

Abstract
We report the fast relaxation dynamics of “native” apomyoglobin (pH 5.3) following a 10-ns, laser-induced temperature jump. The structural dynamics are probed using time-resolved infrared spectroscopy. The infrared kinetics monitored within the amide I absorbance of the polypeptide backbone exhibit two distinct relaxation phases which have different spectral signatures and occur on very different time scales (ν = 1633 cm−1, τ = 48 ns; ν = 1650 cm−1, τ = 132 μs). We assign these two spectral components to discrete substructures in the protein: helical structure that is solvated (1633 cm−1) and native helix that is protected from solvation by interhelix tertiary interactions (1650 cm−1). Folding rate coefficients inferred from the observed relaxations at 60°C are kf(solvated) = (7 to 20) × 106 s−1 and kf(native) = 3.6 × 103 s−1, respectively. The faster rate is interpreted as the intrinsic rate of solvated helix formation, whereas the slower rate is interpreted as the rate of formation of tertiary contacts that determine a native helix. Thus, at 60°C helix formation precedes the formation of tertiary structure by over three orders of magnitude in this protein. Furthermore, the distinct thermodynamics and kinetics observed for the apomyoglobin substructures suggest that they fold independently, or quasi-independently. The observation of inhomogeneous folding for apomyoglobin is remarkable, given the relatively small size and structural simplicity of this protein.
The mechanisms by which a protein searches vast conformational space to attain its native fold in reasonable times and by which the three-dimensional structure is encoded in the primary sequence have not been resolved experimentally. In particular, the critical early-time structural dynamics which carry a protein along the pathway(s) from extended, disordered conformations to a compact fold are poorly characterized. A major impediment has been the conventional solution-mixing approach to initiation of a folding reaction, which imposes a short-time observation limit of greater than 1 ms. Initiation of folding by solution mixing generally produces a temporally unresolved (i.e., within the “dead time” of the mixing) burst phase, such that by NMR, CD, and fluorescence criteria a compact structure having a substantial fraction of the native secondary structural content is already present before the observation of kinetics can begin (refs. 1 and 2 and references therein). Furthermore, high-resolution structural techniques generally lack the time resolution necessary for observation of fast folding dynamics. In the few extant studies of fast folding events, the observation methods used have generally lacked structural specificity (3–6). The obvious importance of early events (1, 2), particularly secondary structure formation, hydrophobic collapse, the development of early tertiary contacts, and the possible formation of intermediates, clearly illustrates the need for fast experimental probes which are sensitive to the structural progress of folding. The complexity of the possible folding pathways (1, 2, 7, 8), the ways in which fast structure formation may guide subsequent events, and the possibility that different substructures within a protein may fold on different time scales can be ascertained only by fast, structure-specific measurements. Here we report such measurements for apomyoglobin (apoMb), an archetype for the folding of small, single-domain, globular proteins.
apoMb is prepared by removing the heme group from myoglobin. The holoprotein contains eight strands of mostly α-helical segments, labeled A–H. Near neutral pH, apoMb adopts a structure that is native-like according to the available NMR, CD, and calorimetric evidence. Thus “native” apoMb has a compact hydrophobic core, consisting of at least the very stable A, G, and H helices, with roughly the same secondary structure content and tertiary fold as the holoprotein (9, 10, 11, 12). The major differences appear to occur in the C, D, and F helices, which are considerably more disordered in the apoprotein. Under conditions of low pH, apoMb forms an equilibrium “molten globule.” Extensive studies of the kinetics of apoMb folding using conventional stopped-flow initiation methods, hydrogen–deuterium pulsed labeling, and small-angle x-ray scattering have revealed the rapid (submillisecond) development of a molten globule intermediate on the kinetic folding pathway (13, 14).
Our approach is to probe the structural evolution of a folding reaction using time-resolved infrared (IR) spectroscopy, which has well-established temporal resolution and structural specificity (15, 16). The reactions are initiated using a laser-induced temperature jump (T-jump) of typically 15°C in 10 ns (17, 18). We have applied this approach to a solution of native apoMb (pH 5.3) that is in equilibrium between folded and unfolded states. The T-jump rapidly changes the equilibrium conditions, in the direction toward the unfolded state; both folding and unfolding kinetics contribute to the observed kinetics of achieving the new equilibrium point (19).
We have determined the dynamics of secondary and tertiary structure formation for the high-temperature thermal denaturation transition of native apoMb. Time-resolved IR measurements reveal that segments of the protein form helical secondary structure from disordered coil in about 100 ns, while tertiary contacts are made much more slowly, on the 100-μs time scale. We have also found that, despite the relative structural simplicity of this protein, apomyoglobin consists of substructures which have different thermodynamic stabilities and kinetic labilities and which may fold independently. These observations have profound implications for the mechanism of protein folding.
METHODS
ApoMb was prepared from horse heart myoglobin (Sigma) by 2-butanone extraction (20). The sample was filtered on a Sephadex G75 column. Monomeric apoMb fractions were extensively dialyzed against deionized water, then lyophilized. The sample was completely homogeneous on sodium dodecyl sulfate/polyacrylamide gel electrophoresis. The 280-nm absorption (A1% = 0.84 cm−1) was used to determine concentration (21). The protein tended to aggregate during equilibrium measurements at temperatures higher than 55°C. No aggregation was observed during kinetics measurements, however, even with T-jumps to temperatures higher than 55°C, suggesting that the aggregation time scale is significantly longer than the duration of the T-jump, which relaxes to ambient in a few milliseconds.
The general approach to study fast folding dynamics developed by our group (17, 18) uses a short-pulsed, laser-induced T-jump to impulsively initiate a folding reaction on a nanosecond time scale, and the use of a structure-specific probe, IR spectroscopy, to follow the evolution of the secondary and tertiary structures that define the folding reaction. The time-resolved IR methods we have developed for this purpose have been described previously (18). Briefly, the T-jump is achieved using a pump laser pulse (1.9 μm, 3.0 mJ) generated from the difference frequency of a Nd:ytrium aluminum garnet (YAG) pumped dye laser and the Nd:YAG fundamental in a LiNbO3 crystal. The pump is focused to a 200 μm spot in the sample. About 10% of the pump beam is absorbed in the 100-μm sample pathlength, resulting in a T-jump of 20°C. A lead-salt IR diode laser serves as the mid-IR probe. The transient absorbtion of the probe beam in the sample is probed using a fast HgCdTe detector (10-ns rise time). Transients are digitized in a variable timebase digitizer and signal averaged (5,000 shots at 10 Hz). Equilibrium Fourier-transform IR (FTIR) spectra are obtained using a Bio-Rad model FTS 60A FTIR spectrometer and a thermostatted IR cell with CaF2 windows.
RESULTS
The amide I region of the IR spectrum used to probe the thermal unfolding of apoMb is shown in Fig. 1. The amide I band, arising primarily from the C=O stretching vibrations of the polypeptide backbone carbonyls, is an established indicator of secondary and tertiary structural changes because of its sensitivity to hydrogen bonding and to structure-dependent vibrational coupling. A broad amide I absorption envelope is observed (Fig. 1A) that contains unresolved components due to peptide groups in helical structures, turns, and disordered coil. Previous studies have shown that in D2O, disordered coil structures generally exhibit amide I frequencies between 1665 and 1675 cm−1 (22–24), while for α-helix the characteristic frequency range is 1650–1655 cm−1 (for native helix protected from solvent by the tertiary fold; ref. 23). More recent studies have shown that, in the absence of protection by tertiary structure, a helix solvated in D2O exhibits amide I frequencies that are considerably lower, between 1630 and 1645 cm−1 (the “solvated” helix; refs. 18, 25, and 26). Resolution enhancement of the equilibrium FTIR spectra by a second-derivative analysis (Fig. 1B) reveals three major subcomponents, at 1675, 1650, and 1633 cm−1. The peak at 1650 cm−1, the characteristic native helix frequency, dominates the second-derivative spectrum as expected for the predominately α-helical apoMb. A significant contribution at 1630 cm−1 indicates that there is also some solvated helical structure in the native form of apoMb. This interpretation is supported by x-ray and light-scattering results which indicate that the structure of native apoMb is considerably more extended (resulting in greater penetration of water) compared with the holoprotein (27). In addition, NMR observations of small or nonexistent hydrogen/deuterium exchange protection factors for the C, D, E, and F helices of the native apoMb structure indicate greater flexibility and solvent exposure for these helices (28).
Figure 1.

(A) FTIR absorbance spectra of apoMb in D2O (2H2O) (4.2 mg/ml in 0.01 M NaCl, uncorrected pH = 5.3) in the region of the amide I band, as a function of temperature from 32° to 55°C. The temperature-dependent D2O background has been subtracted from each FTIR spectrum. (B) Second-derivative spectrum at 40°C; major components of amide I are observed at 1675, 1650, and 1633 cm−1. (C) Difference spectra, generated by subtracting the spectrum at 32.8°C from the spectrum at each temperature. The primary difference peaks occur at 1673 and 1648 cm−1.
The equilibrium changes of the IR spectrum produced by thermal denaturation are not dramatic in the raw spectra (Fig. 1A) due to the strong overlap of the subcomponents. In contrast, the difference spectra in Fig. 1C show that, as temperature is increased, helical structure is lost (the trough at 1648 cm−1) and disordered coil appears (the peak at 1673 cm−1). These observations serve as a guide for the kinetics experiments. The equilibrium changes in the amide I absorbance are plotted versus temperature in Fig. 2. The IR melting curve of the native helix (loss of signal at 1650 cm−1 and gain of signal at 1675 cm−1) is cooperative, with a melting temperature near 59°C, which closely resembles the melting curve derived from CD measurements (29). The endpoint of this melting transition (determined from the fit shown in Fig. 2) corresponds to only about half of the total 1650-cm−1 intensity present; half of the intensity at 1650 cm−1 remains even at the highest temperatures. In contrast, the thermal denaturation of the solvated helical component (loss of signal at 1633 cm−1 and gain of signal at 1675 cm−1) does not show a sharp transition with temperature, instead exhibiting a continuous loss of structure over the entire temperature range studied. This noncooperative melting is similar to that observed previously for short peptides (18).
Figure 2.

Thermal denaturation of apoMb, monitored at 1650 (□, native helix), 1629 (+, solvated helix), and 1676 (○, disordered coil) cm−1. The solid lines are fits obtained as follows. The 1650-cm−1 fit is based on a two-state model, which gives a melting temperature for native helix of 59°C, very close to the value observed in the far-UV CD (220 nm) thermal denaturation curve (29). The 1629-cm−1 fit is linear, showing the monotonic change at this IR frequency. The 1676-cm−1 fit is a convolution of the sigmoidal and linear functions used for the two helix IR frequencies, and it demonstrates that the loss of each helix type is correlated with a concomitant gain of disordered structure.
The equilibrium constant for folding, Keq, was derived from the FTIR data by fitting the absorbance as function of temperature to the function A = C0{1 + (C1 − T)/[(C1 − T)2 + C2T]1/2} + C3, where A is the absorbance at 1650 cm−1, T is the temperature, and Cn are constants, to determine the equilibrium endpoints. For the native helix component, Keq = 0.75 at 60°C (the temperature of the kinetics observations). The same analysis is not possible for the equilibrium between the solvated helix and its unfolded state, because it is difficult to estimate the endpoints of the reaction from the nearly linear variation in the 1633-cm−1 intensity with temperature. A lower limit of Keq = 0.5 can be obtained, however, by assuming that all of the 1633-cm−1 intensity is converted to 1675 cm−1 at high temperature.
The equilibrium FTIR data show that the melting of native apoMb occurs as two independent processes, corresponding to the native and solvated helical portions of the protein. The melting of the solvated helix and that of the native helix follow entirely different curves; moreover, there appears to be no influence of the melting of the native helix over its narrow temperature range on the melting of the solvated helix. The most plausible explanation for this behavior is that apoMb contains two distinct, independent regions primarily composed of solvated and native helix, respectively. Accordingly, it seems reasonable that the kinetics responses of the solvated and native helices correspond to these different substructures within the protein.
The unfolding of apoMb was initiated by a laser-induced T-jump from 45° to 60°C and the kinetics were followed in the amide I region. Fig. 3 shows the three key vibrational frequencies which correspond to changes in the solvated helix (1632 cm−1), the native helix (1655 cm−1) and the disordered structure (1664 cm−1). Two clearly resolved kinetics phases, separated by several orders of magnitude in time, are evident at each of these frequencies. The transient at 1632 cm−1 is dominated (>96%) by a single fast kinetics phase with a relaxation time of 48 ns. The transient at 1655 cm−1 has a small (17%) contribution from the fast phase, followed by the predominant (83%) slower phase with a relaxation time of 132 μs. The small contribution of the fast phase kinetics at 1655 cm−1 arises from spectral overlap of the 1632 and 1655 cm−1 bands. The transient at 1664 cm−1 has contributions from the fast (55%) and slow (45%) phases. Transient absorbance spectra at 10 μs and 500 μs after the T-jump are shown in Fig. 4. The early time spectrum is dominated by a bleach at 1632 cm−1, indicating loss of solvated helix. The longer time spectrum is essentially identical to the equilibrium FTIR difference spectrum, with the major bleach centered at the native helix frequency of 1650 cm−1.
Figure 3.

Kinetic IR response for a T-jump from 45° to 60°C at 1664 cm−1 (near the peak of the positive absorbance in the static difference FTIR spectrum), 1655 cm−1 (near the peak of the bleach in the difference spectrum), and 1632 cm−1. The background D2O response has been subtracted from these data. The region from 2 × 10−7 to 8 × 10−6 s is obscured by a photoacoustic artifact and is not shown. Two clear kinetic phases are present, one on the nanosecond time scale and the other on the microsecond time scale. Single-exponential fits to the data, shown by solid lines, yielded 48 ns and 120 μs for the two lifetimes. All three signals begin to show recovery on the millisecond time scale. This is because heat flows out of the laser interaction volume with a ≈7-ms time constant, returning the sample to its initial temperature (45°C).
Figure 4.

FTIR difference spectrum (55.1°C − 45.9°C) for native apoMb, uncorrected pH = 5.3 (solid line) compared with the time-resolved IR spectra at 10 μs (○) and 500 μs (▪) after reaction initiation.
The transients assigned to the loss of solvated and native helix are well separated in time, and each is dominated by a single relaxation. In addition, the transient assigned to the formation of disordered state has two clearly resolved kinetic phases corresponding exactly to the respective helix relaxations. It is likely that the “disordered state” is composed of a heterogeneous mixture of structures. The gradual melting behavior of the solvated helix (relatively low enthalpy of stability) and the observed fast kinetics (relatively low energy transition barrier) imply that this state also exists as a heterogeneous population. The observed single exponential kinetics therefore indicate that either the heterogeneous population of states is in fast equilibrium or each state has a similar barrier to melting. Therefore, the results for each helix type can be modeled separately as single two-state kinetics relaxations between the respective folded and unfolded structures:
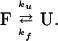 |
This approximation gives kobs = 1/τobs = kf + ku, where τobs is the observed relaxation time, kf is the folding rate coefficient, ku is the unfolding rate coefficient, and Keq = kf/ku. The observed rate constant for the slow phase is kobs = 8.4 ± 1.0 × 103 s−1, and Keq = 0.75, yielding kf(native) = 3.6 × 103 s−1 and ku(native) = 4.8 × 103 s−1 (or τf(native) = 280 μs, τu(native) = 210 μs). The counterintuitive nature of reversible first-order kinetics is evident. Although the process that we are measuring is definitely unfolding, the rate that we measure, because of the position of the equilibrium, has a substantial contribution from folding.
The resolution of the folding and unfolding rate constants for the solvated helix is imprecise, since Keq for this helix type is imprecise. The lower limit of Keq = 0.5 yields a range of kf(solvated) = 7 × 106 to 2 × 107 s−1 (τf(solvated) = 48 ns to 200 ns). This range in folding time is in accord with our previous measurements of the kinetics of the helix–coil transition in a solvated helical peptide composed mostly of alanine [Suc-A5(A3RA)3A-NH2], wherein the folding lifetime is 180 ns at 27°C (18). Essentially the same rate has been observed in two additional systems, the S-peptide of ribonuclease A and the solvated helix component of the molten globule form of apoMb (unpublished results). The close correspondence in rate between the folding kinetics of solvated peptides and the solvated helices of apoMb suggest that this is a typical, intrinsic rate of folding of solvated helix. Furthermore, this rate appears insensitive to temperature, at least over the range from 27°C to 60°C.
DISCUSSION
The IR kinetics data provide clear evidence for two different processes in the folding of native apoMb. The first is very fast helix formation, which we assign to the folding of solvated helix (τf(solvated) = 48–200 ns) on the basis of two lines of evidence: (i) the unusually low amide I frequency (18, 25, 26) of this kinetic phase; and (ii) the similarity of its equilibrium and kinetic properties to those of a solvated model peptide (18). The second process is about three orders of magnitude slower (τobs = 132 μs), from which we derive a folding lifetime, τf = 280 μs. We assign this phase to the formation of native helices, that is, helices with significant tertiary interactions which exclude water, on the basis of the amide I frequency of 1650 cm−1 (23). In addition, our kinetics measurements at long times quantitatively account for the equilibrium IR absorbance changes over the same temperature range. Therefore, the 132-μs relaxation is by the IR criterion the final step in the melting of the protein, and the 280-μs lifetime derived therefrom is the slow step in its folding. We suggest that the rate-limiting step for this process is the formation of the tertiary contacts, which is much slower than the intrinsic helix–coil transition rate. This implies that the early dynamics of the folding of globular, helical proteins involve the formation of secondary structure, while the formation of tertiary structure is a much later event.
The equilibrium and kinetics results together support a model of apoMb containing three substructures which are to a great extent thermodynamically and kinetically independent: (i) labile solvated helices, having few if any stable tertiary contacts, which are responsible for the 48-ns relaxation; (ii) a compact substructure consisting of native helices which is responsible for the 132-μs relaxation; and (iii) a compact substructure consisting of native helices which do not melt under our conditions (i.e., the 1650-cm−1 component that does not change with temperature). Recent fluorescence measurements in our laboratories suggest that this nonmelting substructure comprises the AGH core. These observations of independent regions of the protein are consistent with previous CD and NMR measurements (11, 28, 30). Hydrogen–deuterium exchange NMR experiments show that the helices of the AGH core are most highly protected against exchange, followed by the B and E helices; finally, the C, D, and F helices and the CD loop are very labile to exchange and accessible to solvent. Since much of the AGH core does not melt in our experiments, the B and E helices are then the most likely candidates for the native helix transition observed at 1650 cm−1 (132-μs relaxation), whereas the C, D, and F helices that surround the heme in the holoprotein are all plausible candidates for the labile substructure responsible for the solvated helix transition observed at 1633 cm−1 (48-ns relaxation). Insertion of the heme into this labile structure apparently leads to the formation of tertiary contacts that lock down the solvated helix region of the apoprotein into a more stable (native) helical form. This hypothesis can be tested by IR measurements, given specific isotope labeling of these target helices.
It has been hypothesized for some time, and inferred from recent measurements, that short-range forces dominate the inception of folding, leading to the initial formation of secondary structure (1, 7). In this view such conformations then serve as nucleation structures about which appropriate tertiary contacts are made, with formation of secondary structure guiding subsequent events. Hence the folding process occurs in a reasonable time, because local interactions dominate the process at early times and facilitate the process at longer times. In support of this view, it is found that many native-sequence peptide fragments adopt the correct or nearly correct secondary structure in solution (refs. 31–33 and references therein). This model of protein folding has remained largely hypothetical, however, because how the formation of tertiary structure depends on the formation of secondary structure is generally unknown.
The results reported here provide one of the first experimental glimpses of the structural details of the initial steps in protein folding. At 60°C, the rate of helical structure formation is some three orders of magnitude faster than tertiary contact formation, which means that even a trace concentration of helix is kinetically competent to act as a nucleation site for the much slower formation of native tertiary contacts. This difference is probably even greater at room temperature, because the rate of helix formation appears to be temperature independent, whereas the rate of tertiary structure formation in apoMb is known to be much slower (kf ≈ 1 to 0.1 s−1 for the B, C, D, and E helices) at 5°C (13). Since the rate of secondary structure formation relative to other processes in the folding reaction is so fast, steady-state formation of solvated helix, even if it is thermodynamically unfavorable, can carry the rate of the overall reaction. Furthermore, the sensitivity of IR spectroscopy to secondary and tertiary structure allows us to conclude that protein substructures in different thermodynamic states are encompassed within a single protein molecule. These substructures appear at different times during the folding dynamics; thus folding can be a heterogeneous process involving formation of native-like subdomains as intermediates on the folding pathway (34). This observation is quite significant for small single-domain proteins such as apoMb, which have generally been thought to fold as a single cooperative unit. The formation of substructures could be instrumental in guiding the folding process.
Acknowledgments
We thank M. Jamin and R. L. Baldwin for the opportunity to read their paper prior its publication. This work was supported by the National Institutes of Health, Grants GM53640 (to R.B.D.) and GM45807 (to W.H.W.), and the National Science Foundation, Grant MCB-9417892 (to R.H.C.).
ABBREVIATIONS
- apoMb
apomyoglobin
- T-jump
temperature jump
- FTIR
Fourier-transform IR
References
- 1.Kim P S, Baldwin R L. Annu Rev Biochem. 1990;59:631–660. doi: 10.1146/annurev.bi.59.070190.003215. [DOI] [PubMed] [Google Scholar]
- 2.Evans P A, Radford S E. Curr Opin Struct Biol. 1994;4:100–106. [Google Scholar]
- 3.Ballew R M, Sabelko J, Gruebele M. Proc Natl Acad Sci USA. 1996;93:5759–5764. doi: 10.1073/pnas.93.12.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones C M, Henry E R, Hu Y, Chan C-K, Luck S D, Bhuyan A, Roder H, Hofrichter J, Eaton W A. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pascher T, Chesick J P, Winkler J R, Gray H B. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 6.Nolting B, Golbik R, Fersht A R. Proc Natl Acad Sci USA. 1995;92:10668–10672. doi: 10.1073/pnas.92.23.10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid F. In: Protein Folding. Creighton T E, editor. New York: Freeman; 1992. pp. 197–241. [Google Scholar]
- 8.Ptitsyn O B. Curr Opin Struct Biol. 1995;5:74–78. doi: 10.1016/0959-440x(95)80011-o. [DOI] [PubMed] [Google Scholar]
- 9.Barrik D, Baldwin R L. Protein Sci. 1993;2:869–876. doi: 10.1002/pro.5560020601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cocco M J, Lecomte J T J. Protein Sci. 1994;3:267–281. doi: 10.1002/pro.5560030211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson R S, Walsh K A. Protein Sci. 1994;3:2411–2418. doi: 10.1002/pro.5560031224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin L, Pinker R J, Forde K, Rose G D, Kallenbach N R. Nat Struct Biol. 1994;1:447–452. doi: 10.1038/nsb0794-447. [DOI] [PubMed] [Google Scholar]
- 13.Jennings P A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 14.Eliezer D, Jennings P A, Wright P E, Doniach S, Hodgson K O, Tsuruta H. Science. 1995;270:487–488. doi: 10.1126/science.270.5235.487. [DOI] [PubMed] [Google Scholar]
- 15.Phillips C M, Mizutani Y, Hochstrasser R M. Proc Nat Acad Sci USA. 1995;92:7292–7296. doi: 10.1073/pnas.92.16.7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoutland P O, Dyer R B, Woodruff W H. Science. 1992;257:1913–1917. doi: 10.1126/science.1329200. [DOI] [PubMed] [Google Scholar]
- 17.Woodruff W H, Dyer R B, Callender R H, Paige K, Causgrove T. Biophys J. 1994;66:A397. (abstr.). [Google Scholar]
- 18.Williams S, Causgrove T P, Gilmanshin R, Fang K S, Callender R H, Woodruff W H, Dyer R B. Biochemistry. 1996;35:691–697. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- 19.Cantor C R, Schimmel P R. Biophysical Chemistry. Vol. 3. San Francisco: Freeman; 1980. pp. 907–916. [Google Scholar]
- 20.Teale F W J. Biochim Biophys Acta. 1959;35:543. doi: 10.1016/0006-3002(59)90407-x. [DOI] [PubMed] [Google Scholar]
- 21.Crumpton M J, Polson A. J Mol Biol. 1965;11:722–729. doi: 10.1016/s0022-2836(65)80030-4. [DOI] [PubMed] [Google Scholar]
- 22.Chirgadze Yu N, Shestopalov B V, Venyaminov S Yu. Biopolymers. 1973;12:1337–1351. doi: 10.1002/bip.1973.360120610. [DOI] [PubMed] [Google Scholar]
- 23.Susi H, Byler D M. Methods Enzymol. 1986;130:290–311. doi: 10.1016/0076-6879(86)30015-6. [DOI] [PubMed] [Google Scholar]
- 24.Gilmanshin R, Van Beek J, Callender R H. J Phys Chem. 1996;100:16754–16760. [Google Scholar]
- 25.Haris P I, Chapman D. Biopolymers. 1995;37:251–263. doi: 10.1002/bip.360370404. [DOI] [PubMed] [Google Scholar]
- 26.Reisdorf W C, Jr, Krimm S. Biochemistry. 1996;35:1383–1386. doi: 10.1021/bi951589v. [DOI] [PubMed] [Google Scholar]
- 27.Gast K, Damaschun H, Misselwitz R, Müller-Frohne M, Zirwer D, Damaschun G. Eur Biophys J. 1994;23:297–305. doi: 10.1007/BF00213579. [DOI] [PubMed] [Google Scholar]
- 28.Hughson F M, Wright P E, Baldwin R L. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 29.Nishii I, Kataoka M, Tokunaga F, Goto Y. Biochemistry. 1994;33:4903–4909. doi: 10.1021/bi00182a019. [DOI] [PubMed] [Google Scholar]
- 30.De Sanctis G, Ascoli F, Brunori M. Proc Natl Acad Sci USA. 1994;91:11507–11511. doi: 10.1073/pnas.91.24.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waltho J P, Feher V A, Merutka G, Dyson H J, Wright P E. Biochemistry. 1993;32:6337–6347. doi: 10.1021/bi00076a006. [DOI] [PubMed] [Google Scholar]
- 32.Kippen A D, Sancho J, Fersht A R. Biochemistry. 1994;33:3778–3786. doi: 10.1021/bi00178a039. [DOI] [PubMed] [Google Scholar]
- 33.Baldwin R L. Biophys Chem. 1995;55:127–135. doi: 10.1016/0301-4622(94)00146-b. [DOI] [PubMed] [Google Scholar]
- 34.Jamin M, Baldwin R L. Nat Struct Biol. 1996;3:613–618. doi: 10.1038/nsb0796-613. [DOI] [PubMed] [Google Scholar]