

Abstract
The mouse glucocorticoid receptor-interacting protein (GRIP1) is a member of the ERAP160 family of nuclear receptor (NR) coactivators (including SRC-1 and TIF2) which function as bridging proteins between ligand-activated NRs bound to cognate hormone-response elements (HREs) and the transcription initiation apparatus (TIA). Although these coactivators bind to several NRs, studies overexpressing these coactivators with these NRs in mammalian cells have not uniformly observed a corresponding enhancement of ligand-dependent transactivation. Here, we show that GRIP1 interacts in vitro in a ligand-dependent manner with thyroid receptor, retinoic acid receptor, and retinoid X receptor. Additionally, in yeast (Saccharomyces cerevisiae) GRIP1 coactivator protein markedly increased the ability of these full-length class II NRs to transactivate β-galactosidase reporter genes containing cognate HREs. The magnitude of GRIP1 enhancement of liganded NR homodimer was dependent upon NR subtype and HRE configuration. For most HRE configurations, thyroid receptor and retinoic acid receptor homodimers were essentially unresponsive or very weakly active in the absence of GRIP1, but GRIP1 dramatically restored the ligand-dependent function of these NRs. Although GRIP1 exerted no significant effect on NR homodimers in the absence of their cognate ligands, it increased the transactivation of unliganded NR heterodimers. Whether GRIP1 increased ligand-dependent transactivation of a heterodimer to levels greater than that of the cognate homodimer was determined by HRE configuration and copy number. Compared with the limitations of yeast two-hybrid and mammalian coexpression systems, the yeast HRE-assay systems described in this report facilitated both the detection of putative mammalian NR coactivator function and the elucidation of their mechanisms of transactivational enhancement.
Keywords: cell biology, thyroid hormone, transcriptional regulation, subtype specificity, nuclear receptor-interacting protein
Thyroid hormone (T3) receptor (TR), retinoic acid receptor (RAR), and retinoid X receptor (RXR) are members of the nuclear receptor (NR) superfamily of transcriptional proteins which regulate complex gene networks and thereby control diverse biological aspects of growth, development, and homeostasis (1–5). TR, RAR, and RXR are activated by their cognate ligands l-triiodothyronine (T3), all-trans-retinoic acid (at-RA), and 9-cis-retinoic acid (9c-RA), respectively (1–3). Additionally, there are multiple subtypes derived from different genes that have been detected for TRs (1, 4), RARs, and RXRs (1–3). These nuclear receptors (NRs) modulate gene expression primarily by binding as homodimers or heterodimers to AGGTCA hexameric core motifs with variations in sequence, spacing, and orientation called hormone-response elements (HREs), which are located in the regulatory region of target genes (1–5). The steroid (class I) subgroup of NRs bind to HREs as homodimers, whereas members of class II subgroup—i.e., TR, RAR, vitamin D receptor (VDR), and peroxisome proliferator activator receptor (PPARs) may bind not only as homodimers but also with high affinity as heterodimers with RXR to specific HREs (3). Consequently, RXR not only functions as a homodimer that is responsive to its cognate ligand, 9c-RA, but also, through the formation of heterodimers with other class II NRs, plays a central role in the regulation of many intracellular receptor signaling pathways (1–3).
NRs function as ligand-dependent DNA-binding activator proteins to stimulate efficient transcriptional initiation of RNA polymerase II by either directly or indirectly interacting with components of the transcription initiation apparatus (TIA) assembled at the TATA box (6, 7). TIA consists of RNA polymerase II and general transcription factors, designated as TFIID [a complex of TATA box-binding protein (TBP) plus TBP-associated factors (TAFs)], TFIIA, TFIIB, TFIIE, TFIIF, and TFIIH, which are evolutionarily conserved between yeast and humans. Some DNA-bound transcriptional activator proteins directly interact with general transcription factors, whereas others require additional proteins called coactivators to activate the TIA (6, 7). Although coactivators cannot function directly as transcriptional activators because they lack DNA-binding domains (DBDs) to interact with target genes, they nevertheless can mediate transcriptional activation by linking DNA-bound activator proteins with the TIA (7). Several proteins which function as coactivators are TAFs—i.e., subunits of TFIID, whereas others are not related to TAFs (6, 7). TR and RAR have been shown to interact directly with basal transcription factors TFIIB and TBP as well as specific TAFs (8, 9), but the functional significance of these interactions and the requirement for coactivator proteins are still under investigation. The identification of additional proteins that interact with TR, RAR, and RXR NRs could also provide important insights into the roles played by these transcription factors in the regulation of gene function.
A large number of candidate coactivators have been identified in mammalian cells as proteins that interact with the hormone-binding domain (HBD) of NRs in the presence of cognate ligands (10–21). However, only a few fulfill the criteria of a bona fide ligand-dependent coactivator (ref. 10, and references therein). A 125-kDa coactivator, SRC-1, enhances in vivo transcription activation by not only progesterone receptor (PR) but also glucocorticoid receptor (GR), estrogen receptor (ER), TR, and RXR in mammalian cells (16). Subsequently, a 160-kDa mouse coactivator protein that is 88% identical to SRC-1 in the C terminus and an N-terminal extension has been detected as well as related proteins arising from splice variations (17). An 86-kDa mouse protein fragment with coactivator activity called GR-interacting protein (GRIP1) (18) has partial identity to SRC-1. A 160-kDa coactivator, TIF2, has been subsequently reported (19); it has 94% amino acid sequence identity to full-length GRIP1 and is therefore likely its human orthologue (43). In a yeast two-hybrid system and in vitro, GRIP1 interacted with the HBD of GR, ER, and androgen receptor (AR) in a ligand-regulated manner (18). TIF2 also exhibited ligand-dependent interactions in vitro with the HBD of class I and class II NRs but had detectable enhancement of ligand-dependent transactivation by class I but not class II NRs when overexpressed in HeLa and Cos1 cells (19). A 300-kDa protein (CBP/p300) which binds to the cAMP response element-binding protein (CREB) functions as a coactivator of CREB-mediated signaling pathways. CBP/p300 also binds to the SRC1 coactivator variant and NRs, and thereby functions as a cointegrator of several signaling pathways (17, 20, 44). Proteins that may function as transactivational corepressors for unliganded class II NRs have also been recently reported (10, 22–24).
The recently accumulated evidence indicates that SRC-1, GRIP1, and TIF2 are true coactivators (10). However, when both physical and functional interactions have been examined, a precise correlation of physical interaction with transcriptional activation by multiple NRs in mammalian cells has not been uniformly observed. For example, SRC-1 (16) and TIF2 (19) interacted physically with several class I and II NR-HBDs in vitro, whereas they enhanced the transcriptional activation of only a few of these NRs when overexpressed in mammalian cells (19). One possible explanation of such findings is that the levels of endogenous coactivators in mammalian cells are maximal for some NRs and suboptimal for others. This situation could result from different efficiencies of interaction between NRs and endogenous coactivators. Such conditions indigenous to mammalian cells could mask the detection of additional effects from overexpressed coactivators. In the search for systems wherein endogenous coregulators would not compromise the study of coregulator function, we have explored the utility of yeast as an alternative in vivo coexpression system for determining the molecular mechanisms mediating NR ligand-dependent function (25–28). However, we (28) and others (29–31) have observed concurrently that full-length TR homodimers have very weak ligand-inducible effects on single-copy TREs in yeast and that the actions of cognate ligands could be moderately enhanced by RXR. Similarly, RAR homodimer function in yeast is very weak and requires RXR to achieve a moderate ligand-dependent transactivational response (32–34). A GenBank search with the National Institutes of Health blast program (35) has detected no significant structural homologues in S. cerevisiae for mammalian NR coactivators SRC-1 (16, 17), GRIP1 (18), and TIF2 (19) or mammalian NR corepressors N-CoR (22), SMRT (23), and SMRT/TRAC2 (24). Although these findings support the idea that yeast does not contain such putative NR transcriptional coregulators, it does not exclude the presence in yeast of structurally nonhomologous adaptor proteins with overlapping functions.
We have examined, therefore, the functional effects of the 86-kDa fragment of the mouse GRIP1 coactivator protein (18) in the cellular context of yeast using an in vivo coexpression system of full-length NRs and β-galactosidase (β-gal) reporters containing cognate HREs as previously described (26, 28). To distinguish the “yeast two-hybrid assays,” which directly measure interactions between known proteins by using a reporter gene containing a heterologous promoter (36), from the in vivo coexpression systems used in the present report, which analyze interactions between NRs and reporter genes containing cognate HREs, the descriptive term “yeast-HRE assays” has been assigned. Here, we have analyzed the in vivo and in vitro effects of GRIP1 coactivator on several class II NR homodimers and RXR heterodimers. We show that yeast HRE assay systems can detect GRIP1 coactivator-dependent transactivational responses of TR, RAR, and RXR homodimers to their cognate ligands. The levels of reporter gene activity achieved by TR and RAR homodimers in the presence of GRIP1 were often severalfold greater than those of RXR heterodimers in the absence of GRIP1. The ligand-dependent responses of NR homodimers facilitated by GRIP1 were also regulated by variations in NR subtype and HRE characteristics. The GRIP1-mediated transactivational effects by both unliganded and liganded RXR heterodimers could be regulated by HRE characteristics. Taken together, these studies have shown the utility of yeast HRE assay systems in detecting and characterizing the activation function of putative mammalian NR coactivators.
MATERIALS AND METHODS
Yeast Strains and Media.
The S. cerevisiae strain YPH 499 (MATa, ura3, lys2, ade2, trp1, his3, leu2) was used for transformations. Yeast transformants were grown in minimal medium (0.67% yeast nitrogen base/2% glucose) routinely supplemented with adenine (40 mg/liter), lysine (40 mg/liter), and, when required, histidine (20 mg/liter).
Yeast Plasmids for NRs, GRIP1, and Reporter Genes.
The yeast expression plasmid pGRIP1 encoding the 86-kDa (812-amino acid) fragment of GRIP1 and using a LEU2 selectable marker was as previously described (18). Human TRβ1, TRα1, RARα, and RARγ and mouse RXRα, β, and γ subtypes were cloned downstream of a CUP1 promoter into a 2μ multicopy YEp 46 yeast vector containing a TRP1 selectable marker (25). Human TRβ1 was cloned downstream of a GPD promoter into a 2μ p2HG vector containing a HIS3 selectable marker (29), and human RARα in a 2μ YEp90 yeast vector containing a HIS3 selectable marker (32) under the control of a PGK promoter. The mouse RXRα was under the control of a PGK promoter of a 2μ YEp10 yeast vector containing a TRP1 marker (34). The chicken RXRγ and human RARβ were inserted downstream of a 2μ GPD promoter in a pG1 vector containing the TRP1 marker (29). HREs were prepared as double-stranded oligonucleotides containing a single copy (two hexameric motifs) of a DR4 consensus sequence thyroid response element (TRE) (28), F2 enhancer element of chicken lysozyme promoter as a TRE, PAL sequence of the rat growth hormone gene as a TRE (28), the βRAR gene as a retinoic acid response element (RARE) (34), and the ApoA1 gene as an RXR response element (37). These single-copy or two-copy (when indicated) HREs were placed in the pC2 URA3 reporter plasmid using a unique XhoI site upstream from a cytochrome c promoter (CYC1) linked to the Escherichia coli lacZ gene expressing β-gal as previously described (26, 28).
Interaction of GRIP1 and NRs in Vitro.
Glutathione S-transferase (GST) and GST-GRIP1 fusion proteins containing amino acids 415–812 of the 86-kDa GRIP1 protein were prepared, bound to glutathione-Sepharose beads, and analyzed for binding with labeled proteins in vitro as described previously (18). Briefly, the 35S-labeled full-length TRα1, TRβ1, RARα, and RXRα proteins were synthesized by transcription and translation in vitro, and incubated with Sepharose containing GST or GST-GRIP1; after washing, the labeled proteins bound to the beads were eluted by boiling in SDS sample buffer and analyzed by SDS/PAGE and autoradiography (2-day exposure).
Analysis of NR Transcriptional Activation Function and GRIP1 Coactivator Function in Yeast.
The yeast transformants were isolated and grown in the appropriate minimal medium with added supplement as required. Cells were grown overnight with the appropriate ligand at a final concentration of 1 μM, harvested, washed and resuspended in Z buffer (28), and lysed with glass beads (425–600 μm) before centrifugation. The supernatant was collected, and the protein concentration was determined by the Lowry method (38) using BSA as a standard. Ten or 20 μg of protein was used for β-gal assay (39), and the activities were expressed as Miller units/mg of protein. Data shown were pooled from three independent experiments and calculated as the mean ± standard error.
RESULTS
GRIP1 Interacts in Vitro with Full-Length TR, RAR, and RXR.
GRIP1 was previously shown to interact in a hormone-dependent manner with GR, ER, and AR in vitro and also served as an in vivo transcriptional coactivator for the transactivation domains in the HBD of these steroid receptors using a yeast hybrid protein system (18). In the current report, we measured the in vitro interaction of GRIP1 with several full-length class II NR proteins. Amino acid residues 415–812 of the 86-kDa GRIP1 fragment were expressed in E. coli as a fusion protein with GST (18). This GST/GRIP1 fusion protein was absorbed onto glutathione-Sepharose beads and subsequently incubated with in vitro synthesized 35S-labeled full-length NRs and respective cognate agonists (T3 for TR, at-RA for RAR, and 9c-RA for RXR). Sepharose-bound material was eluted and analyzed by SDS/PAGE and autoradiography. TRα1 and RARα did not bind to GST in the presence (Fig. 1, lane 3) or absence (not shown) of ligand. Binding of TRβ1 and RXRα to GST/GRIP1 in the absence of cognate ligand (Fig. 1, Lane 1) was more specific than to GST in the presence of ligand (Fig. 1, lane 3). Additionally, increased specific and highly hormone-dependent in vitro binding of GST/GRIP1 fusion protein was detected for all these NRs (Fig. 1, lane 2) compared with the activity when ligand was not present (Fig. 1, lane 1). Thus, the GST/GRIP1 415–812 fusion protein previously shown to interact in vitro with GR, ER, and AR steroid receptors (class I NRs) (18), can also interact with TR, RAR, and RXR (class II NRs) in a ligand-dependent manner. These observations obtained using full-length class II NRs were in accord with the results reported by others (19) of class II NR HBD physical interactions with a TIF2.1 (amino acid residues 624-1287) fragment fused to GST.
Figure 1.

Demonstration of ligand-dependent in vitro interaction of GRIP1 with full-length class II NRs. 35S-labeled full-length class II NRs were synthesized in vitro and then incubated with Sepharose beads containing bound GST/GRIP1 or GST protein in the presence (+) or absence (−) of cognate ligands at a concentration of 1 μM. The beads were washed and bound proteins were eluted and analyzed by SDS/PAGE and autoradiography. The percentage of total added radioactivity bound in the absence of ligand (lane 1) < 1% and in the presence of ligand (lane 2) ≈ 10%.
GRIP1 Is a Coactivator for the Ligand-Dependent Function of TR, RAR, and RXR Homodimers in Yeast.
Class II NR homodimers have previously been shown to have little or no ligand-induced transactivational activity on single HREs in yeast, whereas RXR heterodimers are active (27–28). This could indicate either that class II NRs must always function as RXR heterodimers or that yeast is devoid of coactivator proteins essential for activation of TR and RAR homodimers by their cognate ligands. Since GRIP1 functioned as a transcriptional coactivator of steroid receptors HBDs fused to the GAL4 DBD (18), we tested whether it could also enhance the ligand-dependent transactivation by full-length TRβ1 and TRα1 of reporter genes controlled by various TREs. In accord with our previous studies performed in the absence of GRIP1 coactivator (25–28), T3 had only a weak transactivational effect on TRβ1 in a yeast-HRE system with F2 and PAL enhancer elements (Fig. 2 Middle and Bottom, column 3) and no significant effect on a DR4 element (Fig. 2 Top, column 3). As previously reported (29, 30), TRα1 was even less responsive to T3 on all three TREs (Fig. 2, column 5). Although GRIP1 alone had no intrinsic transcriptional activation activity in either the absence or the presence of T3 (Fig. 2, column 2), it could dramatically enhance hormone-dependent transactivational function for both TRα1 and β1 subtypes on the differently configured TREs in the presence of T3 ligand (Fig. 2, columns 4 and 6). The TRα1 and TRβ1 subtypes were equally responsive to T3 ligand on PAL-TRE, whereas TRα1 was slightly less active than TRβ1 on F2 and DR4-TREs. Thus, the ligand-dependent coactivation exerted by GRIP1 on different TR subtypes could be regulated by HRE characteristics.
Figure 2.

Coexpression of GRIP1 in yeast increases the transactivation by TRβ1 and TRα1 homodimers in response to T3 ligand. Yeast transformants expressing TRs alone or with GRIP1 and containing β-gal reporter genes with a single copy of DR4 (Top), F2 (Middle), or PAL (Bottom) response element as TREs were grown for 16 hr in the presence of no ligand (open bars) or 1 μM T3 (shaded bars) and β-gal was assayed [U, Miller unit (39)].
Whether the coexpression of NR coactivators with RARs could restore a transactivational response to at-RA or 9c-RA cognate ligands in yeast without the presence of RXRs had not been previously studied. When a β-gal reporter gene containing the βRARE × 1 response element was used (Fig. 3 Lower), only RARβ had significant transactivational response to at-RA in the absence of GRIP1 (Fig. 3, column 5), whereas both RARα (Fig. 3, column 3) and RARγ (Fig. 3, column 7) were unresponsive. Reporter gene transactivation in either the presence or the absence of ligand was not stimulated by GRIP1 alone (Fig. 3, column 2). However, the presence of GRIP1 and at-RA significantly increased the transactivation activity of all RAR subtypes: RARβ > RARγ > RARα (Fig. 3 Lower, columns 4, 6, and 8). When a βRARE × 2 HRE was used, the at-RA-dependent transactivational responses of all RARs were remarkably enhanced by the presence of GRIP1 (Fig. 3 Upper, columns 4, 6, and 8). Similar results were obtained when 9c-RA was substituted for at-RA (data not shown). Thus, a potent ligand-dependent activation of all RAR subtypes by GRIP1 was demonstrated with βRARE × 1 and βRARE × 2 reporter genes.
Figure 3.

Coexpression of GRIP1 in yeast increases the transactivation by RARs in response to at-RA ligand. Yeast transformants expressing different RAR receptor subtypes alone or with GRIP1 and containing β-gal reporter genes with either βRARE × 2 (Upper) or βRARE × 1 (Lower) enhancer elements were grown for 16 hr in the presence of no ligand (open bars) or 1 μM at-RA (shaded bars) and β-gal was assayed.
Studies of RXR homodimer function in yeast using several RXREs have established that 9c-RA but not at-RA can stimulate the transactivation of RXR homodimers (31, 33). To examine the possibility that GRIP1 could also increase cognate ligand responses of RXRs, a yeast reporter containing the ApoA1 × 1 response element was used. The increases in transactivation exerted by 9c-RA in the absence of GRIP1 also varied according to receptor subtype: RXRα > RXRβ > RXRγ (Fig. 4, columns 3, 5, and 7). As observed for the other class II NRs, GRIP1 caused substantial enhancement of 9c-RA-dependent transactivation by RXRs: RXRβ > RXRα > RXRγ (Fig. 4, columns 4, 6, and 8). However, when the enhancement of ligand-dependent transactivational responses mediated by GRIP1 on single-copy HRE reporters for TR (Fig. 2) and RXR (Fig. 4) were compared, the increases of RXR activity due to GRIP1 (ranging from 3- to 18-fold) were relatively less than those for TRs (10- to 45-fold). Moreover, the ApoA1 × 2 reporter exhibited stronger ligand-dependent activity with RXRs than the corresponding single element, and this activity was only modestly enhanced by GRIP1 (data not shown). Thus, differences in the magnitude of GRIP1-mediated enhancement of ligand-dependent transactivation by RXRs and TRs in yeast likely reflect the relatively greater cognate ligand responsiveness of RXRs in the absence of GRIP1 compared with TRs.
Figure 4.

Coexpression of GRIP1 increases the transactivation by RXR in response to 9c-RA ligand. Yeast transformants expressing RXRα, RXRβ, or RXRγ alone or with GRIP1 and containing a β-gal reporter with a site A1 enhancer element sequence of the apolipoprotein gene (ApoA1 × 1) were grown for 16 hr in the presence of no ligand (open bars) or 1 μM 9c-RA (shaded bars) and β-gal was assayed.
GRIP1 Enhances Transactivational Effects of Unliganded and Liganded RXR Heterodimers in Vivo.
The formation by RXR heterodimers with TR and RAR can enhance DNA binding and transactivation function (refs. 1–3 and references therein). We have therefore examined the influence of GRIP1 coactivators on the transcriptional activation function of TRβ1/RXRα heterodimers (Fig. 5) and RARα/RXRα heterodimers (Fig. 6) in the absence and presence of cognate ligands. Interestingly, GRIP1 caused a substantial enhancement of heterodimer activity on some promoters even in the absence of cognate ligands (Figs. 5 and 6, column 8, open bars). The magnitude of GRIP1-enhanced transactivation by liganded TRβ1/RXRα heterodimer was regulated by HRE configuration—i.e., DR4 > F2 and PAL-TRE (Fig. 5) and liganded RARα/RXRα heterodimer by HRE copy number—i.e., βRARE × 2 > × 1 (Fig. 6). The enhancement of unliganded heterodimer activity is consistent with increased in vitro binding of GRIP1 to RXR compared with TRs and RARs in the absence of cognate ligands (Fig. 1, column 1). Compared with the GRIP1 enhancement of ligand-dependent activity by the TRβ1 homodimer (Fig. 5, column 4) as well as the RAR and RXR homodimers (Fig. 6, columns 4 and 6), RXR heterodimer transactivation by single and dual ligands was not necessarily enhanced by GRIP1 (Figs. 5 and 6, column 8). For the F2 and PAL response elements—i.e., everted and inverted palindrome TREs, respectively, the ligand-dependent β-gal activity levels observed in the presence of GRIP1 were no greater for TRβ1/RXRα heterodimers than for TR homodimer (Fig. 5 Middle and Bottom), whereas, for the DR4 TRE element—i.e., direct hexamer repeat spaced by 4 bp—the heterodimer was much more strongly activated than the homodimer in the presence of GRIP1 (Fig. 5 Top, column 8 versus column 4). Interestingly, in most studies, RXR heterodimers exhibited ½ to 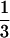 as much transactivation activity by cognate ligands in the absence of GRIP1 as that achieved by TRβ1 and RARα homodimers when GRIP1 was present (see Figs. 5 and 6, column 7 versus column 4). Although the absolute β-gal activity levels achieved by ligands on heterodimers were clearly enhanced severalfold by the presence of GRIP1 (Figs. 5 and 6, column 7 versus column 8) and the levels achieved were often greater than those of corresponding homodimers, these apparent effects could be mediated in part by a GRIP1-mediated enhancement of the basal (unliganded) transactivation of the NR heterodimer but not the homodimer.
as much transactivation activity by cognate ligands in the absence of GRIP1 as that achieved by TRβ1 and RARα homodimers when GRIP1 was present (see Figs. 5 and 6, column 7 versus column 4). Although the absolute β-gal activity levels achieved by ligands on heterodimers were clearly enhanced severalfold by the presence of GRIP1 (Figs. 5 and 6, column 7 versus column 8) and the levels achieved were often greater than those of corresponding homodimers, these apparent effects could be mediated in part by a GRIP1-mediated enhancement of the basal (unliganded) transactivation of the NR heterodimer but not the homodimer.
Figure 5.

Coexpression of GRIP1 in yeast modulates the effects of single and dual cognate ligands on a TRβ1/RXRα heterodimer. Yeast transformants expressing TRβ1 and/or RXRα receptors either alone or with GRIP1 and containing a β-gal reporter gene containing DR4 (Top), F2 (Middle), or PAL (Bottom) enhancer element as TREs were grown for 16 hr in the presence of no ligand (open bars), 1 μM T3 (shaded bars), 1 μM 9c-RA (cross-hatched bars), or 1 μM T3 + 1 μM 9c-RA (solid bars) and β-gal was assayed.
Figure 6.

Coexpression of GRIP1 in yeast modulates the effects of single and dual cognate ligands on RARα/RXRα heterodimer. Yeast transformants expressing RARα and/or RXRα either alone or with GRIP1 and containing β-gal reporters with either βRARE × 2 (Top) or βRARE × 1 (Middle) as RARE or ApoA1 × 1 (Bottom) enhancer elements were grown for 16 hr in the presence of no ligand (open bars), 1 μM at-RA (shaded bars), 1 μM 9c-RA (cross-hatched bars), or 1 μM at-RA + 1 μM 9c-RA (solid bars).
DISCUSSION
Class I and class II NRs function as DNA-bound transcription factors that can activate or repress the function of adjacent genes by crucial protein–protein contacts with basal transcription factors, transcriptional coactivators, or chromatin structures (1–5). In a yeast two-hybrid system, the 86-kDa GRIP1 protein fragment was shown to act as a coactivator of GAL4 DBD-steroid receptor HBD fusion proteins by interacting with the HBDs of GR, ER, and AR in a hormone-regulated manner (18). We show that the GRIP1 fragment coexpressed in a yeast HRE assay system can also serve as a coactivator for the ligand-dependent function of native full-length TR, RAR, and RXR class II NR homodimers. The in vivo coactivator effects exerted by GRIP1 correlated with its observed in vitro binding to these NRs. The GRIP1-mediated ligand-dependent transactivation effects could also be regulated by NR subtype and by HRE configuration and copy number. In the absence of GRIP1, class II NR homodimers were essentially inactive or had weak cognate ligand responses in yeast, and this activity could only be weakly up-regulated by coexpression of RXR (Figs. 5 and 6 and ref. 28). Remarkably, GRIP1 dramatically increased cognate ligand-induced transactivation by TR and RAR homodimers. Recently, the full-length 158-kDa GRIP1 has been identified (43). Compared with the transactivational effects of the GRIP1 activator fragment in the yeast HRE assays reported herein, the substitution of full-length GRIP results in a lower (≈30–40%) but similar pattern of transactivational enhancement of liganded TR, RAR, and RXR homodimers (data not shown), suggesting that the presence of additional N- and C-terminal amino acids may exert a moderate inhibition of the GRIP1 activator fragment. While the precise mechanisms whereby coactivators mediate transcriptional regulation remain to be defined, ligand binding to class I and class II NRs is thought to induce conformational changes that promote interactions among receptor dimers, coactivators, cognate HREs, and the TIA (refs. 1 and 3 and references therein). Our studies in yeast HRE assay systems are consistent with the hypothesis that GRIP1 acts primarily as a bridge (or adaptor) between the DNA-bound ligand-activated NR and the TIA.
The striking enhancement of NR homodimer function by GRIP1 also indicates that the yeast cell lacks the endogenous coactivators essential for the potent transactivation of these NRs by their cognate ligands. These differences between yeast and mammalian cells likely account for the efficacy of yeast HRE systems in detecting GRIP1-dependent regulation of class II NRs. In contrast, HeLa and Cos1 mammalian cells did not detect in vivo coactivator function by TIF2 (a 160-kDa probable human orthologue of GRIP1) on class II NRs (19). Since NRs as well as their cognate ligands and coactivators are widely expressed in mammalian tissues (15–20), the difficulty in detecting ligand-dependent effects in mammalian cell cotransfection systems may be due to nonlimiting endogenous levels of the coactivator. In fact, it is possible that the higher the endogenous coactivator binding for specific NRs in mammalian cells, the more difficult it will be to detect functional effects. Thus, the absence of these indigenous complexities in yeast provides an ideal eukaryotic HRE coexpression system to study the functional characteristics of mammalian NR coregulatory proteins. In this regard, the concurrent absence of NR corepressor proteins could result in higher effects or lesser ligand dependence by NR coactivators in yeast. It is also very likely that NR coactivators and corepressors are ubiquitously expressed and have interactive regulatory effects on NR transactivation function which are difficult to unravel in mammalian cells. It will be of interest therefore to reconstruct the interplay between corepressors and coactivators on the regulation of transcription by NRs in yeast HRE assay systems.
Because RXRs are indigenous to mammalian cell systems, it is difficult to determine whether TRs and RARs can activate target genes as homodimers or, alternatively, must always function as a heterodimer partner with RXR (refs. 1 and 3 and references therein). The absence of all NRs in yeast allowed us to test the transactivation activity of TR and RAR homodimers. We have shown, using yeast–HRE systems, that TR and RAR homodimers can activate some target genes more efficiently than RXR heterodimers when appropriate mammalian coactivators are present. These observations suggest the possibility that NR class II homodimers may also have physiologically relevant activity in mammalian cells. Compared with TR and RAR, RXR homodimers could express in yeast quite high hormone-dependent transactivaton. Such observations suggest the presence of a mechanism for transactivation by RXR in yeast which cannot be efficiently utilized by several other NRs. This mechanism could account for the relatively weak activation of TR/RXR heterodimer in the absence of GRIP1 by cognate ligands in yeast (27–29). Since the transactivation activity of both RXR homodimer and TR or RAR/RXR heterodimers could be clearly enhanced by the presence of GRIP1 (see Figs. 5 and 6, column 7 versus column 8), we would predict that the transactivation activity of RXR in yeast could be supported by multiple mechanisms. Thus, the unique cellular context of the yeast provides a useful in vivo transactivational assay system to detect novel molecular mechanisms of coactivator-dependent activation of NR homodimers and heterodimers on differently configured HREs.
Yeast two-hybrid assays (36) have been used to detect mammalian coregulatory protein interactions with NRs (17–20). This system analyzes the protein–protein interactions of HBD fragments of NRs and coactivator proteins fused to the DBD or activational domain (AD) of GAL4 or LexA. These interactions are measured by the activation of β-gal reporter plasmids containing GAL4- or LexA-responsive promoters (36). The transactivational effects resulting from interactions of the coactivator with full-length NRs and their cognate HREs cannot be deciphered in the yeast two-hybrid system. Consequently, the anticipated regulation of transcription in vivo by receptor dimerization and folding (40–42) and the modulating effects on coactivator protein interactions exerted by different NR subtypes and HRE configurations cannot be directly assessed by yeast two-hybrid assays. However, our studies reveal that such interactions between an NR coactivator such as GRIP1 and liganded full-length NRs can be experimentally evaluated in yeast HRE assays.
To our knowledge, this is the first report which has demonstrated the potential utility of yeast–HRE systems for both detecting and characterizing the functional activities of NR coactivator proteins. Thus, compared with the complexities of mammalian cells and the limitations of yeast two-hybrid systems, we show that the yeast HRE assay as applied to the study of GRIP1 provides a valuable in vivo transactivation assay system to elucidate the potential functional properties of putative mammalian NR coactivators.
Acknowledgments
We thank R. Y. Wang, E. Pisano, I. Frenkel, D. Grynspan, and K. Kohli for technical assistance; E. Klugerman for computer graphic assistance; C. Walfish for the preparation of the manuscript; A. Levin of Hoffmann–La Roche for supplies of 9c-RA; R. Lossant and P. Chambon for the mouse RXRα yeast vector; B. L. Hall, Z. Smit-McBride, and M. L. Privalsky for yeast vectors; P. Brickell for the chicken RXRγ cDNA; R. M. Evans for human RARα cDNAs; and P. Mak for the ApoA1 yeast reporter. This work was funded by grants to P.G.W. from the Medical Research Council of Canada (Grant MT 12604), an Isabel Silverman Canadian International Exchange Program project of the Mount Sinai Hospital Department of Medicine Research Fund, Saul A. Silverman Family Foundation, Temmy Latner/Dynacare and The Meadowcroft Group; funding to M.R.S. was from National Institute of Diabetes and Digestive and Kidney Diseases Grant DK 43093 and American Cancer Society Grant BE-253. T.Y was supported by a Research Fellowship from the Thyroid Foundation of Canada, and H.H. was the recipient of a predoctoral traineeship from the University of California Breast Cancer Research program.
ABBREVIATIONS
- GR
glucocorticoid receptor
- GRIP1
glucocorticoid receptor-interacting protein
- TR
thyroid receptor
- TRE
thyroid response element
- T3
thyroid hormone (l-triiodothyronine)
- RAR
retinoic acid receptor
- RARE
retinoic acid response element
- at-RA
all-trans-retinoic acid
- RXR
retinoid X receptor
- RXRE
retinoid X receptor response element
- 9c-RA
9-cis-retinoic acid
- AR
androgen receptor
- ER
estrogen receptor
- DBD
DNA-binding domain
- HBD
hormone-binding domain
- HRE
hormone-response element
- NR
nuclear receptor
- GST
glutathione S-transferase
- β-gal
β-galactosidase
- TIA
transcription initiation apparatus
- TBP
TATA box-binding proteins
- TAFs
TBP-associated factors
References
- 1.Glass C K. Endocr Rev. 1994;15:391–407. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- 2.Giguere V. Endocr Rev. 1994;15:61–79. doi: 10.1210/edrv-15-1-61. [DOI] [PubMed] [Google Scholar]
- 3.Mangelsdorf D J, Evans R M. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 4.Lazar M A. Endocr Rev. 1993;14:184–193. doi: 10.1210/edrv-14-2-184. [DOI] [PubMed] [Google Scholar]
- 5.Leid M, Kastner P, Chambon P. Trends Biochem Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 6.Tjian R, Maniatis T. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 7.Roeder R G. Trends Biochem Sci. 1996;21:327–334. [PubMed] [Google Scholar]
- 8.Baniahmad A, Ha I, Reinberg D, Tsai S, Tsai M-J, O’Malley B W. Proc Natl Acad Sci USA. 1993;90:8832–8836. doi: 10.1073/pnas.90.19.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulman I G, Chakravarti D, Juguilon H, Romo A, Evans R M. Proc Natl Acad Sci USA. 1995;92:8288–8292. doi: 10.1073/pnas.92.18.8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horwitz K B, Jackson T A, Bain D L, Richer J K, Takimoto G S, Tung L. Mol Endocrinol. 1996;10:1167–1177. doi: 10.1210/mend.10.10.9121485. [DOI] [PubMed] [Google Scholar]
- 11.Cavailles V, Dauvois S, L’Horset F, Lopez G, Hoare S, Kushner P J, Parker M G. EMBO J. 1995;14:3741–3751. doi: 10.1002/j.1460-2075.1995.tb00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halachmi S, Marden E, Martin G, MacKay H, Abbondanza C, Brown M. Science. 1994;264:1455–1458. doi: 10.1126/science.8197458. [DOI] [PubMed] [Google Scholar]
- 13.Lee J W, Choi H-S, Gyuris J, Brent R, Moore D D. Nature (London) 1995;374:91–94. [Google Scholar]
- 14.Seol W, Choi H-S, Moore D D. Mol Endocrinol. 1995;9:72–85. doi: 10.1210/mend.9.1.7760852. [DOI] [PubMed] [Google Scholar]
- 15.Le Douarin B, Zechel C, Garnier J-M, Lutz Y, Tora L, Pierrat B, Heery D, Gronemeyer H, Chambon P, Losson R. EMBO J. 1995;14:2020–2033. doi: 10.1002/j.1460-2075.1995.tb07194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onate S A, Tsai S Y, Tsai M-J, O’Malley B W. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 17.Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin S-C, Heyman R A, Rose D W, Glass C K, Rosenfeld M G. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 18.Hong H, Kohli K, Trivedi A, Johnson D L, Stallcup M R. Proc Natl Acad Sci USA. 1996;93:4948–4952. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voegel J J, Heine M J S, Zechel C, Chambon P, Gronemeyer H. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- 20.Chakravarti D, LaMorte V J, Nelson M C, Nakajima T, Schulman I G, Juguilon H, Montminy M, Evans R M. Nature (London) 1996;383:99–103. doi: 10.1038/383099a0. [DOI] [PubMed] [Google Scholar]
- 21.Yeh S, Chang C. Proc Natl Acad Sci USA. 1996;93:5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horlein A J, Naar A M, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryen A, Kamei Y, Soderstrom M, Glass C K, Rosenfeld M G. Nature (London) 1995;377:397–403. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 23.Chen J D, Evans R M. Nature (London) 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 24.Sande S, Privalsky M. Mol Endocrinol. 1996;7:813–825. doi: 10.1210/mend.10.7.8813722. [DOI] [PubMed] [Google Scholar]
- 25.Lu C, Yang Y-F, Ohashi H, Walfish P G. Biochem Biophys Res Commun. 1990;171:138–142. doi: 10.1016/0006-291x(90)91367-2. [DOI] [PubMed] [Google Scholar]
- 26.Ohashi H, Yang Y-F, Walfish P G. Biochem Biophys Res Commun. 1991;178:1167–1175. doi: 10.1016/0006-291x(91)91015-5. [DOI] [PubMed] [Google Scholar]
- 27.Butt T R, Walfish P G. Gene Expression. 1996;5:225–268. [PMC free article] [PubMed] [Google Scholar]
- 28.Walfish P G, Yang Y F, Yoganathan T, Chang L A, Butt T R. Gene Expression. 1996;6:169–184. [PMC free article] [PubMed] [Google Scholar]
- 29.Hall B L, Smit-McBride Z, Privalsky M L. Proc Natl Acad Sci USA. 1993;90:6929–6933. doi: 10.1073/pnas.90.15.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smit-McBride Z, Privalsky M L. Oncogene. 1992;8:1465–1475. [PubMed] [Google Scholar]
- 31.Lee J W, Moore D D, Heyman R A. Mol Endocrinol. 1994;8:1245–1252. doi: 10.1210/mend.8.9.7838157. [DOI] [PubMed] [Google Scholar]
- 32.Heery D M, Zacharewski T, Pierrat B, Gronemeyer H, Chambon P, Losson R. Proc Natl Acad Sci USA. 1993;90:4281–4285. doi: 10.1073/pnas.90.9.4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allegretto E A, McClurg M R, Lazarchik S B, Clemm D L, Kerner S A, Elgort M G, Boehm M F, White S K, Pike J W, Heyman R A. J Biol Chem. 1993;268:26625–26633. [PubMed] [Google Scholar]
- 34.Heery D M, Pierrat B, Gronemeyer H, Chambon P, Losson R. Nucleic Acids Res. 1994;22:726–731. doi: 10.1093/nar/22.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul S F, Gish W, Miller E W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 36.Fields S, Song O-K. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 37.Mak P, Fuernkranz H A, Ge R, Karathanasis S K. Gene. 1994;145:129–133. doi: 10.1016/0378-1119(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 38.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 39.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. pp. 352–355. [Google Scholar]
- 40.Petersen J M, Skalicky J J, Donaldson L W, McIntosh L P, Alber T, Graves B J. Science. 1995;269:1866–1869. doi: 10.1126/science.7569926. [DOI] [PubMed] [Google Scholar]
- 41.Rastinejad F, Perlman T, Evans R M, Sigler P B. Nature (London) 1995;375:203–211. doi: 10.1038/375203a0. [DOI] [PubMed] [Google Scholar]
- 42.Gronemeyer H, Moras D. Nature (London) 1995;375:190–191. doi: 10.1038/375190a0. [DOI] [PubMed] [Google Scholar]
- 43.Hong, H., Kohli, K., Garabedian, M. J. & Stallcup, M. R. (1997) Mol. Cell. Biol., in press. [DOI] [PMC free article] [PubMed]
- 44.Yao T P, Ku G, Zhou N, Scully R, Livingston D M. Proc Natl Acad Sci USA. 1996;93:10626–10631. doi: 10.1073/pnas.93.20.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]