

Abstract
Unrepaired or erroneously repaired DNA lesions drive genomic instability and contribute to cellular and organ decline. Since delayed neuropathologies are common in survivors of smoke inhalation injuries, we asked whether the integrity of brain DNA might be compromised by acute exposure to combustion smoke. Although many studies demonstrate that the brain is equipped to repair oxidatively damaged DNA, to date, the capacity for accurate DNA repair under conditions of disrupted oxygenation and oxidative stress has not been defined. We show that DNA adducts detectable by their ability to block PCR amplification form in the rat hippocampus after acute exposure to smoke. To identify the different types of adducts and to dissect their temporal formation and repair profiles in vivo in the brain, we used DNA modifying enzymes to convert specific adducts into strand breaks prior to PCR amplification. Using this strategy, we detected formation of oxidative DNA adducts early on after smoke inhalation, while mismatched bases emerged at the later recovery times, potentially due to erroneous DNA repair process. Erroneous repair can be mutagenic and because the initial smoke-induced oxidative damage to DNA is extensive, compromised fidelity of DNA repair may underlie neurotoxicity and contribute to delayed death of hippocampal neurons.
Keywords: oxidative DNA lesions, base excision repair, mismatched DNA bases, DNA repair fidelity, combustion smoke inhalation, brain, neuronal injury, 8-oxodG, OGG1, APE1, malondialdehyde
INTRODUCTION
Since delayed neuropathologies persist in survivors of severe carbon monoxide and combustion smoke inhalation [1–5] and because neurons are particularly susceptible to insults involving disrupted oxygenation and excessive generation of free radicals, we asked to what extent the integrity of brain DNA is compromised after acute inhalation of combustion smoke. Both, nuclear and mitochondrial DNA are oxidation targets for free radicals and unrepaired or erroneously repaired oxidative DNA lesions lead to mutations and cellular dysfunction [6, 7]. Since the highly energetic and long lived neuronal cells are at increased risk of oxidation damage [8–10], it is expected that CNS cells are equipped to repair both, nuclear and mitochondrial DNA damage and in fact, oxidation damage DNA repair activities have been documented in the mammalian brain [11–14]. Interestingly, in humans the relevance of integrity of brain DNA to brain function was suggested in a recent report showing elevated oxidative damage in nuclear and mitochondrial DNA in brains of human subjects with mild cognitive impairments [15].
In mammals, the major pathway for repair of oxidative DNA lesions is the base excision repair (BER) process. BER is initiated by DNA glycosylases which cleave aberrant bases. The abasic sites generated by glycosylases are then processed for the most part, by the apurinic/apyrimidinic endonuclease APE1, to become substrates for the gap filling repair synthesis by DNA polymerase β in nuclei and DNA polymerase γ in mitochondria. This synthesis step is a critical determinant of fidelity of DNA repair and erroneous incorporation of nucleotides is mutagenic [16]. Notably, recent studies underscore the importance of mutation-free DNA by demonstrating that genetically engineered mice, lacking Neil1, a DNA glycosylase targeting oxidation products of cytosine, manifest metabolic syndrome and mtDNA deletions [17], while mice engineered to express an error prone mitochondrial polymerase γ accumulate mutations and present accelerated aging [18]. Furthermore, mitochondrial mutations have been implicated in etiology of human diseases, including neurodegeneration and neuromuscular disorders [19, 20].
We have developed a model of combustion-smoke inhalation in the conscious rat and demonstrated that changes in gene expression and in the nitric oxide system that are indicative of excessive oxidative stress, are induced in the rat brain by acute exposure to smoke [21]. Here we report that early on after inhalation of smoke, oxidative DNA adducts are formed in rat hippocampal DNA. Since we have used DNA modifying enzymes to convert selected adducts into strand breaks prior to PCR amplification, we were able to categorize the different adduct types and monitor their temporal formation and clearance. Consistent with the prediction that DNA is repaired in the brain, frequencies of oxidatively generated DNA adducts were significantly reduced in the course of recovery after inhalation of smoke. In contrast however, within 24 hours post smoke injury, we began to detect formation of mismatched DNA bases, which persisted at the delayed recovery times and were accompanied by loss of hippocampal neurons. These findings provide novel insights into events underlying the maturation of smoke inhalation injury in the brain and for the first time implicate differential accumulation of DNA damage as a mechanism of neurotoxicity.
METHODS
Smoke inhalation model
Sprague Dawley rats (250–300 g) were exposed to combustion smoke generated by burning wood shavings for three successive 10-minute periods separated by 30 seconds in ambient air, as we described [21]. Sham-controls were given similar treatment without smoke. Rats were allowed to recover for either short (2, 6 and 24 hours) or prolonged (1, 8, 10 weeks) periods of time. Brains were harvested, dissected and tissues snap frozen in liquid nitrogen prior to storage at −80°C. All experiments were conducted in accordance with mandated standards of humane care and were approved by the UTMB Institutional Animal Care and Use Committee.
Malondialdehyde (MDA) measurements
Tissue MDA content was determined using the Bioxytech MDA-586 kit (OxisResearch, Portland, OR), which is based on a reaction of the chromogenic N-methyl-2-phenylindole with MDA and was done according to manufacturer’s instructions. Briefly, snap frozen brain tissues from control rats and rats sacrificed at different time points after exposure to smoke (n=3), were dissected and homogenized in four volumes of 20 mM Tris, 10 mM EDTA, pH 7.6 buffer containing 5 mM butylated hydroxytoluene. Homogenates were divided for parallel measurement of free and total MDA. For free MDA, homogenates were incubated 30 minutes on ice, spun down and supernatants saved for measurements. For total MDA homogenates were made 0.25 M NaCl, adjusted to pH 1 with HCl and incubated for 80 minutes at 60°C to hydrolyze and release MDA. Supernatants were collected by centrifugation for total MDA measurements. Absolute concentrations were determined from standard curves generated with known amounts of MDA and calculated per mg protein. Protein-bound MDA content was obtained by subtraction.
Enzyme assays
Frozen hippocampal tissues were chopped with a razor blade, homogenized briefly with a loose glass pestle in cold hypotonic buffer (10 mM Hepes, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 2 μg/ml Pepstatin, and Protease Inhibitor Cocktail, Roche) and left on ice for 30 minutes. Tissues were then homogenized with a tight pestle and spun 5′ at 800 g to obtain nuclear pellets. Nuclear proteins were extracted in high salt (20 mm Hepes, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 2 μg/ml Pepstatin) for 45′ on ice, made 30% glycerol and stored at −80°C [13]. Glycosylase and endonuclease activities were measured in in vitro assays assembled with the above nuclear extracts and synthetic end labeled double stranded oligonucleotide substrates containing 8-oxodG/C (*5′-CTGCTCACACTCGACACCAC[8-oxodG]AATTGTGTAGTAGTGC-3′), targeted by OGG1 or an abasic site AP/A (*5-CCTGCCCTG[THF]GCAAGTGTGGC-3′) targeted by APE1 like activities, respectively, as we described previously [14]. To ensure incision specificity, tetrahydrofuran (THF), an analog of abasic site was used. Briefly, for 8-oxodG, reactions were assembled in 20 μl with 20 μg nuclear extracts, 1 nM end-labeled substrate and reaction buffer (10 mM Tris-HCl pH 7.6, 0.5 mM DTT, 1.5% glycerol, 2.25 mM EDTA, 80 mM NaCl). Incubation was at 37°C for indicated times and was terminated with alkaline loading buffer [22]. For abasic site incision assays, reactions were done with 10 ng nuclear extract in buffer containing MgCl2 (66 mM Tris-HCl pH 7.6, 1 mM DTT, 1 mM MgCl2, 0.2 mM EDTA, 0.1 mg/ml BSA). Positive control reactions were assembled using recombinant human OGG1 and APE1 enzymes (Trevigen). Reaction products were resolved in 15% polyacrylamide-7 M urea gels, visualized by autoradiography and quantified on a Phosphorimager (Molecular Dynamics). Phosphorimager values were used to determine percent of substrate cleavage over time.
DNA damage detection
DNA damage was measured using the PCR-mediated detection method as described previously [23–25]. Briefly, DNA was isolated from rat hippocampal tissue using QIAamp Tissue Kit (Qiagen), DNA quality was assessed on agarose gels by EtBr staining. DNA was quantified by PicoGreen (Molecular Probes) fluorescence with Tecan’s GENIOS microplate reader (excitation: 485, emission: 518 nm). PCR amplifications were carried out with MJ PTC200 thermal cycler (MJ Research) with sense 5′-CCGACCTTGCCGTGGTTGAGCTTTT-3′ and antisense 5′-TGTGGTGCTTGCTGGGGTCTGTCCT-3′ primers spanning a 5-kb region of the rat MutY homolog (MYH) gene locus (Genebank #NC_005104). Human DNA and primers spanning the 2.2-kb human β-globin gene (sense 5′-AAGAGACCATTGTGGCAGTGATTGC -3′ and antisense: 5′-GGTTATGGGCTCTGGTTCTGGACTC -3′, Genebank # U01317) were included in each reaction and served as an internal control for PCR amplification. Amplification linearity range was predetermined to ensure that under selected PCR conditions, amplification was linearly dependent on template availability. Accordingly, amplification reactions were assembled with 40 ng rat and 30 ng human DNA. Amplification was by 23 cycles (15 sec at 94°C, 30 sec at 68°C, 5 min at 72°C) in a 20 μl volume assembled with 0.75 μCi of 3,000 Ci/mM [α-32P]dCTP, 300 μM dNTPs, 400 nM primers and 2.0 U enzyme using the Expand 20 kb plus PCR System (Roche Diagnostics). Products were resolved in 1.0% agarose gels and incorporated label quantified using the ImageQuant software on a Phosphorimager. Phosphorimager values for controls and rats sacrificed at 6, 24 hours, and 8 weeks after exposure to smoke (n=4), were generated by quantification of 3 PCR reactions per sample, normalized to amplification of the human β-globin locus and expressed as mean ± SD. Amplification values obtained with post-smoke DNA samples were divided by values obtained with control samples to express the relative amounts of intact template in each reaction. Thus, this approach quantifies levels of intact DNA in test samples relative to respective controls, which for quantitation purposes are considered intact. Lesion frequencies were calculated assuming random distribution and using the Poisson equation [f(X)=e−λλ/x!]. Lesion frequency λ, was derived from the equation λ=−lnAT/AC, where AT is amplification yield for each test and AC for each control sample. Lesion frequency (λ) was normalized to 10 kb and presented as mean ± SD [23, 26]. To determine relative abundance of a specific type of adduct, DNA was treated prior to PCR amplification with a DNA modifying enzyme (E.coli Formamidopyrimidine [fapy]-DNA glycosylase, Fpg; E. coli Endonuclease III or E. coli Endonuclease V, from Trevigen). The Fpg DNA glycosylase cleaves 8-oxoG and to a lesser degree other oxidized purines and because it has lyase activity it converts the resultant abasic sites into single strand breaks which block Taq polymerase mediated DNA synthesis and render subsequent PCR amplification inversely proportional to the frequency of Fpg targets in template DNA. Amplification products obtained with control samples were assigned the value of 100%. The E. coli Endo III enzyme targets oxidized pyrimidines, including thymine glycol and uracil glycol as well as abasic sites and Endo V cuts next to mismatched DNA bases. Reaction conditions were predetermined individually for each enzyme with respect to incubation time and concentration by incubating 200 ng aliquots of control DNA template with incrementally increasing enzyme concentrations. The highest concentration, which did not affect subsequent PCR amplifications levels of control templates was determined (0.1 units Fpg, 6 units Endo III and 2×10−6 units Endo V) and used to treat DNA samples prior to PCR amplification. Following treatment, DNA samples preincubated with/without enzyme were subjected to PCR amplification as described above.
Immunohistochemistry, histochemistry and TUNEL
Brains were fixed in 4% paraformaldehyde; coronal paraffin embedded sections (Bregma ~3.14) were prepared for controls and smoke exposed rats (n=4). Sections were processed as we described [13]. Antigen retrieval was for 10 minutes at 98°C using the Daco retrieval solution (DacoCytomation, Carpinteria, CA). Nonspecific binding was blocked with 5% horse serum in PBS and 1% BSA followed by incubation with the anti 8-oxoG monoclonal antibody (Clone 15A3, QED Biosciences, San Diego, CA) at 2.5 μg/ml at 4°C overnight. Following washing, sections were incubated with biotinylated anti-mouse IgG antibody, washed and incubated for 30 minutes with the ABC reagent (Vectastatin, Vector Lab) followed by incubation with the DAB chromogen (DacoCytomation). Sections were lightly counterstained with hematoxylin and sections incubated without the primary antibody served as negative control. Cleared sections were mounted with Permount (Fisher Scientific) and analyzed with a Nikon Eclipse 600 microscope. Images were captured by Nikon DXM1200 digital camera using ACT-1 (v. 2.63) program. Hematoxylin and eosin staining was used to assess neuronal injury. Non-adjacent sections through the hippocampus were examined for presence of dark/eosinophilic (dark pink), shrunk neurons. Total (at least 300 neurons) and shrunk/dark neurons were counted in the specified hippocampal region separately in the left and right side of the brain. Counting was done in a blinded fashion and each section was counted at least three times. Counts were expressed as mean ± SD of the ratio between dark and total neurons for smoke and for sham treated age matched controls. Death of neuronal cells was assessed by counting apoptotic bodies by TUNEL - terminal deoxynucleotide transferase-mediated dUTP nick end labeling with fluorescein-dUTP according to the manufacturer (Roche Biochemicals, Kit #1684817). Fluorescein-dUTP was visualized by immunostaining and positive cells were counted in non-adjacent sections. For both assessments counts were analyzed by ANOVA for a two factorial experiment; the two factors were treatment (smoke inhalation or sham) and harvest time (1, 8 and 10 weeks).
RESULTS
Smoke inhalation-induces lipid peroxidation in the brain
To assess consequences of combustion smoke-associated oxidative stress, we measured changes in the brain tissue content of malondialdehyde (MDA), a major product of lipid peroxidation. Within two hours after smoke exposure, brain MDA content sharply increased, remained elevated at 24 hours and moderately declined by seven days post smoke (Fig 1). MDA can oxidize and modify different cellular components, including DNA and accumulation of mutagenic lipid peroxidation-derived DNA lesions has been reported [27–29].
Fig 1.

Smoke inhalation-induced MDA formation in the brain. Free and total MDA levels were determined; values for protein bound MDA were obtained by subtraction. Values represent pmoles of MDA per mg protein extract expressed as means±SD, n=3.
Evidence for induction of 8-oxodG immunoreactivity in hippocampus after inhalation of smoke
To assess the extent of smoke inhalation induced oxidation of DNA in the brain, immunoreactivity levels of the salient biomarker of oxidative stress, 8-oxodG, were examined in sections through the hippocampus (Fig 2). Intensified staining (dark brown) in the hippocampal formation was noted at 6 and 24 hours post smoke, with a reduction in staining by 7 days.
Fig 2.

Smoke inhalation induces 8-oxodG immunoreactivity in the hippocampus. Representative photomicrographs of sections (Bregma -3.14) from control rats and rats harvested at 6, 24 hours and 7 days post smoke stained with the 8-oxodG antibody. Staining is brown and nuclei are counterstained light blue with hematoxylin. Significantly elevated 8-oxodG immunoreactivity is observed at 6 and 24 hours post smoke with a decline in staining by 7 days.
Smoke inhalation alters base excision repair (BER) enzyme activity
In mammalian cells, most oxidative DNA damage is repaired by the BER pathway. To assess to what extent the pathway might be affected by oxidative stress associated with acute exposure to smoke [21], we measured two major BER activities (Fig 3), excision of the oxidatively generated adduct, 8-oxo-7,8-dihydroguanine (8-oxoGua), likely by the mammalian oxoguanine DNA glycosylase (OGG1) and incision of abasic sites most likely by the mammalian apurinic/apyrimidinic endonuclease, APE1, which facilitates conversion of abasic sites into substrates for the gap filling DNA repair synthesis. Activities were measured for controls and rats harvested 2, 6, 24 hours, and 7 days post smoke (n=4). Assays were done with end labeled oligonucleotide substrates carrying adducts (*8-oxodG/C or *AP/A) targeted either by OGG1- or by APE1-like activities, respectively [14]. Mean cleavage activities calculated from product yields in the linear range of reaction (Fig 3A) show that while the OGG1 like activity remained mostly unchanged, APE1 like activity was significantly elevated at 6 hours post smoke (Fig 3B). Increased APE1 activity may generate excess of non-instructive repair intermediates to imbalance the repair process; imbalanced repair pathway can be mutagenic.
Fig 3.

Smoke inhalation modulates BER in the hippocampus. In vitro cleavage of oxidative adducts by hippocampal nuclear extracts from control rats and rats at 2, 6, 24 hours and 7 days post smoke is shown: (A) Autoradiograms of incision products (P) generated over time by cleavage of end labeled double stranded oligonucleotide substrates (S) carrying either the *8-oxodG/C adduct or the abasic site *AP[THF]/A. [−] is a negative control in absence of extract; OGG1 and APE1 lanes show reference reactions assembled with the respective recombinant enzymes. (B) Values from Phosphorimager quantitation of products generated with extracts from 4 rats per group and 3 cleavage assays per extract were averaged and plotted as mean ± SD. * indicates cleavage significantly different from control for each respective time point P<0.05.
Evidence for formation of oxidative adducts and mismatched bases in hippocampal DNA after acute exposure to combustion smoke
To define the spectrum of DNA adducts formed after exposure to smoke, DNA templates purified from hippocampi of control rats or rats at 6, 24 hours, and 8 weeks after smoke were preincubated in presence or absence of DNA modifying enzymes, which target distinct types of DNA adducts. It is noteworthy, that contrary to the case of conventional PCR-mediated DNA damage detection method, where reduced amplification yields reflect abundance of directly arising PCR-blocking adducts, PCR amplification yields obtained after template preincubation with DNA modifying enzymes, reveal abundance of lesions, which in their original form do not block PCR. Specifically, the E. coli Fpg DNA glycosylase cleaves the oxidized guanine and because it has lyase activity it converts the resultant abasic sites into single strand breaks. Thus, adducts which are originally invisible to Taq polymerase, are converted in vitro, into strand breaks, rendering subsequent PCR amplification inversely proportional to the frequency of Fpg targets in template DNA (Fig 4). The E. coli Endo III enzyme converts oxidized pyrimidines, including thymine glycol and uracil glycol as well as abasic sites into strand breaks, while the E. coli Endo V cleaves next to mismatched DNA bases, thereby registering mismatches and potential for mutagenesis. Interestingly, the directly arising PCR blocking adducts as well as adducts revealed following incubation with Fpg or Endo III were detected at 6 and 24 hours but not by 8 weeks post smoke. In sharp contrast, adducts targeted by Endo V, ie, mismatches were absent at 6 hours, emerged at 24 hours and persisted at the 8 weeks recovery following inhalation of smoke. Frequencies of the various adduct types (Fig 4B) were calculated from quantification of ratios of PCR products obtained after incubation of DNA with/without modifying enzymes, while frequencies of the directly arising PCR blocking lesions were calculated from amplification ratios between DNA from smoke exposed groups and sham controls.
Fig 4.

Smoke inhalation-induced DNA damage in the rat hippocampus. Representative autoradiograms show PCR products generated by amplification of the MYH gene locus in hippocampal DNA from control rats and rats after exposure to combustion smoke. DNA was preincubated with/without the DNA modifying enzymes, Fpg, Endo III or Endo V, prior to amplification. Amplification levels were reduced at 6 and 24 hours and restored within 8 weeks after exposure to smoke for DNA preincubated without DNA modifying enzymes, reflecting formation and subsequent clearance of the directly arising PCR-blocking adducts. Amplification product of the human β-globin locus (2.2 kb) that serves as an internal control for each PCR reaction is indicated. Amplification levels of DNA preincubated with/without Fpg (+/−) were similar for controls but reduced for rats harvested at 6 and 24 hours post smoke. Likewise, in the case of Endo III, the +/− yields were reduced at 6 and 24 hours and restored by 8 weeks post smoke indicating that levels of Fpg and Endo III targets return to near normal. In contrast, while no reduced amplification was detected for Endo V treated DNA at 6 hours, reduction was seen at 24 hours with a further decrease by 8 weeks, indicative of an increase in the number of Endo V targets. (B) Bar graphs show relative frequencies of adducts generated in hippocampal DNA following smoke inhalation: Relative amplification values for enzyme treated/non-treated DNA were averaged (n=5, triplicate PCR reactions), converted into lesions frequencies and normalized to 10 kb (expressed as mean ± SD). Frequency of directly arising PCR blocking adducts was determined from values obtained following preincubation in absence of DNA modifying enzymes (blank bars). Frequency of sites targeted by Fpg is depicted as dotted bars, by Endo III as hatched bars and Endo V as solid bars.
Assessments of hippocampal neuronal injury after smoke inhalation
Hippocampus is involved in learning and memory tasks and is particularly vulnerable to insults associated with disrupted oxygen supply and perturbed energy metabolism, including ischemia, carbon monoxide and toxic gases. In humans, following ischemia, neurons become eosinophilic and shrunken with pyknotic hyperchromatic nuclei (dark neurons) [30]. We examined to what extent hippocampal neurons were affected by acute exposure to smoke in the rat smoke inhalation model (Fig 5). While no obvious neuronal injury was observed at the early recovery times, an increase in the number of dark neurons as well as the number of TUNEL positive cells was observed in the hippocampus by 8–10 weeks after exposure to smoke when compared to age matched sham-controls (Table 1). Fig 5 shows hematoxylin and eosin staining of sham-control and rats at 1, 8, and 10 weeks after smoke injury (n=3). Total and shrunk-dark neurons were counted in regions captured with the 20x objective; values were expressed as means ± SD of the ratio between shrunk and total number of neurons based on cells counted separately in the left and right side of the brain. The number of dark neurons increased ~2.5 fold by 8–10 weeks but not one week after inhalation of smoke indicative of a delayed manifestation of smoke inhalation injury. Cell death in same regions as assessed by TUNEL also revealed significantly higher number of apoptotic TUNEL positive cells (Table 1).
Fig 5.
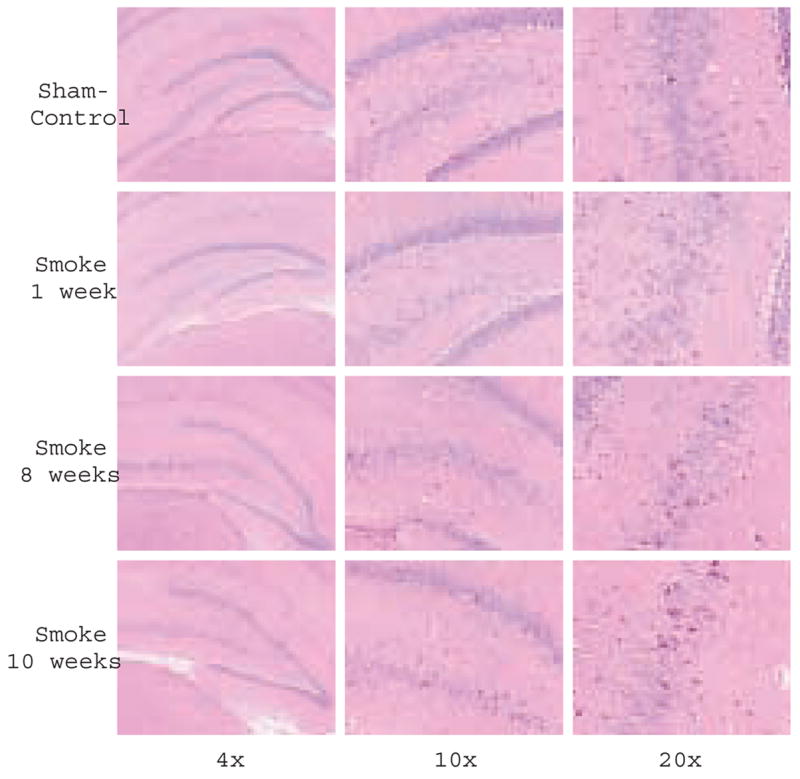
Delayed neuronal injury after inhalation of smoke. Histochemical analysis of coronal sections from smoke and sham-smoke treated rats: Representative photomicrographs of hematoxylin and eosin staining through the hippocampus (Bregma -3.14) captured with 4x, 10x and 20x objectives, from control rats and rats sacrificed 1, 8 and 10 weeks post smoke exposure. An increase in number of dark neurons (dark pink) is observed at 8 and 10 weeks.
Table 1.
Shrunk neurons and TUNEL positive cell counts after Inhalation of smoke
| Treatment | Time Post Treatment | Shrunk/Total Neuronsa | TUNEL positiveb |
|---|---|---|---|
| Sham | 1 week | 0.022 ± 0.0015 | 2.0 ± 1.0 |
| Smoke | 1 week | 0.019 ± 0.0004 | 2.3 ± 1.3 |
| Sham | 8 weeks | 0.026 ± 0.0015 | 1.6 ± 1.3 |
| Smoke | 8 weeks | 0.060 ± 0.0030* | 5.7 ± 1.6* |
| Sham | 10 weeks | 0.025 ± 0.0015 | 2.3 ± 1.6 |
| Smoke | 10 weeks | 0.071 ± 0.0012* | 6.2 ± 0.3* |
Values are means ± SD given as
ratios of shrunk to total neurons and
counts of TUNEL positive cells (n=3). Counting was done in a blinded fashion and each section was counted three times. Counts were analyzed by ANOVA for a two factorial experiment; the two factors were treatment (smoke inhalation or sham) and harvest time (1, 8, and 10 weeks).
P<0.05 vs. sham control
DISCUSSION
To gain insights into progression of events contributing to the complex molecular pathophysiology of smoke inhalation injury to the brain, we developed a combustion-smoke inhalation model in the awake rat [21]. Now, we report marked increase in lipid peroxidation and generation of oxidatively damaged DNA in the rat brain. Lipid peroxidation serves a biomarker of oxidative stress under diverse pathological conditions [31–33] and is associated with a broad range of effects ranging from changes in gene expression [34] through induction of DNA damage [35]; distinct lipid peroxidation-derived oxidative DNA lesions have been described [27–29]. Interestingly, while lipid peroxidation was evident shortly after acute exposure to smoke and was accompanied by induction of oxidative lesions in hippocampal DNA, the formation of mismatched DNA bases and loss of hippocampal neurons became apparent only at the delayed recovery times. Although the base excision repair (BER) pathway, which is the major process for repair of oxidative lesions is clearly active in the brain [14], an important question is, to what extent disrupted oxygenation and resultant oxidative stress may undermine BER capacity for faithful repair of oxidatively damaged DNA to compromise genomic integrity. This question merits investigation, particularly in view of previous reports showing that excessive oxidative stress can inhibit the DNA mismatch repair process [36] and that inflammation inhibits the process of global DNA repair [37].
Repair of DNA damage via BER is a stepwise process initiated by DNA glycosylases, which generate abasic sites that are processed into substrates for the gap filling DNA synthesis step, a critical determinant of fidelity of DNA repair. Erroneous incorporation of DNA bases might be mutagenic and destabilize the genome [16]. Interestingly, recent studies show that imbalanced BER pathway can be deleterious and that in cultured cells overexpression of certain BER enzymes leads to genomic instability and declining survival rates [38–42]. Furthermore, imbalance in BER has been implicated in microsatellite instability and human disease [43, 44]. To assess to what extent the BER pathway might become imbalanced by acute inhalation of combustion smoke, we measured BER activities and found that while excision activity for the oxidative adduct 8-oxodG remains unchanged, APE1-like activity that incises abasic sites to generate substrates for gap filling repair synthesis, was significantly elevated at 6 hours post smoke. Stimulation of APE1 by oxidative stress as redox responsive transcription factor, has been documented and is generally considered protective [45–47]. However, it is plausible that in the context of the BER pathway, APE1 activation may generate high levels of non-instructive repair intermediates and transiently overwhelm and imbalance the tightly regulated BER process.
Because a significantly elevated 8-oxodG immunoreactivity was detected in the rat hippocampal region after acute inhalation of smoke, the levels of 8-oxodG and other oxidation adducts in hippocampal DNA were further assessed using specific DNA modifying enzymes. As expected based on the immunohistochemical analysis, increased levels of 8-oxodG and other oxidation adducts in brain DNA were measured within 6 hours after exposure to smoke. Specifically, the frequency of 8-oxodG, the salient biomarker of oxidative DNA damage, as revealed by cleavage with Fpg was ~0.6/10 kb and the frequency of oxidized pyrimidines and abasic sites, targeted by Endo III, was ~0.4/10 kb. These adducts frequencies were somewhat reduced by 24 hours post smoke, indicative of partial DNA repair, with subsequent return to normal levels within 8 weeks of recovery. In sharp contrast, however, mismatched bases, revealed by cleavage with Endo V, first emerged at 24 hours post smoke at frequency of ~0.27/10 kb, which by 8 weeks post injury further increased to ~0.6/10 kb, suggesting that mismatched bases have been generated in the course of the repair process.
When considered together, these data point to the possibility that in the brain, under pathological conditions, the BER pathway becomes imbalanced resulting in reduced repair fidelity that may compromise the genome and in a long term contribute to cellular demise. It is plausible that high levels of non-instructive repair intermediates burden the repair system and contribute to generation of mismatches that persist at the late recovery times when we begin to observe delayed neuronal injury. Although, DNA mismatch repair has been reported in the brain [48], the magnitude and scope of mismatch repair activities have not been fully characterized. Our findings suggest that in the brain, under conditions of disrupted oxygenation caused by acute inhalation of smoke [21] the BER pathway becomes imbalanced. Imbalanced BER process owing for example, to transiently elevated APE1 activity may fail to sustain the fidelity of DNA repair. Since we find that the direct, smoke inhalation-induced hippocampal oxidative DNA damage is extensive, it is plausible that under the heavy load of oxidative DNA lesions, the consequences of imbalanced BER are amplified and neuronal genomic integrity is significantly compromised.
Acknowledgments
Support: Shriners Hospitals for Children 8670 and NIH NINDS 39449 grants
Abbreviations
- 8-oxodG
8-oxo-7,8-dihydro-2′-deoxyguanosine
- APE1
apurinic/apyrimidinic endonuclease 1
- BER
base excision repair
- Fpg
E. coli Formamidopyrimidine glycosylase
- Endo
endonuclease
- OGG1
oxoguanine DNA glycosylase 1
- MDA
malondialdehyde
- THF
tetrahydrofuran
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fitzpatrick JC, Cioffie WG., Jr . Diagnosis and treatment of inhalation injury. In: Herndon DN, editor. Total Burn Care, volume. Philadelphia: W.B. Saunders; 1996. pp. 184–192. [Google Scholar]
- 2.Rossi J, 3rd, Ritchie GD, Macys DA, Still KR. An overview of the development, validation, and application of neurobehavioral and neuromolecular toxicity assessment batteries: potential applications to combustion toxicology. Toxicology. 1996;115:107–117. doi: 10.1016/s0300-483x(96)03498-1. [DOI] [PubMed] [Google Scholar]
- 3.Raub JA, Benignus VA. Carbon monoxide and the nervous system. Neurosci Biobehav Rev. 2002;26:925–940. doi: 10.1016/s0149-7634(03)00002-2. [DOI] [PubMed] [Google Scholar]
- 4.Weaver LK, Hopkins RO, Chan KJ, Churchill S, Elliott CG, Clemmer TP, Orme JF, Jr, Thomas FO, Morris AH. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347:1057–1067. doi: 10.1056/NEJMoa013121. [DOI] [PubMed] [Google Scholar]
- 5.Prockop LD. Carbon monoxide brain toxicity: clinical, magnetic resonance imaging, magnetic resonance spectroscopy, and neuropsychological effects in 9 people. J Neuroimaging. 2005;15:144–149. doi: 10.1177/1051228404273819. [DOI] [PubMed] [Google Scholar]
- 6.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 7.Lindahl T, Wood RD. Quality Control by DNA Repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 8.Bogenhagen DF, Pinz KG, Perez-Jannotti RM. Enzymology of mitochondrial base excision repair. Prog Nucleic Acid Res Mol Biol. 2001;68:257–271. doi: 10.1016/s0079-6603(01)68105-4. [DOI] [PubMed] [Google Scholar]
- 9.Nouspikel T, Hanawalt PC. Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol Cell Biol. 2000;20:1562–1570. doi: 10.1128/mcb.20.5.1562-1570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viswanathan A, You HJ, Doetsch PW. Phenotypic change caused by transcriptional bypass of uracil in nondividing cells. Science. 1999;284:159–162. doi: 10.1126/science.284.5411.159. [DOI] [PubMed] [Google Scholar]
- 11.Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen D, Lan J, Pei W, Chen J. Detection of DNA base-excision repair activity for oxidative lesions in adult rat brain mitochondria. J Neurosci Res. 2000;61:225–236. doi: 10.1002/1097-4547(20000715)61:2<225::AID-JNR13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 13.Lee HM, Hu Z, Ma H, Greeley GH, Jr, Wang C, Englander EW. Developmental changes in expression and subcellular localization of the DNA repair glycosylase, MYH, in the rat brain. J Neurochem. 2004;88:394–400. doi: 10.1046/j.1471-4159.2003.02164.x. [DOI] [PubMed] [Google Scholar]
- 14.Englander EW, Ma H. Differential modulation of base excision repair activities during brain ontogeny: Implications for repair of transcribed DNA. Mech Ageing Dev. 2006;127:64–69. doi: 10.1016/j.mad.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem. 2006;96:825–832. doi: 10.1111/j.1471-4159.2005.03615.x. [DOI] [PubMed] [Google Scholar]
- 16.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 17.Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U S A. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 19.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 20.Saada-Reisch A. Deoxyribonucleoside kinases in mitochondrial DNA depletion. Nucleosides Nucleotides Nucleic Acids. 2004;23:1205–1215. doi: 10.1081/NCN-200027480. [DOI] [PubMed] [Google Scholar]
- 21.Lee HM, Greeley GH, Herndon DN, Sinha M, Luxon BA, Englander EW. A rat model of smoke inhalation injury: influence of combustion smoke on gene expression in the brain. Toxicol Appl Pharmacol. 2005;208:255–265. doi: 10.1016/j.taap.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 22.Ma H, Lee HM, Englander EW. N-terminus of the rat adenine glycosylase MYH affects excision rates and processing of MYH-generated abasic sites. Nucleic Acids Res. 2004;32:4332–4339. doi: 10.1093/nar/gkh758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yakes FM, van Houten B. PCR-based assays for the detection and quantitation of DNA damage and repair. In: Preifer GP, editor. Technologies for Detection of DNA Damage and Mutations, volume. New York: Plenum Press; 1997. pp. 171–183. [Google Scholar]
- 24.Englander EW, Greeley GH, Wang G, Perez-Polo JR, Lee HM. Hypoxia-induced mitochondrial and nuclear DNA damage in the rat brain. J Neurosci Res. 1999;58:262–269. [PubMed] [Google Scholar]
- 25.Wang G, Hazra TK, Mitra S, Lee HM, Englander EW. Mitochondrial DNA damage and a hypoxic response are induced by CoCl(2) in rat neuronal PC12 cells. Nucleic Acids Res. 2000;28:2135–2140. doi: 10.1093/nar/28.10.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G, Hallberg LM, Englander EW. Rapid SINE-mediated detection of cisplatin: DNA adduct formation in vitro and in vivo in blood. Mutat Res. 1999;434:67–74. doi: 10.1016/s0921-8777(99)00021-x. [DOI] [PubMed] [Google Scholar]
- 27.Bartsch H, Nair J. Accumulation of lipid peroxidation-derived DNA lesions: Potential lead markers for chemoprevention of inflammation-driven malignancies. Mutat Res. 2005;591:34–44. doi: 10.1016/j.mrfmmm.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Otteneder MB, Knutson CG, Daniels JS, Hashim M, Crews BC, Remmel RP, Wang H, Rizzo C, Marnett LJ. In vivo oxidative metabolism of a major peroxidation-derived DNA adduct, M1dG. Proc Natl Acad Sci U S A. 2006;103:6665–6669. doi: 10.1073/pnas.0602017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou X, Taghizadeh K, Dedon PC. Chemical and biological evidence for base propenals as the major source of the endogenous M1dG adduct in cellular DNA. J Biol Chem. 2005;280:25377–25382. doi: 10.1074/jbc.M503079200. [DOI] [PubMed] [Google Scholar]
- 30.Davis RL, Robertson DM. Textbook of Neuropathology. 3. Baltimore: Williams & Wilkins; 1997. pp. 547–576. [Google Scholar]
- 31.Garcia YJ, Rodriguez-Malaver AJ, Penaloza N. Lipid peroxidation measurement by thiobarbituric acid assay in rat cerebellar slices. J Neurosci Methods. 2005;144:127–135. doi: 10.1016/j.jneumeth.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 32.Choi YB, Kim YI, Lee KS, Kim BS, Kim DJ. Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Res. 2004;1019:47–54. doi: 10.1016/j.brainres.2004.05.079. [DOI] [PubMed] [Google Scholar]
- 33.Smith AM, Zeve DR, Grisel JJ, Chen WJ. Neonatal alcohol exposure increases malondialdehyde (MDA) and glutathione (GSH) levels in the developing cerebellum. Brain Res Dev Brain Res. 2005;160:231–238. doi: 10.1016/j.devbrainres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Marini H, Altavilla D, Bellomo M, Adamo EB, Marini R, Laureanti F, Bonaccorso MC, Seminara P, Passaniti M, Minutoli L, Bitto A, Calapai G, Squadrito F. Modulation of IL-1 beta gene expression by lipid peroxidation inhibition after kainic acid-induced rat brain injury. Exp Neurol. 2004;188:178–186. doi: 10.1016/j.expneurol.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181–182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 36.Chang CL, Marra G, Chauhan DP, Ha HT, Chang DK, Ricciardiello L, Randolph A, Carethers JM, Boland CR. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002;283:C148–154. doi: 10.1152/ajpcell.00422.2001. [DOI] [PubMed] [Google Scholar]
- 37.Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 38.Coquerelle T, Dosch J, Kaina B. Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents--a case of imbalanced DNA repair. Mutat Res. 1995;336:9–17. doi: 10.1016/0921-8777(94)00035-5. [DOI] [PubMed] [Google Scholar]
- 39.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria; targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63:608–615. [PubMed] [Google Scholar]
- 40.Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Res. 2005;33:4660–4671. doi: 10.1093/nar/gki759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shokolenko IN, Alexeyev MF, Robertson FM, LeDoux SP, Wilson GL. The expression of Exonuclease III from E. coli in mitochondria of breast cancer cells diminishes mitochondrial DNA repair capacity and cell survival after oxidative stress. DNA Repair (Amst) 2003;2: 471–482. doi: 10.1016/s1568-7864(03)00019-3. [DOI] [PubMed] [Google Scholar]
- 42.Cabelof DC, Raffoul JJ, Nakamura J, Kapoor D, Abdalla H, Heydari AR. Imbalanced base excision repair in response to folate deficiency is accelerated by polymerase beta haploinsufficiency. J Biol Chem. 2004;279:36504–36513. doi: 10.1074/jbc.M405185200. [DOI] [PubMed] [Google Scholar]
- 43.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou X, Mechanic LE, Zurer I, Rotter V, Samson LD, Harris CC. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan CY, Liu KL, Huang HY, Barnes EL, Swalsky PA, Bakker A, Woods J, Finkelstein SD. Frequent allelic imbalance and loss of protein expression of the DNA repair gene hOGG1 in head and neck squamous cell carcinoma. Lab Invest. 2001;81:1429–1438. doi: 10.1038/labinvest.3780356. [DOI] [PubMed] [Google Scholar]
- 45.Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- 46.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci U S A. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ziel KA, Campbell CC, Wilson GL, Gillespie MN. Ref-1/Ape is critical for formation of the hypoxia-inducible transcriptional complex on the hypoxic response element of the rat pulmonary artery endothelial cell VEGF gene. Faseb J. 2004;18:986–988. doi: 10.1096/fj.03-1160fje. [DOI] [PubMed] [Google Scholar]
- 48.Brooks PJ, Marietta C, Goldman D. DNA mismatch repair and DNA methylation in adult brain neurons. J Neurosci. 1996;16:939–945. doi: 10.1523/JNEUROSCI.16-03-00939.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]