

Abstract
Periplasmic SER (selenate reductase) from Thauera selenatis is classified as a member of the Tat (twin-arginine translocase)-translocated (Type II) molybdoenzymes and comprises three subunits each containing redox cofactors. Variable-temperature X-band EPR spectra of the purified SER complex showed features attributable to centres [3Fe–4S]1+, [4Fe–4S]1+, Mo(V) and haem-b. EPR-monitored redox-potentiometric titration of the SerABC complex (SerA–SerB–SerC, a hetero-trimetric complex of αβγ subunits) revealed that the [3Fe–4S] cluster (FS4, iron-sulfur cluster 4) titrated as n=1 Nernstian component with a midpoint redox potential (Em) of +118±10 mV for the [3Fe–4S]1+/0 couple. A [4Fe–4S]1+ cluster EPR signal developed over a range of potentials between 300 and −200 mV and was best fitted to two sequential Nernstian n=1 curves with midpoint redox potentials of +183±10 mV (FS1) and −51±10 mV (FS3) for the two [4Fe–4S]1+/2+ cluster couples. Upon further reduction, the observed signal intensity of the [4Fe–4S]1+ cluster decreases. This change in intensity can again be fitted to an n=1 Nernstian component with a midpoint potential (Em) of about −356 mV (FS2). It is considered likely that, at low redox potential (Em less than −300 mV), the remaining oxidized cluster is reduced (spin S=1/2) and strongly spin-couples to a neighbouring [4Fe–4S]1+ cluster rendering both centres EPR-silent. The involvement of both [3Fe–4S] and [4Fe–4S] clusters in electron transfer to the active site of the periplasmic SER was demonstrated by the re-oxidation of the clusters under anaerobic selenate turnover conditions. Attempts to detect a high-spin [4Fe–4S] cluster (FS0) in SerA at low temperature (5 K) and high power (100 mW) were unsuccessful. The Mo(V) EPR recorded at 60 K, in samples poised at pH 6.0, displays principal g values of g3∼1.999, g2∼1.996 and g1∼1.965 (gav 1.9867). The dominant features at g2 and g3 are not split, but hyperfine splitting is observed in the g1 region of the spectrum and can be best simulated as arising from a single proton with a coupling constant of A1 (1H)=1.014 mT. The presence of the haem-b moiety in SerC was demonstrated by the detection of a signal at g∼3.33 and is consistent with haem co-ordinated by methionine and lysine axial ligands. The combined evidence from EPR analysis and sequence alignments supports the assignment of the periplasmic SER as a member of the Type II molybdoenzymes and provides the first spectro-potentiometric insight into an enzyme that catalyses a key reductive reaction in the biogeochemical selenium cycle.
Keywords: iron-sulfur (Fe–S) clusters, molybdoenzyme, selenate reductase, X-band electron paramagnetic resonance (EPR) spectroscopy
Abbreviations: DMSDH, dimethyl sulfide dehydrogenase; DMSOR, dimethyl sulfoxide reductase; DmsABC, DMSOR from Escherichia coli (where DmsB is the electron transfer subunit); EBDH, ethylbenzene dehydrogenase; FDH, formate dehydrogenase; Fe–S/FS0–FS5, iron-sulfur clusters; ICPMS, inductively coupled plasma MS; MGD, molybdopterin guanine dinucleotide; NAP, periplasmic nitrate reductase; NAR, membrane-bound nitrate reductase; NarG, bacterial membrane-bound nitrate reductase; SER, selenate reductase; SerA, SerB and SerC, subunits of SER; Tat, twin-arginine translocase
INTRODUCTION
The microbial reduction of selenium oxyanions is a key process in the transformation of selenium in the biosphere [1,2]. The oxidized and bio-available forms of selenium [selenate (SeO42−) and selenite (SeO32−)] can be utilized as respiratory substrates by some bacteria, resulting in their reduction to elemental selenium (Se0). Elemental selenium readily precipitates and is largely unavailable for assimilation into selenoproteins [3]. Although a number of organisms respire selenium oxyanions, little is known about the biochemistry of the selenate reduction process. To date, the purification of only two periplasmic SERs (selenate reductases) has been reported [4,5]. The enzyme isolated from Enterobacter cloacae SLD1a-1 is membrane-bound and comprises three subunits of ∼100, ∼55 and ∼36 kDa, with the 100 kDa protein functioning as the catalytic subunit [5]. Although it was reported that the enzyme complex contained Mo, haem and non-haem iron, low protein yields have subsequently prevented any detailed spectroscopic characterization of these redox cofactors [5]. The dissimilatory periplasmic SER isolated from the selenate-respiring bacterium Thauera selenatis [6], in contrast, is a soluble periplasmic enzyme and is much more abundantly produced than the membrane-bound enzyme in E. cloacae SLD1a-1. The periplasmic enzyme, termed SER, comprises three subunits, SerA (96 kDa), SerB (40 kDa) and SerC (23 kDa) and catalyses the two electron reduction of selenate to selenite, as shown in the reaction:
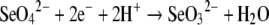 |
The SerA subunit has an N-terminal cysteine-rich motif (HX3CX3CX34C, where X is any amino acid), probably co-ordinating a [4Fe–4S] cluster and also contains the Mo active site in the form of the bis-MGD (molybdopterin guanine dinucleotide) cofactor [4,7,8]. The SerB subunit also has four cysteine-rich motifs, which again suggest the presence of a number of Fe–S (iron–sulfur) clusters. The SerABC complex also contains a b-type cytochrome, as shown by absorption spectroscopy [4], and this is presumed to be co-ordinated with the SerC subunit. The periplasmic SER shows substrate selectivity and does not reduce nitrate, arsenate or sulfate, but does reduce chlorate (S. Bydder and J. M. Santini, unpublished work). Amino acid sequence alignment of SerA with other Mo-containing enzymes has shown that it is closely related to the Type II molybdoproteins [9], a group that also includes the well-characterized respiratory NAR (membrane-bound nitrate reductase) [10,11]. The highly conserved aspartate residue, shown to co-ordinate the Mo in the NAR X-ray structures [12,13] is also conserved in SerA. However, unlike the bacterial NAR (NarG), the SER SerA component has a leader sequence that targets the protein complex via the Tat (twin-arginine translocase) apparatus to the periplasmic (potential positive, ψ+) side of the membrane [14,15] and, as such, places SER as a member of a distinct subgroup of Tat-translocated D-group (Type II) molybdoenzymes that also includes; chlorate reductase (Ideonella dechloratans [16]), DMSDH (dimethyl sulfide dehydrogenase) (Rhodovulum sulfidophilum [17]), perchlorate reductase (Dechloromonas species [18]) and EBDH (ethylbenzene dehydrogenase) (Aromatoleum aromaticum [19,20]). A number of putative enzyme complexes that are predicted to be unusual Tat-dependent nitrate reductases (pNARs) [21,22] have recently been identified in some archaeal genomes and are also included in this Type II group [11]. Owing to the relatively recent identification of this unusual sub-group of molybdoenzymes, few spectroscopic studies of their redox centres have been reported, with the notable exception of the detailed study by McEwan and co-workers on the DMSDH from Rhodovulum sulfidophilum [17]. Consequently, in the present work we have undertaken the first EPR spectroscopic analysis of the SerABC complex from T. selenatis, and probed the centres under oxidized, reduced and turnover conditions. Signals attributed to Fe–S clusters, haem-b and the Mo centre are reported and the assignment of these signals is discussed in relation to the recent crystal structure of EBDH [20].
EXPERIMENTAL
Growth of T. selenatis and production of periplasmic fractions
T. selenatis was grown anaerobically at 30 °C in mineral-salts medium [6] containing yeast extract (0.1%), selenate (10 mM) and acetate (10 mM) in 10 litre batch cultures. Cultures were harvested during late exponential phase (after 16–18 h growth) at an attenuance (D600) of 0.6–0.7 and spheroplasts were prepared as described previously [4]. The spheroplasts were removed by centrifugation (25000 g, 20 min) and the supernatant, containing periplasm, was retained.
The purification of the SER SerABC complex
Periplasmic SER was concentrated by 50–80% satd. (NH4)2SO4 as described previously [4]. After centrifugation (25000 g, 20 min), the precipitated proteins were suspended in 50 mM Pipes buffer, pH 6.0, containing 1 M (NH4)2SO4. The solution was directly loaded on to a Phenyl-Sepharose high-performance hydrophobic-interaction column, which had been equilibrated with 1 M piperazine/HCl pH 6.0, containing 1 M (NH4)2SO4. All chromatography procedures were performed at room temperature (approx. 20 °C). Protein concentration was monitored by measuring the A280. After washing the column with one volume of 1 M piperazine/HCl pH 6.0 containing 1 M (NH4)2SO4, the periplasmic SER was eluted from the column with a 1–0 M (NH4)2SO4 gradient in 1 M piperazine/HCl pH 6.0. Following elution, fractions were assayed for SER activity using a Viologen-based anaerobic microtitre plate assay [23], and those showing SER activity were pooled and concentrated using a 30-kDa-cut-off Amicon ultrafiltration cell. The resulting fraction was loaded on to a Superdex 200 gel-filtration column (Amersham) that had been equilibrated with 2 column vol. of 50 mM Pipes (pH 6.0). The periplasmic SER was eluted in the same buffer, with the fractions being assayed for SER activity as described above, and those containing the activity were concentrated using a 30-kDa-cut-off Vivaspin centrifugal concentrator and stored at −80 °C. Purity was determined by SDS/PAGE. N-terminal sequencing was performed as described previously [5] in the Molecular Biology Facility, University of Newcastle upon Tyne. In order to determine the metal (Fe and Mo) content of the purified periplasmic SER, samples were analysed by ICPMS (inductively coupled plasma MS). Standards of Fe and Mo were prepared at concentrations of 1, 5, 10, 25, 50 and 100 p.p.b. All samples and standards were treated with 6.5% nitric acid and analysed using a Thermo X-series ICPMS spectrometer in accordance with the manufacturer's instructions.
EPR spectroscopy
All EPR spectra were measured using a Bruker EMX spectrometer (X-band 9.38 GHz) equipped with an ER4112HV liquid-helium flow-cryostat system. Samples were prepared as detailed in the appropriate Figure legends. Fe–S EPR spectra were recorded at a range of temperatures (5–25 K) and microwave powers (2–100 mW) at 9.38 GHz microwave frequency and 0.5 mT field modulation amplitude. Mo(V) EPR spectra were recorded at 60 K, approx. 9.38 GHz microwave frequency, 2 mW microwave power and 0.5 mT field modulation amplitude. Spin concentration of samples was determined from integrations of their EPR absorption spectra by comparison with those of a 2 mM Cu(II)-EDTA standard [24,25]. EPR-monitored redox potentiometric titrations were performed as described previously [26]. Spectral simulations were carried out using WINEPR Simfonia Version 1.25 (Bruker). Where simulations are presented, experimental and simulated spectra are aligned with a magnetic field range corresponding to a microwave frequency of 9.38 GHz.
Homology modelling
Homology modelling was carried out using the homology tools in the software package MOE™ (Molecular Operating Environment). The sequence of T. selenatis SerB was aligned and modelled against the structure of the EBDH β-subunit from A. aromaticum (PDB: 2IVF B [20]). An ensemble of 30 intermediate models was created and the best intermediate model minimized using the CHARM22 force field to an rms (root mean square) gradient of 0.01. The quality of the model was assessed by PROCHECK [27,28]. Molecular pictures were created using PyMol [29].
RESULTS
EPR studies of the Fe–S clusters
Periplasmic SER purifies as a three subunit complex (Figure 1, inset) and metal analysis has confirmed the presence of 24±2 mol of Fe and 0.5±0.1 mol of Mo per mol of enzyme. Further purification to achieve homogeneity resulted in a significant loss of activity and a concomitant decrease in the Mo content, and, consequently, the spectroscopic analysis presented here was performed on a sample considered to be >80% pure. The only significant contaminant present is a protein of molecular mass ∼28 kDa which we have confirmed by N-terminal sequence analysis to be uridine phosphorylase and, as such, co-ordinates no EPR-detectable clusters [30]. The variable-temperature X-band (9.38 GHz) EPR spectra of as prepared ‘resting’ SerABC (Figure 1) at 10 K was dominated by a large signal centred at gav∼2.01, a small isotropic signal at g∼4.3 and a feature at g∼3.3 (Figure 1). The signal at g∼4.3 is typical of that routinely observed for adventitious high-spin Fe(III). The lineshape of the large signal at gav∼2.01 is, in general, similar to that observed from proteins containing an oxidized (spin S=1/2) [3Fe–4S]1+ cluster; notably the signal displays a low-field sharp positive peak followed by a less intense broad negative tail to high field [31]. The temperature-dependence of the signal (Figure 2) shows that the signal broadens rapidly above 15 K, such that at 20 K the signal is barely detectable. The power dependence of the signal displays a P1/2∼50 mW (where P1/2 is the microwave power that causes the amplitude of an EPR signal to be half of that predicted in the absence of power saturation) at 10 K (re-sults not shown). Simulation of the spectrum recorded at 10 K gives principal g values of g1∼1.991, g2∼2.004 and g3∼2.026 (gav=2.007) (Figure 2). Assuming a ground state of spin S=1/2, double integration of the signal recorded under non-saturating conditions, when compared with a Cu(II)-EDTA standard, yields a spin concentration representing a population of ∼0.9 [3Fe–4S] clusters per SerABC unit. On reduction of the sample by the addition of excess sodium dithionite the signal at gav∼2.01 is no longer detectable and is now replaced by a number of features resolvable in the g∼2 region (Figure 3). The principal signal (see simulation) with g values g1,2∼1.882 and g3∼2.026 (gav∼1.930) can be best described as an axial signal. It again shows temperature-dependence and is no longer detectable at 40 K, and as such is indicative of a [4Fe–4S]2+ cluster rather than a HIPIP (high-potential iron-sulfur protein)-type [4Fe–4S]3+ or a reduced [2Fe–2S]1+ cluster, which are often detected at temperatures up to 60 K or higher [31]. Power saturation studies (results not shown) demonstrated that this signal is difficult to saturate within the available power range (P1/2 > 100 mW at 10 K). This observed behaviour is consistent with other studies of [4Fe–4S] clusters in related enzymes. The signal shows resemblance to the axial signal observed for the minor conformation of the FS1 cluster in nitrate reductase NarH (Table 1). Since the gav value (1.930) is less than ge (2.00232), the electronic structure of the [4Fe–4S] cluster is more likely to be [4Fe–4S]1+ than [4Fe–4S]3+ [32] and is again in agreement with the observed temperature dependence. Assuming a ground state of spin S=1/2, double integration of the [4Fe–4S]1+ signal, when compared with a Cu(II)-EDTA standard, yielded a spin concentration representing a population of ∼0.5 [4Fe–4S] clusters per SerABC unit. An additional signal is also observed at g∼1.999, but is not subject to the same temperature dependence and can probably be assigned to the g3 feature of the Mo(V) species (see below). Also, on reduction, a number of low-intensity broad resonances are observed in the g∼1.96 region which probably arise due to spin–spin interactions of other reduced centres within the SerABC complex. In order to demonstrate the involvement of both [3Fe–4S] and [4Fe–4S] clusters in electron transfer during selenate turnover, the reduced sample was thawed under nitrogen, treated with selenate and subsequently rapidly (within 5 s) refrozen. The selenate re-oxidized spectrum recorded at 10 K clearly shows selenate-dependent re-oxidation of the [4Fe–4S]1+ cluster, rendering most of its signal EPR-silent with the concomitant re-oxidation of the [3Fe–4S] cluster regenerating the spin S=1/2 signal at gav∼2.01 (Figure 4). This suggests that both types of Fe–S clusters play a functional role in electron transfer to the selenate reductase active site and that the [3Fe–4S] cluster observed is not due to iron loss from a more typical [4Fe–4S] cluster. The SerABC samples were electrochemically poised at a range of potentials between +400 and −400mV. The [3Fe–4S]1+ cluster EPR signal was lost between the potential range 400–0 mV, and the titration could be fitted with a simple n=1 Nernstian curve, from which a midpoint potential of +118±10 mV for the [3Fe–4S]0/1+ couple could be derived (Figure 5B). The [4Fe–4S]1+ cluster EPR signal developed over a range of potentials between +300 mV and −300mV and was best fitted with two sequential Nernstian n=1 curves with midpoint redox potentials of +183±10 mV and −51±10 mV for the two [4Fe–4S]1+/2+ cluster couples. Double integration of this signal yielded a spin concentration representing a population of ∼1.5 [4Fe–4S] clusters per SerABC unit. Upon further reduction to less than −300 mV, the observed signal intensity of the [4Fe–4S]1+ cluster decreased. This change in intensity could again be fitted to an n=1 Nernstian component with an Em of about −356±10 mV (Figure 5B).
Figure 1. X-band EPR spectrum of the SER complex as prepared.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT; temperature 10 K. One scan each. The inset shows SDS/PAGE-resolved SER subunits (20 μg of total protein) stained with Coomassie Blue stain.
Figure 2. Temperature dependence of the [3Fe-4S]1+ EPR spectrum.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). Spectra recorded at the following temperatures; 10 K (——); 12.5 K (– – –); 15 K (····); 20 K (–·–·) and 25 K (–··–). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT. One scan each. Offset spectrum shows the simulation of the experimental spectrum recorded at 10 K.
Figure 3. Temperature dependence of the [4Fe–4S]1+ EPR spectrum.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). Samples were reduced by the addition of 10 mM sodium dithionite. Spectra were recorded at the following temperatures; 10 K (——); 20 K (– – –); 30 K (····) and 40 K (–·–·). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT. One scan each. Offset spectrum shows the simulation of the experimental spectrum recorded at 10 K.
Table 1. Spectroscopic parameters for the redox centres of periplasmic SER and their comparison with other related molybdoenzymes.
Δg (ge − gav), anisotropy (g3 − g1) and rhombicity [(g3 − g2)/(g3 − g1)] parameters are provided to allow comparisons to be made between signals. Membrane orientation (+/−) distinguishes proteins that are exported to the membrane potential positive side (Δψ+) via the Tat-translocase. Samples are as follows: 1DMSDH from Rhodovulum sulfidophilum; 2NarGH from E. coli; 3NarGH from Haloarcula marismortui; 4DMSOR from Rhodobacter capsulatus; 5FDH, formate dehydrogenase from Desulfovibrio desulfuricans; 6NAP, periplasmic nitrate reductase from Paracoccus pantotrophus; 7DmsABC, DMSOR from E. coli. Two different [4Fe–4S] signals have been observed for E. coli NAR and are represented as the †major conformation and ‡minor conformation [35,41].
| Enzyme class | Membrane orientation | Enzyme/sample | Redox centre | g1 | g2 | g3 | gav | Δg for [4Fe–4S] | Anisotropy | Rhombicity | Reference(s) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II | + | SerABC | [3Fe–4S]1+ | 1.9910 | 2.0046 | 2.0260 | 2.0072 | – | 0.035 | 0.611 | This work |
| II | + | DMSDH1 | [3Fe–4S]1+ | 1.9650 | 1.9870 | 2.0180 | 1.9900 | – | 0.053 | 0.585 | [17] |
| II | + | SerABC | [4Fe–4S]1+ | 1.8820 | 1.8820 | 2.0260 | 1.930 | 0.072 | 0.144 | 1.000 | This work |
| II | + | DMSDH1 | [4Fe–4S]1+ | 1.8620 | 1.8870 | 2.0158 | 1.9216 | 0.081 | 0.154 | 0.837 | [17] |
| II | + | NarGH2† | [4Fe–4S]1+ | 1.88 | 1.95 | 2.05 | 1.960 | 0.042 | 0.17 | 0.647 | [35,41] |
| II | − | NarGH2‡ | [4Fe–4S]1+ | 1.88 | 1.88 | 2.01 | 1.923 | 0.079 | 0.13 | 1.000 | [35,41] |
| II | − | NarGH3 | [4Fe–4S]1+ | 1.8710 | 1.8850 | 2.0100 | 1.9220 | 0.080 | 0.139 | 0.899 | [42] |
| III | + | DMSOR4 | [4Fe–4S]1+ | 1.8660 | 1.9300 | 2.0100 | 1.9353 | 0.067 | 0.144 | 0.556 | [43] |
| I | + | FDH5 | [4Fe–4S]1+ | 1.8650 | 1.9260 | 2.0710 | 1.9500 | 0.052 | 0.206 | 0.643 | [44] |
| I | + | NAP6 | [4Fe–4S]1+ | 1.8900 | 1.9400 | 2.030 | 1.9500 | 0.052 | 0.14 | 0.643 | [24] |
| II | + | SerABC | Mo(V) | 1.9650 | 1.9960 | 1.9990 | 1.9867 | – | 0.0340 | 0.09 | This work |
| II | + | DMSDH1 | Mo(V)-H2O | 1.9650 | 1.9846 | 2.0006 | 1.9834 | – | 0.0356 | 0.45 | [17] |
| II | + | DMSDH1 | Mo(V)-X | 1.9600 | 1.9805 | 1.9989 | 1.9798 | – | 0.0389 | 0.47 | [17] |
| II | + | DMSDH1 | Mo(V)-OH | 1.9627 | 1.980 | 1.9914 | 1.9785 | – | 0.0287 | 0.40 | [17] |
| II | − | NarGH2 | Mo(V) low pH | 1.9642 | 1.9851 | 1.9997 | 1.9830 | – | 0.0355 | 0.41 | [45,46] |
| II | − | DmsABC7 | Mo(V) | 1.960 | 1.980 | 1.984 | 1.9746 | – | 0.0240 | 0.167 | [36] |
| III | + | DMSOR4 | Mo(V) unsplit | 1.9611 | 1.9833 | 1.9906 | 1.9783 | – | 0.0238 | 0.16 | [47] |
| I | + | FDH5 | Mo(V) | 1.9630 | 1.9880 | 2.0190 | 1.9900 | – | 0.0560 | 0.55 | [44] |
| I | + | NAP6 | Mo(V) | 1.9806 | 1.9902 | 1.9985 | 1.9898 | – | 0.0180 | 0.46 | [25] |
Figure 4. Selenate-dependent re-oxidation of the [3Fe–4S] and [4Fe–4S] clusters.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0) prior to the addition of reactants. Both sodium dithionite and selenate where added to a final concentration of 10 mM. The enzyme was reduced by dithionite (——) or re-oxidized by selenate under anoxic conditions (–··–··). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT; temperature 10 K. One scan each.
Figure 5. EPR monitored redox-potentiometric titration of the [3Fe–4S] and [4Fe–4S] clusters.

(A) EPR spectra recorded from samples poised at different redox potentials from +400 to −400 mV. The enzyme concentration was ∼10 μM in 50 mM Pipes (pH 6.0) prior to the addition of reductant. (B) Change in signal intensity as a function of redox potential. Normalized signal intensities (%) were determined by the change in intensity at the spectral positions indicated by the g values in (A) relative to the maximum intensity (100%) measured. The continuous line in each case shows the best fit with n=1 Nernstian components. Δ, Change in intensity at g∼1.991 and ▼, change in intensity at g∼1.882.
The N-terminal domain of NarG displays a conserved cysteine-rich motif (HX3CX3CX35C) that is involved in the co-ordination of an Fe-S cluster, FS0, of the [4Fe–4S] type, yet attempts to detect the cluster in early EPR studies were unsuccessful [33,34]. The involvement of this motif in the co-ordination of a [4Fe–4S] cluster was only demonstrated in the recent crystal structures of NarG from Escherichia coli [12,13]. A similar structural co-ordination for FS0 has now also been shown in A. aromaticum EbdA [20]. The FS0 cluster has novel co-ordination provided by the three cysteine ligands with an additional histidine ligand positioned at residue 49. It was suggested that such co-ordination might give rise to unusual spectroscopic properties [12]. Subsequent EPR studies of NarG [35] have now shown that FS0 has an unusual high-spin (S=3/2) ground state, giving rise to an EPR signal in the g∼5 region of the spectrum. By recording the EPR spectrum at low temperature (5 K) and high power (100 mW), signals at g∼5.023 and g∼5.556 were readily detected in dithionite-reduced NarG [35]. A similar cluster in SerA is also presumed to be co-ordinated by one histidine and three cysteine ligands since the motif is conserved. Assuming that FS0 in SerA might also have an unusual high-spin ground state we have also investigated the periplasmic SER complex under similar conditions. Whilst a broad feature is observed between 160–180 mT, no peaks at g∼5 or g∼5.5 are observed typical of a high-spin cluster (results not shown). Whilst it was noted by Rothery et al. [35] that the FS0 was not observed in apomolybdo-NarGHI, low Mo occupancy (∼50%) in SerA might also point to low occupancy of FS0, but the total metal content would suggest otherwise. Given that the metal analysis indicates Fe occupancy consistent with 5×Fe–S cluster and that the selenate-dependent re-oxidation of the redox centres of SerB are observed, it would suggest that FS0 is probably present, but in an undetectable state, possibly having a lower equilibrium potential than can be reached by reduction with sodium dithionite.
EPR studies of haem-b
The full-sweep EPR spectrum of the ‘as prepared’ selenate reductase complex revealed a small positive peak at g∼3.33. Closer inspection of the selenate re-oxidized sample (Figure 6) showed that this signal had increased in intensity and its lineshape is considered typical of the g3 feature of a rhombic trio, possibly with the g2 and g1 resonances either hidden under those arising from the [3Fe–4S]1+ cluster or are too broad to be observed at X-band. The g3 signal is similar to a feature observed in the DMSDH that has been studied further by magnetic CD spectroscopy and been assigned to the g3 feature of the spectrum of low-spin ferric haem-b in the γ-subunit [17]. It would thus seem appropriate that the same g3 signal can be attributed to a low-spin ferric haem-b in SerC.
Figure 6. EPR spectrum from selenate-oxidized periplasmic SER complex revealing low-spin Fe(III) haem-b.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 1 mT; temperature 10 K.
EPR studies of the Mo centre of SerA
The EPR spectrum of the as prepared SerABC complex recorded at 60 K is dominated by resonances, with g-anisotropy consistent with a Mo(V) centre containing a single d1 unpaired electron (spin S=1/2) (Figure 7) (Table 1). The spectrum has principal g values of g3∼1.999, g2∼1.996 and g1∼1.965 (gav 1.9867). Comparison of the spectroscopic parameters with other molybdoenzymes shows that the anisotropy of the Mo(V) signal is similar to values reported for other members of the Type II group (Table 1). However, the rhombicity of the signal is notably lower than that from most other members of this group except for the value reported [36] for DmsA from E. coli. The sample was poised at pH 6.0 and the dominant features at g2 and g3 do not resolve splittings consistent with proton hyperfine coupling. Hyperfine splitting is observed in the g1 region of the Mo(V) spectrum and can be best simulated as arising from a single proton nucleus (I=1/2, where I is the nuclear spin value) with a coupling constant of A1 (1H)=1.014 mT. A splitting of 1.014 mT is consistent with that caused by a proton of an equatorial hydroxy ligand to the Mo(V) centre [37] and is in agreement with the postulated structure of the active site derived from EXAFS [8], that places a bound hydroxy ligand with a short Mo–O bond of 1.81 Å (1 Å=0.1 nm). A second, much less intense, spectrum is also observed with resonances at higher field (A∼4 mT) and probably arises from a mixture of natural Mo isotopes (95,97Mo, I=5/2). Upon reduction with dithionite (Figure 8) there is a decrease in the intensity of the g3∼1.999 signal and a loss of the feature at g1∼1.965. Treatment of the sample with selenate appears to again enhance features at g3∼1.999 and g1∼1.965, suggesting that the observed Mo(V) species is formed as an intermediate during selenate turnover. However, the selenate re-oxidized spectrum clearly shows a sharper feature at g∼2.008, and the g1∼1.965 feature is no longer split by a proton, and as such might represent an SeO42−-bound intermediate. Experiments to determine the redox potential for the Mo(IV)/Mo(V) and Mo(V)/Mo(VI) couples were not possible at this stage due to insufficient material to produce a series of electrochemically poised samples at concentrations required to observe a resolvable Mo(V) signal.
Figure 7. Mo(V) EPR spectrum from SER complex as prepared.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT; temperature 60 K. A total of 20 scans were made. Spectra (a) experimentally recorded; (b) simulation of unsplit spectrum; (c) simulation with the presence of a single proton (I=1/2) A1 (1H)=1.014mT.
Figure 8. Mo(V) EPR spectra from SER complex under turnover conditions.

The enzyme concentration was 70.4 μM in 50 mM Pipes (pH 6.0). (a) As prepared oxidized; (b) dithionite reduced and (c) selenate re-oxidized. Conditions of measurement: microwave frequency, 9.38 GHz; microwave power, 2 mW; modulation amplitude, 0.5 mT; temperature 60 K. A total of 20 scans were made in each case.
DISCUSSION
In the present work we have undertaken a detailed spectroscopic study of the redox cofactors of the periplasmic SER from T. selenatis, and the results presented have resolved signals attributed to centres [3Fe–4S]1+, [4Fe–4S]1+, Mo(V) and haem-b. The periplasmic SER is a member of an expanding group of molybdoenzymes, which, based upon sequence alignments, have tentatively been assigned to the Type II clade. The Type II members are a distinct subgroup that include; the nitrate reductases nNAR and pNAR, EBDH, DmsABC (DMSOR from E. coli) and DMSDH. This group all have a number of common features. Sequence analysis of the catalytic subunits, for example, shows that an aspartate residue is highly conserved amongst this group and, in the recent crystal structures of the nitrate reductases NarGH and NarGHI [12,13] and EBDH [20], it has been shown to co-ordinate directly to the Mo centre. This is in contrast with members of the Type I and Type III molybdoenzymes. In the Type I enzymes, the Mo centre either has no ligand, as is the case for arsenite oxidase, or is liganded via a cysteine (e.g. NAP) or selenocysteine (e.g. FDH) residue. In the Type III enzymes, the Mo centre is co-ordinated via a serine ligand (e.g. DMSO reductase). The distinction of the different co-ordination types observed in the structures of the active sites of these molybdoenzymes is also evident in the EPR parameters recorded for their Mo(V) species [17]. Consequently, it was expected that the Mo(V) EPR signals for periplasmic SER would be similar to those of other Type II molybdoenzymes. The combined evidence from all the EPR parameters gav, anisotropy and rhombicity, show that periplasmic SER is indeed better grouped as a Type II enzyme. In light of the recently resolved crystal structures of NarGH (and NarGHI) from E. coli and EBDH from A. aromaticum, the present spectroscopic work provides strong evidence that periplasmic SER is correctly grouped and is also likely to have the Mo centre co-ordinated directly by the highly conserved aspartic acid residue at position 209 in the SerA sequence. However, in the EXAFS analysis of periplasmic SER by Maher et al. [8], no evidence was presented in support of an aspartate ligand to Mo and the authors speculated that the structure of the active site might resemble that of the Type I enzyme, arsenite oxidase, with a bis-MGD cofactor in which Mo is not ligated directly via an amino acid ligand [38]. Unfortunately, no comparisons can be made between the Mo(V) EPR spectra of periplasmic SER and arsenite oxidase, since the Mo(V) species in arsenite oxidase is inherently unstable [39]. Consequently, the one-electron-reduced Mo(V) state does not accumulate and no Mo(V) EPR spectrum has yet been detected [39]. The fact that periplasmic SER loses the Mo cofactor during purification suggests that any amino acid ligand present might readily dissociate. The flexible co-ordination of the aspartic acid ligand may be evident when considering the structures obtained for nitrate reductase; in the structure reported by Bertero et al. [12], the Mo is co-ordinated by six ligands, four cis-dithiolene sulfur atoms from the bis-MGD and a bidentate ligand from both side chain oxygen atoms from the carboxylate group of Asp222. In contrast, the structure reported by Jormakka et al. [13] has the Mo again co-ordinated by four cis-dithiolene sulfur atoms, but only a single carboxylate oxygen from Asp222.
The amino acid sequence of SerB shows four potential Fe–S binding motifs (Figure 9A). It is also predicted that a Fe–S is co-ordinated to the N-terminal region of SerA. EPR spectroscopy in the present study has clearly resolved two distinct signals that can be attributed to clusters of the [3Fe–4S] and [4Fe–4S] type. The [3Fe–4S] cluster is resolved in the oxidized protein and its EPR signal is lost upon reduction, with redox titration revealing an Em of about +118mV. An axial signal typical of a [4Fe–4S] cluster is observed upon reduction from +400 to −200 mV, and the signal intensity can be fitted to two sequential n=1 Nernstian components with Em values of about +183 mV and −51 mV. On further reduction to −400 mV, the observed signal intensity of the [4Fe–4S] cluster decreases. This change in intensity can again be fitted to an n=1 Nernstian component with an Em of about −356 mV. It would appear from the redox titration that in SerB the three [4Fe–4S] clusters have very similar overlapping spectral features with a strong feature (g1,2) at g∼1.882. Upon reduction, two of the [4Fe–4S] clusters are clearly resolved, one at high potential (Em about +183 mV) and the other at considerably lower potential (Em about −51 mV). The resolution of the third cluster is apparent only by the decrease in signal intensity. It is considered likely that, at very low Em of less than −300 mV, the remaining oxidized cluster is reduced (spin S=1/2) and strongly spin-couples to a neighbouring [4Fe–4S]1+ cluster, rendering both centres EPR-silent (net spin S=0), thus decreasing the observed signal intensity at g∼1.882. Can these EPR signals be assigned to any particular clusters? If we consider the recently resolved crystal structure of EBDH [20], we see that the β-subunit has a structure that resembles an N-terminal ferredoxin, containing two Fe–S clusters (termed FS1 and FS2) followed by a tandem repeat duplication, containing the second pair of Fe–S clusters (termed FS3 and FS4). The main difference between the two domains is that cluster FS4 is a [3Fe–4S] type co-ordinated by only three cysteine ligands. SerB shares high sequence identity (55%) with the EBDH β-subunit, and modelling the SerB structure on the EBDH β-subunit provides convincing evidence that the conserved cysteine-rich motifs co-ordinate similar clusters (Figure 9B). Hence, we consider it highly likely that the [3Fe–4S]1+ EPR spectrum detected at gav∼2.01 arises from the oxidized cluster-FS4-co-ordinated cysteine residues at positions 145, 166 and 172 (Figure 9C). The other [4Fe–4S] clusters form a sequentially numbered (FS3-FS2-FS1) electron conduit through the protein transferring electrons to FS0 and MGD in SerA. SerB and the EBDH β-subunit also show homology with the NarH component of the respiratory nitrate reductase, which has been the subject of extensive spectroscopic analysis. The Em values for the clusters in NarH have been resolved, and are +130 mV (FS1), −420 mV (FS2), −55 mV (FS3) and +180 mV (FS4, the [3Fe–4S] cluster) [40]. Therefore, by analogy, we suggest that, in SerB, the cluster potentials are +183 mV (FS1), −356 mV (FS2), −51 mV (FS3) and +118 mV (FS4) (Figure 10). So which centres are spin-coupled in the fully reduced sample? In the crystal structure of EBDH, all the Fe–S clusters (FS1, FS2, FS3 and FS4) are in close contact (<6.9 Å apart). However, the structure shows that clusters FS2 and FS3 (5.4 Å apart) are closer together by 1.2 Å than clusters FS1 and FS2 (6.6 Å apart). It would thus seem most likely that, in the fully reduced sample, FS2 and FS3 would be spin-coupled. Consequently, we assign the detectable [4Fe–4S] cluster (with EPR features at g1,2∼1.882 and g3∼2.026) in the fully reduced sample to cluster FS1 and speculate that, in SerB, it is co-ordinated by cysteine residues at positions 15, 18, 21 and 212 (Figure 9C).
Figure 9. Co-ordination of Fe–S clusters in SerB.

(A) SerB sequence showing cysteine-rich motifs (underlined); (B) predicted structure of SerB (right) modelled upon the crystal structure of EBDH β-subunit (left) [20]; (C) putative SerB Fe–S cluster ligands.
Figure 10. Electron transfer through SerB (Δ), DmsB (■) and NarH (●).

The Em values of each of the [Fe–S] clusters is shown. Potentials displayed as substrate represent the mid-point potentials for the couples; selenate→selenite (E0′∼+475 mV), DMSO→dimethyl sulfide (E0′∼+160 mV) and nitrate→nitrite (E0′∼+430 mV). Q-pool indicates ubiquinol (E0′∼+65 mV) and menaquinol (E0′∼−55 mV) [48]. Arrows indicate direction of electron transfer. Data for DmsB (the electron transfer subunit of E. coli DMSOR) and NarH are taken from reference [49]. The symbols α, β and γ indicate the interface between the three subunits of the enzyme complex.
The EPR signal seen at g∼3.33 in the ‘as prepared’ sample increased in intensity on selenate re-oxidation. This signal is very similar to a feature observed in DMSDH that, based upon subsequent magnetic CD analysis, has been assigned to the g3 feature of the spectrum of low-spin ferric haem-b with histidine and methione axial ligands [17]. We therefore suggest that the g∼3.33 signal seen in periplasmic SER also arises due to the presence of the haem-b in SerC, but, given the close sequence similarity among a number of Tat-translocated D-group molybdoenzymes [11], it is considerably more likely that the Fe(III) is co-ordinated by the highly conserved Met137 and Lys228 residues, as recently demonstrated in the structure of the γ subunit of EBDH [20]. On purification, the SerC haem is partially reduced whereas other detectable redox centres appear oxidized, indicating that the b-haem in SerC might have a relatively high redox potential. Complete oxidation is achieved by the addition of selenate, demonstrating its involvement of electron transfer to SerAB (results not shown). The similar haem moiety in DMSDH also has a high-redox potential (+315 mV) [17], and these studies have suggested that this will help facilitate its role in electron transfer out of the enzyme to a periplasmic electron acceptor. Clearly, periplasmic SER works in the reverse direction, transferring electrons via the redox centres to the active site of SerA. The means by which periplasmic SER obtains its electrons is currently unknown, but a c-type cytochrome (23 kDa) co-purifies with the reductase complex following ammonium sulfate precipitation and hydrophobic interaction chromatography and has been demonstrated to act as an electron donor to the reductase in vitro (E. C. Lowe, S. Bydder, J. M. Santini, and C. S. Butler, unpublished work). It is presumed that electrons enter the periplasmic SER via SerC, and the presence of a high-potential b-haem might facilitate electron transfer from a number of different redox partners.
In summary, the results of the present study provide the first spectro-potentiometric insight into an enzyme that catalyses a key reductive reaction in the biogeochemical selenium cycle. The variable-temperature EPR spectra of the purified periplasmic SER complex has revealed features attributable to centres [3Fe–4S]1+, [4Fe–4S]1+, Mo(V) and haem-b. The involvement of the [3Fe–4S] and [4Fe–4S] clusters in electron transfer to the active site of periplasmic SER was demonstrated by the re-oxidation of both clusters under selenate turnover conditions. The combined evidence from EPR spectroscopy and redox potentiometry has revealed four of the five Fe–S clusters in SerABC, with only FS0 in SerA remaining undetected. The Mo(V) EPR recorded at 60 K, gives rise to spectral features similar to those obtained for other aspartate-liganded (D-group) molybdoenzymes and provides good evidence in support of the assignment of the periplasmic SER as a member of the Type II molybdoenzymes.
Acknowledgments
This work was funded in part by research grants from the BBSRC (Biotechnology and Biological Sciences Research Council) to C. S. B. and D. J. R. E. J. D. was a recipient of a BBSRC PMS (Plant and Microbial Sciences) committee studentship. We thank Ann Reilly (University of East Anglia) and Helen Ridley (University of Newcastle) for technical assistance. We are grateful to Dr Megan Maher (Division of Molecular and Microbial Biosciences, Imperial College, University of London, London, U.K.), Professor Alastair McEwan and Dr Paul Bernardt (Molecular and Microbial Sciences School, University of Queensland, Brisbane, Australia) for helpful discussions.
References
- 1.Stolz J. F., Oremland R. S. Bacterial respiration of arsenic and selenium. FEMS Microbiol. Rev. 1999;23:615–627. doi: 10.1111/j.1574-6976.1999.tb00416.x. [DOI] [PubMed] [Google Scholar]
- 2.Stolz J. F., Basu P., Santini J. M., Oremland R. S. Arsenic and selenium in microbial metabolism. Annu. Rev. Microbiol. 2006;60:107–130. doi: 10.1146/annurev.micro.60.080805.142053. [DOI] [PubMed] [Google Scholar]
- 3.Heider J., Bock A. Selenium metabolism in micro-organisms. Adv. Microb. Physiol. 1993;35:71–109. doi: 10.1016/s0065-2911(08)60097-1. [DOI] [PubMed] [Google Scholar]
- 4.Schröder I., Rech S., Krafft T., Macy J. M. Purification and characterization of the selenate reductase from Thauera selenatis. J. Biol. Chem. 1997;272:23765–23768. doi: 10.1074/jbc.272.38.23765. [DOI] [PubMed] [Google Scholar]
- 5.Ridley H., Watts C. A., Richardson D. J., Butler C. S. Resolution of distinct membrane-bound enzymes from Enterobacter cloacae SLD1a-1 responsible for the selective reduction of nitrate and selenate. Appl. Environ. Micro. 2006;72:5173–5180. doi: 10.1128/AEM.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macy J. M., Rech S., Auling G., Dorsch M., Stackenbrandt E., Sly L. I. Thauera selenatis gen-nov, sp-nov, a member of the β-subclass of proteobacteria with a novel type of anaerobic respiration. Int. J. Syst. Bacteriol. 1993;43:135–142. doi: 10.1099/00207713-43-1-135. [DOI] [PubMed] [Google Scholar]
- 7.Krafft T., Bowen A., Theis F., Macy J. M. Cloning and sequencing of the genes encoding the periplasmic-cytochrome B-containing selenate reductase of Thauera selenatis. DNA Seq. 2000;10:365–377. doi: 10.3109/10425170009015604. [DOI] [PubMed] [Google Scholar]
- 8.Maher M. J., Santini J., Pickering I. J., Prince R. C., Macy J. M., George G. N. X-ray absorption spectroscopy of selenate reductase. Inorg. Chem. 2004;43:402–404. doi: 10.1021/ic035136n. [DOI] [PubMed] [Google Scholar]
- 9.Trieber C. A., Rothery R. A., Weiner J. H. Engineering a novel iron-sulfur cluster into the catalytic subunit of Escherichia coli dimethyl-sulfoxide reductase. J. Biol. Chem. 1996;271:4620–4626. doi: 10.1074/jbc.271.9.4620. [DOI] [PubMed] [Google Scholar]
- 10.McDevitt C. A., Hugenholtz P., Hanson G. R., McEwan A. G. Molecular analysis of dimethyl sulphide dehydrogenase from Rhodovulum sulfidophilum: its place in the dimethyl sulphoxide reductase family of microbial molybdopterin-containing enzymes. Mol. Microbiol. 2002;44:1575–1587. doi: 10.1046/j.1365-2958.2002.02978.x. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Espinosa R. M., Dridge E. J., Bonete M. J., Butt J. N., Butler C. S., Sargent F., Richardson D. J. Look on the positive side! The orientation, identification and bioenergetics of “Archaeal” membrane-bound nitrate reductases. FEMS Microbiol. Lett. 2007 doi: 10.1111/j.1574-6968.2007.00887.x. 10.1111/j.1574-6968.2007.00887.x. [DOI] [PubMed]
- 12.Bertero M. G., Rothery R. A., Palak M., Hou C., Lim D., Blasco F., Weiner J. H., Strynadka N. C. Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 2003;10:681–687. doi: 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- 13.Jormakka M., Richardson D. J., Byrne B., Iwata S. The architecture of NarGH reveals a structural classification of MGD enzymes. Structure. 2004;12:95–104. doi: 10.1016/j.str.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Sargent F. Constructing the wonders of the bacterial world: biosynthesis of complex enzymes. Microbiology. 2007;153:633–651. doi: 10.1099/mic.0.2006/004762-0. [DOI] [PubMed] [Google Scholar]
- 15.Berks B. C., Sargent F., Palmer T. The Tat protein export pathway. Mol. Microbiol. 2000;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- 16.Thorell H. D., Stenklo K., Karlsson J., Nilsson T. A gene cluster for chlorate metabolism in Ideonella dechloratans. Appl. Environ. Microbiol. 2003;69:5585–5592. doi: 10.1128/AEM.69.9.5585-5592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDevitt C. A., Hanson G. R., Noble C. J., Cheesman M. R., McEwan A. G. Characterization of the redox centers in dimethyl sulfide dehydrogenase from Rhodovulum sulfidophilum. Biochemistry. 2002;41:15234–15244. doi: 10.1021/bi026221u. [DOI] [PubMed] [Google Scholar]
- 18.Bender K. S., Shang C., Chakraborty R., Belchik S. M., Coates J. D., Achenbach L. A. Identification, characterization, and classification of genes encoding perchlorate reductase. J. Bacteriol. 2005;187:5090–5096. doi: 10.1128/JB.187.15.5090-5096.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson H. A., Pelletier D. A., Spormann A. M. Isolation and characterization of anaerobic ethylbenzene dehydrogenase, a novel Mo-Fe-S enzyme. J. Bacteriol. 2001;183:4536–4542. doi: 10.1128/JB.183.15.4536-4542.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kloer D. P., Hagel C., Heider J., Schulz G. E. Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure. 2006;14:1377–1388. doi: 10.1016/j.str.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Richardson D. J., Berks B. C., Spiro S., Russell D. A., Taylor C. J. The biochemical, genetic and physiological diversity of prokaryotic nitrate reduction. Cell. Mol. Life Sci. 2001;58:165–178. doi: 10.1007/PL00000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dridge E. J., Richardson D. J., Lewis R. J., Butler C. S. Developing structure-based models to predict substrate specificity of D-group (Type II) molybdenum enzymes: application to a molybdo-enzyme of unknown function from Archaeoglobus fulgidus. Biochem. Soc. Trans. 2006;34:118–121. doi: 10.1042/BST0340118. [DOI] [PubMed] [Google Scholar]
- 23.Ridley H., Watts C. A., Richardson D. J., Butler C. S. Development of a viologen based microtitre plate assay for the analysis of oxyanion reductase activity: application to the membrane bound selenate reductase from Enterobacter cloacae SLD1a-1. Anal. Biochem. 2006;358:289–294. doi: 10.1016/j.ab.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Bennett B., Berks B. C., Ferguson S. J., Thomson A. J., Richardson D. J. Mo(V) electron paramagnetic resonance signal from the periplasmic nitrate reductase of Thiosphaera pantotropha. Eur. J. Biochem. 1994;226:789–798. doi: 10.1111/j.1432-1033.1994.00789.x. [DOI] [PubMed] [Google Scholar]
- 25.Butler C. S., Charnock J. M., Bennett B., Sears H. J., Reilly A. J., Ferguson S. J., Garner C. D., Lowe D. J., Thomson A. J., Berks B. C., Richardson D. J. Models for molybdenum co-ordination during the catalytic cycle of periplasmic nitrate reductase from Paracoccus denitrificans derived from EPR and EXAFS spectroscopy. Biochemistry. 1999;38:9000–9012. doi: 10.1021/bi990402n. [DOI] [PubMed] [Google Scholar]
- 26.Jepson B., Anderson L., Rubio L. M., Taylor C. J., Butler C. S., Flores E., Herrero A., Butt J., Richardson D. J. Tuning a nitrate reductase for function: the first spectro-potentiometric characterisation of a bacterial assimilatory nitrate reductase reveals novel redox properties. J. Biol. Chem. 2004;279:32212–32218. doi: 10.1074/jbc.M402669200. [DOI] [PubMed] [Google Scholar]
- 27.Morris A. L., MacArthur M. W., Hutchinson E. G., Thornton J. M. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 29.DeLano W. L. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA. 2002 http://www.pymol.org.
- 30.Caradoc-Davies T. T., Cutfirld S. M., Lamont I. L., Cutfield J. F. Crystal structures of Escherichia coli uridine phosphorylase in two native and three complexed forms reveal basis of substrate specificity, induced conformational changes and influence of potassium. J. Mol. Biol. 2004;337:337–354. doi: 10.1016/j.jmb.2004.01.039. [DOI] [PubMed] [Google Scholar]
- 31.Cammack R. Iron-sulphur clusters in enzymes – themes and variations. Adv. Inorg. Chem. 1992;38:281–322. [Google Scholar]
- 32.Mouesca J. M., Noodleman L., Case D. A., Lamotte B. Spin densities and spin coupling in iron-sulfur cluster: a new analysis of hyperfine coupling constants. Inorg. Chem. 1995;34:4347–4359. [Google Scholar]
- 33.Magalon A., Asso M., Guigliarelli B., Rothery R. A., Bertrand P., Giordano G., Blasco F. Molybdenum cofactor properties and [Fe-S] cluster coordination in Escherichia coli nitrate reductase A: investigation by site-directed mutagenesis of the conserved His-50 residue in the NarG subunit. Biochemistry. 1998;37:7363–7370. doi: 10.1021/bi972858f. [DOI] [PubMed] [Google Scholar]
- 34.Guigliarelli B., Magalon A., Asso M., Bertrand P., Frixon C., Giordano G., Blasco F. Complete coordination of the four Fe-S centres of the β-subunit from Escherichia coli nitrate reductase. Physiological, biochemical and EPR characterisation of site-directed mutants lacking the highest or lowest potential [4Fe–4S] clusters. Biochemistry. 1996;35:4828–4836. doi: 10.1021/bi952459p. [DOI] [PubMed] [Google Scholar]
- 35.Rothery R. A., Bertero M. G., Cammack R., Palak M., Blasco F., Strynadka N. C. J., Weiner J. H. The catalytic subunit of Escherichia coli nitrate reductase A contains a novel [4Fe–4S] cluster with a high-spin ground state. Biochemistry. 2004;43:5324–5333. doi: 10.1021/bi049938l. [DOI] [PubMed] [Google Scholar]
- 36.Rothery R. A., Trieber C. A., Weiner J. H. Interactions between the molybdenum cofactor and iron-sulfur clusters of Escherichia coli dimethylsulfoxide reductase. J. Biol. Chem. 1999;274:13002–13009. doi: 10.1074/jbc.274.19.13002. [DOI] [PubMed] [Google Scholar]
- 37.Bray R. C. The inorganic biochemistry of molybdoenzymes. Q. Rev. Biophys. 1988;21:299–329. doi: 10.1017/s0033583500004479. [DOI] [PubMed] [Google Scholar]
- 38.Conrads T., Hemann C., George G. N., Pickering I. J., Prince R. C., Hille R. The active site of arsenite oxidase from Alcaligenes faecalis. J. Am. Chem. Soc. 2002;124:11276–11277. doi: 10.1021/ja027684q. [DOI] [PubMed] [Google Scholar]
- 39.Hoke K. R., Cobb N., Armstrong F. A., Hille R. Electrochemical studies of arsenite oxidase: an unusual example of a highly cooperative two-electron molybdenum center. Biochemistry. 2004;43:1667–1674. doi: 10.1021/bi0357154. [DOI] [PubMed] [Google Scholar]
- 40.Rothery R. A., Magalon A., Giordano G., Guigliarelli B., Blasco F., Weiner J. H. The molybdenum cofactor of Escherichia coli nitrate reductase A (NarGHI). Effect of a mobAB mutation and interactions with [Fe-S] clusters. J. Biol. Chem. 1998;273:7462–7469. doi: 10.1074/jbc.273.13.7462. [DOI] [PubMed] [Google Scholar]
- 41.Guigliarelli B., Asso M., More C., Augher V., Blasco F., Pommier J., Giordano G., Bertrand P. EPR and redox characterization of iron-sulfur centers in nitrate reductases A and Z from Escherichia coli. Evidence for a high-potential and a low-potential class and their relevance in the electron-transfer mechanism. Eur. J. Biochem. 1992;207:61–68. doi: 10.1111/j.1432-1033.1992.tb17020.x. [DOI] [PubMed] [Google Scholar]
- 42.Yoshimatsu K., Iwasaki T., Fujiwara T. Sequence and electron paramagnetic resonance analyses of nitrate reductase NarGH from a denitrifying halophilic euryarchaeote Haloarcula marismortui. FEBS Lett. 2002;516:145–150. doi: 10.1016/s0014-5793(02)02524-3. [DOI] [PubMed] [Google Scholar]
- 43.Cammack R., Weiner J. H. Electron paramagnetic resonance spectroscopic characterization of dimethyl sulfoxide reductase of Escherichia coli. Biochemistry. 1990;29:8410–8416. doi: 10.1021/bi00488a030. [DOI] [PubMed] [Google Scholar]
- 44.Costa C., Teixeira M., LeGall J., Moura J. J. G., Moura I. Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: isolation and spectroscopic characterization of the active sites (heme, iron-sulfur centers and molybdenum) J. Biol. Inorg. Chem. 1997;2:198–208. [Google Scholar]
- 45.George G. N., Bray R. C., Morpeth F. F., Boxer D. H. Complexes with halide and other anions of the molybdenum centre of nitrate reductase from Escherichia coli. Biochem. J. 1985;227:925–931. doi: 10.1042/bj2270925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.George G. N., Turner N. A., Bray R. C., Morpeth F. F., Boxer D. H., Cramer S. P. X-ray-absorption and electron-paramagnetic-resonance spectroscopic studies of the environment of molybdenum in high-pH and low-pH forms of Escherichia coli nitrate reductase. Biochem. J. 1989;259:693–700. doi: 10.1042/bj2590693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennett B., Benson N., McEwan A. G., Bray R. C. Multiple states of the molybdenum centre of dimethylsulphoxide reductase from Rhodobacter capsulatus revealed by EPR spectroscopy. Eur. J. Biochem. 1994;225:321–331. doi: 10.1111/j.1432-1033.1994.00321.x. [DOI] [PubMed] [Google Scholar]
- 48.McEwan A. G., Pride J. P., McDevitt C. A., Hugenholtz P. The DMSO reductase family of microbial molybdenum enzymes; molecular properties and role in the dissimilatory reduction of toxic elements. Geomicrobiol. J. 2002;19:3–21. [Google Scholar]
- 49.Cheng V. W. T., Rothery R. A., Bertero M. G., Strynadka N. C. J., Weiner J. H. Investigation of the environment surrounding iron-sulfur cluster 4 of Escherichia coli dimethylsulfoxide reductase. Biochemistry. 2005;44:8068–8077. doi: 10.1021/bi050362p. [DOI] [PubMed] [Google Scholar]