated with ATP binding, as might be expected from the results of earlier work [50]. None of these possibilities can be discarded at this point, and all are worthy of further investigation.
Acknowledgments
This work has been supported by the NSERC (Natural Sciences and Engineering Research Council of Canada). We thank Bruce Stewart (Department of Biochemistry, Dalhousie University, Halifax, NS, Canada) for his assistance with the isolation and purification of cyt c variants. We are also grateful to Professor Jack Stewart (Department of Biology, Mount Allison University, Sackville, NB, Canada) and Professor Paavo Kinnunen (Department of Medicinal Chemistry, University of Helsinki, Helsinki, Finland) for introducing us to this line of inquiry.
References
- 1.Pettigrew W. G., Moore R. G. Berlin: Springer-Verlag; 1987. Cytochromes c: Biological Aspects. [Google Scholar]
- 2.Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicol. Lett. 2004;149:19–23. doi: 10.1016/j.toxlet.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 3.Tuominen E. K. J., Wallace C. J. A., Kinnunen P. K. J. Phospholipid–cytochrome c interaction. Evidence for the extended lipid anchorage. J. Biol. Chem. 2002;277:8822–8826. doi: 10.1074/jbc.M200056200. [DOI] [PubMed] [Google Scholar]
- 4.Cortese J. D., Voglino A. L., Hackenbrock C. R. Persistence of cytochrome c binding to membranes at physiological mitochondrial intermembrane space ionic strength. Biochim. Biophys. Acta. 1995;1228:216–228. doi: 10.1016/0005-2728(94)00178-8. [DOI] [PubMed] [Google Scholar]
- 5.Martinou J.-C., Desagher S., Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat. Cell Biol. 2000;2:41–43. doi: 10.1038/35004069. [DOI] [PubMed] [Google Scholar]
- 6.Ott M., Robertson J. D., Gogvadze V., Zhivotovsky B., Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berezhna S., Wohrlab H., Champion M. P. Resonance Raman investigations of cytochrome c conformational change upon interaction with the membranes of intact and Ca2+-exposed mitochondria. Biochemistry. 2003;42:6149–6158. doi: 10.1021/bi027387y. [DOI] [PubMed] [Google Scholar]
- 8.Jemmerson R., Liu J., Hausauer D., Lam K. P., Mondino A., Nelson D. R. A conformational change in cytochrome c of apoptotic and necrotic cells is detected by monoclonal antibody binding and mimicked by association of the native antigen with synthetic phospholipid vesicles. Biochemistry. 1999;38:3599–3609. doi: 10.1021/bi9809268. [DOI] [PubMed] [Google Scholar]
- 9.Nicholls P. Cytochrome c binding to enzymes and membranes. Biochim. Biophys. Acta. 1974;346:261–310. doi: 10.1016/0304-4173(74)90003-2. [DOI] [PubMed] [Google Scholar]
- 10.Oellerich S., Leconte S., Patemostre M., Heimburg T., Hildebrandt P. Peripheral and integral binding of cytochrome c to phospholipids vesicles. J. Phys. Chem. B. 2004;108:3871–3878. [Google Scholar]
- 11.Huang Y.-Y., Kimura T. Thermodynamic parameters for the reduction reaction of membrane-bound cytochrome c in comparison with those of the membrane-free form: spectropotentiostatic determination with use of an optically transparent thin-layer electrode. Biochemistry. 1984;23:2231–2236. doi: 10.1021/bi00305a021. [DOI] [PubMed] [Google Scholar]
- 12.Piccotti L., Buratta M., Giannini S., Gresele P., Roberti R., Corazzi L. Binding and release of cytochrome c in brain mitochondria is influenced by membrane potential and hydrophobic interactions with cardiolipin. J. Membr. Biol. 2004;198:43–53. doi: 10.1007/s00232-004-0654-2. [DOI] [PubMed] [Google Scholar]
- 13.Zhang F., Rowe E. S. Calorimetric studies of the interactions of cytochrome c with dioleoylphosphatidylglycerol extruded vesicles: ionic strength effects. Biochim. Biophys. Acta. 1994;1193:219–225. doi: 10.1016/0005-2736(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 14.Heimburg T., Marsh D. Protein surface-distribution and protein–protein interactions in the binding of peripheral proteins to charged lipid membranes. Biophys. J. 1995;68:536–546. doi: 10.1016/S0006-3495(95)80215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nantes I. L., Zucchi M. R., Nascimento O. R., Faljoni-Alario A. Effect of heme iron valence state on the conformation of cytochrome c and its association with membrane interfaces. J. Biol. Chem. 2001;276:153–158. doi: 10.1074/jbc.M006338200. [DOI] [PubMed] [Google Scholar]
- 16.Gorbenko G. P., Domanov Y. A. Cytochrome c location in phosphatidylcholine/cardiolipin model membranes: resonance energy transfer study. Biophys. Chem. 2003;103:239–249. doi: 10.1016/s0301-4622(02)00319-8. [DOI] [PubMed] [Google Scholar]
- 17.Kawai C., Prado M. F., Nunes G. L. C., Di Mascio P., Carmona-Ribeiro A. M., Nantes I. L. pH-dependent interaction of cytochrome c with mitochondrial mimetic membranes. The role of an array of positively charged amino acids. J. Biol. Chem. 2005;280:34709–34717. doi: 10.1074/jbc.M412532200. [DOI] [PubMed] [Google Scholar]
- 18.Kostrzewa A., Pali T., Froncisz W., Marsh D. Membrane location of spin-labeled cytochrome c determined by paramagnetic relaxation agents. Biochemistry. 2000;23:6066–6074. doi: 10.1021/bi992559l. [DOI] [PubMed] [Google Scholar]
- 19.Gorbenko G. P., Molotkovsky J. G., Kinnunen P. K. J. Cytochrome c interaction with cardiolipin/phosphatidylcholine model membranes: effect of cardiolipin protonation. Biophys. J. 2006;90:4093–4203. doi: 10.1529/biophysj.105.080150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soussi B., Bylund-Fellenius A.-C., Schersten T., Angstrom J. 1H-n.m.r evaluation of the ferricytochrome c–cardiolipin interaction. Effect of superoxide radicals. Biochem. J. 1990;265:227–232. doi: 10.1042/bj2650227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinhiero T. J. T., Watts A. Lipid specificity in the interaction of cytochrome c with anionic phospholipid bilayers revealed by solid-state 31P NMR. Biochemistry. 1994;33:2451–2458. doi: 10.1021/bi00175a013. [DOI] [PubMed] [Google Scholar]
- 22.Pinhiero T. J. T., Elove G. A., Watts A., Roder H. Structural and kinetic description of cytochrome c unfolding induced by the interaction with lipid vesicles. Biochemistry. 1997;36:13122–13132. doi: 10.1021/bi971235z. [DOI] [PubMed] [Google Scholar]
- 23.Cortese J. D., Voglino A. L., Hackenbrock C. R. Multiple conformations of physiological membrane-bound cytochrome c. Biochemistry. 1998;37:6402–6409. doi: 10.1021/bi9730543. [DOI] [PubMed] [Google Scholar]
- 24.Zucchi M. R., Nascimento O. R., Faljoni-Alario A., Prieto T., Nantes I. L. Modulation of cytochrome c spin states by lipid acyl chains: a continuous-wave electron paramagnetic resonance (CW-EPR) study of haem iron. Biochem. J. 2003;370:671–678. doi: 10.1042/BJ20021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salamon Z., Tollin G. Interaction of horse heart cytochrome c with lipid bilayer membranes: effects on redox potentials. J. Bioenerg. Biomembr. 1997;29:211–221. doi: 10.1023/a:1022401825287. [DOI] [PubMed] [Google Scholar]
- 26.De Kruijff B., Cullis P. R. Cytochrome c specifically induces non-bilayer structures in cardiolipin-containing model membranes. Biochim. Biophys. Acta. 1980;602:477–490. doi: 10.1016/0005-2736(80)90327-2. [DOI] [PubMed] [Google Scholar]
- 27.Stewart J. M., Blakely J. A., Johnson M. D. The interaction of ferrocytochrome c with long-chain fatty acids and their CoA and carnitine esters. Biochem. Cell Biol. 2000;78:675–681. [PubMed] [Google Scholar]
- 28.Iwase H., Takatoi T., Nagao M., Iwadate K., Nakajimia M. Monoepoxide production from linoleic acid by cytochrome c in the presence of cardiolipin. Biochem. Biophys. Res. Commun. 1996;222:83–89. doi: 10.1006/bbrc.1996.0701. [DOI] [PubMed] [Google Scholar]
- 29.Rytomaa M., Mustonen P., Kinnunen P. K. J. Reversible, nonionic and pH-dependent association of cytochrome c with cardiolipin-phosphatidylcholine liposomes. J. Biol. Chem. 1992;267:22243–22248. [PubMed] [Google Scholar]
- 30.Baldari C., Cesareni G. Plasmids pEMBLY: new single stranded shuttle vectors for the recovery and analysis of yeast DNA sequences. Gene. 1985;35:27–32. doi: 10.1016/0378-1119(85)90154-4. [DOI] [PubMed] [Google Scholar]
- 31.Gao Y., Boyd J., Williams P. J. R., Pielak J. G. Assignment of proton resonances, identification of secondary structural elements, and analysis of backbone chemical shifts for the C102T variant of yeast iso-1-cytochrome c and horse cytochrome c. Biochemistry. 1990;1:95–99. doi: 10.1021/bi00482a007. [DOI] [PubMed] [Google Scholar]
- 32.Bowler E. B., May K., Zaragoza T., York P., Dong A., Caughey S. W. Destabilizing effects of replacing a surface lysine of cytochrome c with aromatic amino acids: implications for the denatured state. Biochemistry. 1993;32:183–190. doi: 10.1021/bi00052a024. [DOI] [PubMed] [Google Scholar]
- 33.Herrmann L., Bowler E. B., Dong A., Caughey S. W. The effects of hydrophilic to hydrophobic surface mutations on the denatured state of iso-1-cytochrome c: investigation of aliphatic residues. Biochemistry. 1995;34:3040–3047. doi: 10.1021/bi00009a035. [DOI] [PubMed] [Google Scholar]
- 34.Pollock R. B. W., Rosell I. F., Twitchett B. M., Dumont E. M., Mauk G. A. Bacterial expression of mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry. 1998;37:6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 35.Corradin G., Harbury A. H. Cleavage of cytochrome c with cyanogen bromide. Biochim. Biophys. Acta. 1970;221:489–496. doi: 10.1016/0005-2795(70)90219-9. [DOI] [PubMed] [Google Scholar]
- 36.Wallace C. J. A., Clark-Lewis I. Functional role of heme ligation in cytochrome c. J. Biol. Chem. 1992;267:3852–3861. [PubMed] [Google Scholar]
- 37.Cutler R. L., Pielak G. J., Mauk A. G., Smith M. Replacement of cysteine-107 of Saccharomyces cerevisiae iso-1-cytochrome c with threonine: improved stability of the mutant protein. Protein Eng. 1987;1:95–99. doi: 10.1093/protein/1.2.95. [DOI] [PubMed] [Google Scholar]
- 38.Rytomaa M., Kinnunen P. K. J. Evidence for two distinct acidic phospholipid-binding sites in cytochrome c. J. Biol. Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- 39.Polastro E. T., Deconinck M. M., Devogel R. M., Mailier E. L., Looze Y. R., Schnek G. A., Leonis J. Evidence that trimethylation of iso-1-cytochrome c from Saccharomyces cerevisiae affects interaction with the mitochondrion. FEBS Lett. 1978;86:17–20. doi: 10.1016/0014-5793(78)80088-x. [DOI] [PubMed] [Google Scholar]
- 40.Kluck M. R., Ellerby M. L., Ellervy M. H., Naiem S., Yaffe P. M., Margoliash E., Bredesen D., Mauk G. A., Sherman F., Newmeyer D. D. Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J. Biol. Chem. 2000;275:16127–16133. doi: 10.1074/jbc.275.21.16127. [DOI] [PubMed] [Google Scholar]
- 41.Yu T., Wang X., Purring-Koch C., Wei Y., McLendon L. G. A mutational epitope for cytochrome c binding to the apoptosis protease activation factor. J. Biol. Chem. 2001;276:13034–13038. doi: 10.1074/jbc.M009773200. [DOI] [PubMed] [Google Scholar]
- 42.Blouin C., Guillemette J. G., Wallace C. J. A. Resolving the individual components of a pH-Induced conformational change. Biophys. J. 2001;81:2331–2338. doi: 10.1016/S0006-3495(01)75879-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louie G. V., Brayer E. D. High-resolution refinement of yeast iso-1-cytochrome c and comparisons with other eukaryotic cytochromes c. J. Mol. Biol. 1990;214:527–555. doi: 10.1016/0022-2836(90)90197-T. [DOI] [PubMed] [Google Scholar]
- 44.Bertini I., Turano P., Vasos R. P., Bondon A., Chevance S., Simmoneaux G. Cytochrome c and SDS: a molten globule protein with altered ligation. J. Mol. Biol. 2004;336:489–496. doi: 10.1016/j.jmb.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 45.Pinheiro T. J. T., Cheng H., Seeholzer H. S., Roder H. Direct evidence for the cooperative unfolding of cytochrome c in lipid membranes from H–2H exchange kinetics. J. Mol. Biol. 2000;303:617–626. doi: 10.1006/jmbi.2000.4159. [DOI] [PubMed] [Google Scholar]
- 46.Sinibaldi F., Mei G., Polticelli F., Piro C. M., Howes D. B., Smulevich G., Santucci R., Ascoli F., Fiorucci L. ATP specifically drives refolding of non-native conformations of cytochrome c. Protein Sci. 2005;14:1049–1058. doi: 10.1110/ps.041069405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McIntosh D. B., Parrish J. C., Wallace C. J. A. Definition of a nucleotide binding site on cytochrome c by photoaffinity labeling. J. Biol. Chem. 1996;271:18379–18386. doi: 10.1074/jbc.271.31.18379. [DOI] [PubMed] [Google Scholar]
- 48.Craig B. D., Wallace C. J. A. Studies of 8-azido-ATP adducts reveal two mechanisms by which ATP binding to cytochrome c could inhibit respiration. Biochemistry. 1995;34:2686–2693. doi: 10.1021/bi00008a036. [DOI] [PubMed] [Google Scholar]
- 49.Bushnell G. W., Lowe G. V., Brayer G. D. High-resolution three-dimensional structure of horse heart cytochrome c. J. Mol. Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 50.Tuominen E. K. J., Zhu K., Wallace C. J. A., Clark-Lewis I., Craig D. B., Rytomaa M., Kinnunen P. K. J. ATP induces a conformational change in lipid-bound cytochrome c. J. Biol. Chem. 2001;276:19356–19362. doi: 10.1074/jbc.M100853200. [DOI] [PubMed] [Google Scholar]
- 51.Craig D. B., Wallace C. J. A. The specificity and Kd at physiological ionic strength of an ATP-binding site on cytochrome c suit it to a regulatory role. Biochem. J. 1991;279:781–786. doi: 10.1042/bj2790781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chandra D., Bratton S. B., Person M. D., Tian Y., Martin A. G., Ayres M., Fearnhead H. O., Gandhi V., Tang D. G. Intracellular nucleotides act as critical prosurvival factors by binding to cytochrome c and inhibiting apoptosome. Cell. 2006;125:1333–1346. doi: 10.1016/j.cell.2006.05.026. [DOI] [PubMed] [Google Scholar]