

Abstract
Important among the responses of endothelial cells to flow stimuli are cytosolic-free calcium transients. These transients are mediated by several factors, including blood-borne agonists, extracellular calcium, and fluid-imposed shear forces. A mathematical model has been developed describing the recognition and transduction of shear stress to the second messenger cytosolic calcium. Shear stress modulates the calcium response via at least two modalities. First, mass transfer of agonist to the cell surface is enhanced by perfusion and is thus related to shear stress. Second, the permeability of the cell membrane to extracellular calcium increases upon exposure to shear stress. A mass balance for agonist in the perfusate is coupled to a previously published calcium dynamics model. Computations indicate a flow region where the transient moves from transport limited to kinetically limited. Parametric studies indicate distinct contributions to the time course by each step in the process. These steps include the time to develop the concentration boundary layer of agonist, receptor activation, and the mobilization of calcium from intracellular stores. Exogenous calcium is presumed to enter the cell via shear stress-gated ion channels. The model predicts a sigmoidal dependence of calcium influx upon shear stress. The peak value of the transient is determined largely by the agonist pathway, whereas the plateau level is governed by calcium influx. The model predicts the modulation of the calcium transient in the physiologically relevant range of flow and the associated shear stress. This implies that hemodynamics is important in regulating endothelial biology.
Keywords: calcium influx, agonist, mechanotransduction
Intracellular transients in Ca2+ are a typical cellular response to many different stimuli, including shear stress (1). Flow enhances the delivery of blood-borne agonists to the cell surface (2). These agonists in turn mediate the release of calcium from intracellular stores into the cytosol. Shear stress increases the permeability of the cell membrane to extracellular calcium, thereby increasing the cytosolic concentration (3, 4). A third transduction mechanism is a direct shear stress effect upon the generation of inositol trisphosphate, which could mediate the release of calcium from intracellular stores (5). This last would operate via a putative mechanoreceptor behaving as a biological “strain gauge.” It is likely that all three pathways act in concert to elicit the calcium transient.
The response of cytosolic calcium to a fluid shear stress has been illuminated to a large degree by experimental work. It is therefore natural to propose mathematical models that account for this behavior. Several models have been proposed that relate cytosolic-free calcium dynamics to stimulation by agonists (6, 7). The concentration profiles of agonists in flow chambers containing cell monolayers have been described (8). There has been at least one report of a mathematical model linking calcium transients to mechanical stimulation (9). There have been no reports of a model encompassing the signal recognition and transduction cascade from flow to cytosolic calcium.
This work presents a model that integrates previous models on agonist stimulation with a new model of shear stress-facilitated calcium ion transport into cells. It builds upon the earlier lumped parameter model of intracellular calcium dynamics by Wiesner et al. (6) by adding flow effects. In this earlier model, a balance on cytosolic-free calcium ion consists of four fluxes.
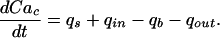 |
1 |
In this expression, Cac is the cytosolic-free calcium concentration, qs is the net flux released from intracellular stores into the cytosol in response to stimulation by agonists, qin is the rate of influx of extracellular calcium into the cytosol, qb is the rate of buffering of calcium ion by soluble cytosolic proteins, and qout is the rate of extrusion and exchange of calcium to the extracellular space. The complete calcium dynamics model, with its parameter values and detailed equations, is given in ref. 6.
An illustration of the flow effects in a capillary tube flow chamber is given in Fig. 1. A confluent monolayer of endothelial cells has been cultured on the bottom surface. The chamber is perfused with media in steady laminar flow with known concentrations of the agonist thrombin and extracellular calcium. Agonist is transported to the cell surface by coupled convection and diffusion. Flow of the perfusate exerts a viscous shear force upon the monolayer. The effect of flow upon mobilization of calcium from intracellular stores is described through its influence on the surface concentration of agonist, cs. This is an effect of shear rate. Shear stress itself affects extracellular calcium influx, qin. This is described via a model of mechanosensitive ion channels. Direct mechanical effects triggering the release of calcium from intracellular stores are not included.
Figure 1.

Effects of flow upon an endothelial cell monolayer in a square capillary tube. Cells are cultured to confluence on the bottom surface, whereas the upper and lateral surfaces are cell free. The tube is perfused with media in laminar flow containing known quantities of thrombin and calcium. Knowledge of the perfusion rate and tube geometry permits calculation of the shear stress and mass transfer of agonist to the monolayer.
Mass Transfer of Agonist in the Capillary Flow Chamber
The coupled convection and diffusion of agonist to the endothelium may be described by a species balance on thrombin in the perfusate.
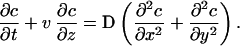 |
2 |
In this equation, v is the z-component of velocity, t is time, c is the local concentration of agonist, and D is the diffusivity.
The species balance (Eq. 2) is subject to the conditions that no agonist is present initially (c[x, y, z, 0] = 0), and that the inlet agonist concentration is fixed (c[x, y, 0, t] = co). Two other boundary conditions are required. There is no agonist consumption at cell free surfaces.
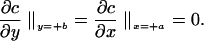 |
3 |
In the experiment, the cells cover only the bottom surface at y = −b. At this surface, the agonist binds to cell surface receptors. The rate of binding depends upon the surface concentration of agonist. Therefore, the consumption of agonist at y = −b is
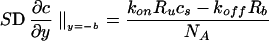 |
4 |
Here Rb is the number of bound receptors, Ru is the number of unbound receptors, and cs is the concentration of agonist at the cell surface. kon, koff, and kd are on, off, and degradation rate constants, respectively. NA is Avogadro’s number, and the quantity S is the en face area of the cell.
The steady-state velocity profile in a rectangular duct undergoing a step increase in pressure gradient is established almost immediately (10). Therefore, an analytical steady-state expression for the profile is used (11). Differentiation of this profile yields the shear stress as a function of position and perfusion rate.
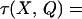 |
5 |
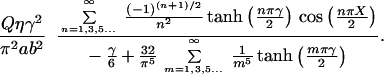 |
In this equation, the dimensionless coordinates X = x/a and Y = y/b have been introduced. The quantity γ = b/a is the aspect ratio of the channel and Q is the perfusion rate. τ is the shear stress and η is viscosity of the perfusate.
Using the bulk agonist concentration, c0, shear stress (τ), and flow chamber geometry as inputs, the species continuity balance is solved to obtain the surface concentration of agonist, cs, which in turn drives the activation of cell surface receptors.
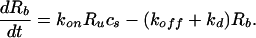 |
6 |
The number of bound receptors governs the generation of inositol trisphosphate, which in turn drives the release of calcium from intracellular stores, qs. Thus, the calcium transient is linked dynamically to the applied shear stress, τ.
Influx Pathway
Shear stress-activated calcium channels have been identified in human umbilical vein endothelial cells (HUVECs) (3) and distal renal tubule cells (4). Receptor-operated channels are also expressed in endothelial cells, and are opened by the binding of extracellular ligands such as thrombin. The increase in calcium influx due to receptor-operated channels (2.53 μM/sec, ref. 12) is quantitatively smaller than that due to shear stress (138 μM/sec, ref. 3). Therefore, the transduction mechanism between agonists and calcium influx is not included in this model. Calcium-activated calcium channels and voltage-gated ion channels are not expressed in cultured endothelial cells (13, 14).
Passive influx of calcium ions has also been established in endothelial cells (12). Shear stress augments this passive leak by inducing local transient defects in the structure of the plasma membrane. The increase in passive calcium transport as a function of shear stress has been measured in liposomes (15). The increase at a shear stress of 3.6 N/m2 is five orders of magnitude less than the basal passive leak of calcium into endothelial cells. Therefore, increased influx of calcium due to shear stress-induced defects in membrane structure is neglected in this study.
The steady-state flux of an ion across the membrane is described by the Goldman–Hodgkin–Katz equation (16), which depends in part upon the membrane permeability coefficient P. In this model, the value of the permeability coefficient is reflected by the fraction of ion channels that are in the open state. From Kirchhoff’s law for parallel conductors and the relationship between the membrane permeability coefficient and membrane conductance (16), it can be shown that the permeability is linearly related to the open channel fraction fo and the permeability of the membrane when all channels are open, Pmax: i.e., P = foPmax.
The application of shear stress induces a uniaxial tension gradient in the membrane and deforms it. Part of the applied load is borne by the membrane and part is borne by underlying cellular structures. The fraction borne by submembranous structures is denoted by ɛ. The deformation of the membrane imparts a strain energy density that depends upon the applied shear stress W(τ). The fraction of open channels has a Boltzmann dependence upon the level of gating energy in the membrane (17). The gating energy in turn can be related to the strain energy density in the membrane. A fraction of the elastic strain energy stored within the membrane is considered to gate the shear stress-activated channel. This fraction is designated as fe. Thus, the fraction of channels in the open state dependsupon the strain energy density as indicated in the following equation:
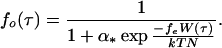 |
7 |
Here N is area channel density, k is the Boltzmann constant, T is the absolute temperature, and α is a measure of the probability that a channel is in the open state in the no-load case.
The strain energy density in the membrane is now related to the applied shear stress. The equations of mechanical equilibrium are used in conjunction with a constitutive relationship to develop this dependence. Employing a simplifying analysis, Fung and Liu (18) demonstrated that the equations of mechanical equilibrium for the endothelial cell membrane reduce to:
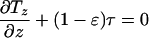 |
8 |
and
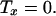 |
9 |
The tensions in the direction of the flow and orthogonal to flow in the plane of the membrane are denoted by Tz and Tx, respectively. In Eq. 8, the tension gradient in the direction of flow is related to the load fraction borne by the membrane, (1 − ɛ)τ. The quantity ɛ is the fraction of the stress borne by the interior of the cell, which is assumed to have a solid-like content. The tension in the direction perpendicular to flow in the plane of the membrane is taken as zero by virtue of the tension field hypothesis. This hypothesis states that the membrane cannot support compressive stresses in its own plane because its thickness is small compared with its length and width. Considering the stresses acting in the plane of the endothelial membrane, Fung and Liu argue that the stress orthogonal to the direction of flow is small and, thus, the simplifying assumption of the stress in the membrane being a uniaxial tension field in the direction of flow is valid. This same assumption is used here.
A well-known constitutive relation for cell membranes in pure shear deformation under uniaxial tension is given by Evans and Skalak (19).
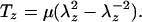 |
10 |
The quantity λz is the stretch ratio in the flow direction. The elastic constant μ is known as the membrane shear modulus.
Skalak et al. (20) give a strain energy density function, W, for a two-dimensional membrane. Integration of that expression and Eq. 8 yields the relationship between strain energy density and the applied shear stress.
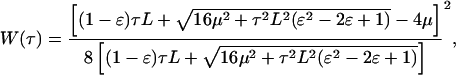 |
11 |
where L is the length of the cell in the flow direction.
The foregoing treatment assumes a steady state. In Schwarz et al. (3), the calcium current is not established immediately, but rather over a period of ≈70 sec. The dynamics are associated with changes in membrane potential, kinetics of ion channel activation, and viscoelasticity of the membrane. The membrane potential hyperpolarizes, i.e., becomes more negative in response to shear stress (21). Membrane potential depends upon transmembrane currents of potassium, sodium, and chloride ions. For the sake of simplicity, an empirical relationship between shear stress and membrane potential, Δφ(t, τ), is used herein. The temporal aspect of membrane permeability, P(t, τ), is also described empirically. The expressions for Δφ(t, τ) and P(t, τ) are given in ref. 22.
Estimation of Parameters in the Model
The model derived in the preceding section describes the effects of flow upon the surface concentration of agonist, cs, and on the influx of extracellular calcium, qin. The constants required to calculate the effect of flow on cs are readily available from handbooks and the experimental setup. The effect of flow upon calcium influx is not as well characterized.
The influx of calcium as a function of the applied fluid shear stress has been measured at only one level of shear stress, 1.0 N/m2, in endothelial cells (3). In the absence of experimental data, a prediction of the magnitude of the flow-induced calcium influx is offered. The load fraction, ɛ, is given a value of 0.90. This initial estimate is based upon the micropipette experiments of Sato et al. (23), in which the main load bearing component of the cell was found to be the cytoskeleton. With ɛ fixed a priori, values for fe, α, and Pmax are obtained by satisfaction of the following three criteria: qin(τ = 0) = 5.33 μM/sec, qin(τ = 3) = 10 μM/sec, qin,max = 23 μM/sec.
The value of qin(τ = 0) is that for passive leak of calcium into the cytosol (12). The value of qin(τ = 3) was selected to obtain a value for the plateau level of calcium corresponding to an agonist-free experiment (24). The maximum value of calcium influx, qin,max, was chosen because influxes above this value removed the experimentally observed biphasic character of the calcium response to agonists when used in Eq. 1. All parameter values are tabulated in Table 1, along with their respective sources. The equations of the model were coded in fortran and integrated numerically on a CRAY-EL computer.
Table 1.
Parameters for calcium model
| Quantity | Value | Units | Source |
|---|---|---|---|
| α | 3 | Dimensionless | Initial estimate |
| ɛ | 0.90 | Dimensionless | Initial estimate |
| η | 0.0012 | kg per m/sec | 25 |
| μ | 10−5 | N/m | 23 |
| Caex | 1.5 | mM | 26 |
| D | 8.75 × 10−11 | m2/sec | 27 |
| L | 35 | μm | 28 |
| Lc | 0.05 | m | Flow chamber dimension |
| N | 1 | μm−2 | 17 |
| Pmax | 2.815*10−8 | m/sec | Initial estimate |
| Ru + Rb | 4.4 × 104 | no. per cell | 29 |
| S | 611 | μm2 | 28 |
| a | 0.0005 | m | Flow chamber dimension |
| b | 0.0005 | m | Flow chamber dimension |
| fe | 0.013351 | Dimensionless | Initial estimate |
| koff | 142.8 | sec−1 | 6 |
| kon | 5316.7 | μM−1 sec−1 | 6 |
RESULTS
The calcium influx, qin, is illustrated as a function of shear stress in Fig. 2. The calculation assumes constant intracellular calcium concentration, extracellular calcium concentration, and transmembrane potential. A sigmoidal relationship is seen. For a value of ɛ = 0.90, there is a threshold value of ≈0.5 N/m2 below which the influx is negligible. In the range of shear stress from ≈0.5 N/m2 to ≈6 N/m2, the flux is shear-sensitive. For shear stresses higher than the shear-sensitive range, the influx is maximal. This corresponds to the situation in which all ion channels are open.
Figure 2.

Dependence of calcium influx upon shear stress as calculated from the Goldman–Hodgkin–Katz equation. A sigmoidal dependence of influx upon shear stress is predicted. The parameter is the load fraction, ɛ, borne by nonmembranous structures. As ɛ increases, less strain energy is available to gate mechanosensitive ion channels.
The effect of ɛ, the fraction of the load borne by the cytoplasm, is also shown in Fig. 2. The threshold value of shear stress and the shear-dependent range are altered. As more of the stress is borne by the membrane, the threshold level of influx decreases. Also, the shear-sensitive range narrows.
The response of cytosolic-free calcium is shown in Fig. 3. In this calculation, the perfusate is agonist free (cs = 0) and contains physiologic calcium (Caex = 1.5 mM). Note that no peak in the transient is observed when agonists are absent. The plateau level increases with increasing shear stress. This is due to enhanced transplasmalemmal calcium influx. Both the lack of a peak and the graded response to the level of shear stress in serum-free media was observed in bovine aortic endothelial cells (25).
Figure 3.

Response of calcium transient to agonist-free media (cs = 0, Caex = 1.5 mM. The calculation indicates a lack of the peak associated with agonist stimulation. Variation of flow rate indicates that the plateau level increases with increasing shear stress. These results are in qualitative agreement with experiment (25).
The agonist pathway is now examined by setting Caex to zero, and the bulk agonist concentration, co, to 1 unit per ml. When an endothelial cell monolayer is exposed to a step change in flow, a concentration profile is established over time by coupled convection and diffusion. The surface concentration of agonist reaches its steady-state value after more than 100 sec (simulation not shown).
In Fig. 4, the effects of mass transfer limitations and receptor dynamics are illustrated. The fastest curve shows the response of the calcium dynamics model to a step change in receptor activity. The peak of this curve is in excess of 18 mM, a nonphysiologic limiting case. The second curve shows the response of the calcium dynamics model coupled to receptor activation. The stimulus here is a step change in surface-agonist concentration. The slowest curve shows the response of the complete agonist model to a step change in shear stress to a level of 0.1 N/m2. This curve contains the effects of mass transfer limitations, receptor dynamics, and intracellular calcium dynamics. As more dynamic lags are included in the simulation, the time to peak increases, and the peak value is attenuated. It can be seen that receptor dynamics and the development of the concentration profile introduce significant delays.
Figure 4.

Receptor dynamics and mass transport limitations introduce significant delay into calcium dynamics. Simulation of Eq. 1 only yields a very high and rapid transient. Dotted line, stimulus is a step increase in receptor activity. Solid line, simulation of Eqs. 1 and 6 coupled via the phosphoinositide pathway. Addition of receptor dynamics significantly attenuates and delays the transient. Broken line, stimulus as a step increase in cs. Simulation of Eqs. 1 and 6 coupled to thrombin balance in the perfusate further attenuates and delays the transient (cs = 1 unit per ml, c0 = 1 unit per ml, Caex = 0, τ = 0.1 N/m2).
Given that mass transfer limitations can govern the response, it is of interest to vary the flow rate parametrically. In Fig. 5 one sees an acceleration in the response as the shear stress is increased from 0.01 N/m2 to 3 N/m2. This is due to enhanced mass transfer of agonist to the surface at higher flow rates. Noteworthy here is the fact that there is little difference between the responses at 0.5 N/m2 and 3 N/m2. This indicates that the agonist-mediated calcium mobilization process is controlled by the development of the concentration profile in the perfusate at low shears (<0.5 N/m2). On the other hand, the response is controlled by the intrinsic kinetics of receptor activation at shear stresses above 3 N/m2. This modulation of the agonist response by the level of shear stress has been observed experimentally (24). In contrast to the no-agonist simulation, the peak here is very prominent. Also, there is no elevated plateau in the absence of extracellular calcium.
Figure 5.

Response of calcium transient to agonist-bearing media. As the perfusion rate (and shear stress) increases, the response accelerates. The influence is significant at low shear stresses (<0.5 N/m2), where mass transfer is rate-limiting. Above 0.5 N/m2, perfusion rate has diminishing effects because receptor dynamics are rate-limiting (C0 = 1 unit per ml, Caex = 0).
Synergistic effects of thrombin and physiologic extracellular calcium are shown in Fig. 6. The calcium transient is calculated for shear stresses from 0.1 N/m2 to 5 N/m2. Both the peak value and the plateau level increase at elevated shear stresses. The effects of shear stress become pronounced only at values higher than 1 N/m2. This is above the threshold level for which calcium influx becomes appreciable. At shear stresses below 1 N/m2, the primary modulation of the response is by mass transfer of agonist to the cell. This modulation is minor and becomes kinetically limited between 0.1 and 0.5 N/m2. It is only when calcium influx becomes large that shear stress begins to significantly alter the peak value and the plateau level.
Figure 6.

Synergistic effects of calcium influx and agonist stimulation upon the calcium transient. For 0.1 < τ < 1 N/m2, shear stress exerts a relatively small influence upon the calcium transient. This influence is exerted through modulation of mass transfer of agonist. Above 1 N/m2, the influence becomes larger through enhanced transplasmalemmal calcium influx. Also plotted is a comparison of an experimental calcium transient to the model prediction (τ = 3 N/m2, c0 = 1 unit per ml thrombin, Caex = 1.5 mM). The time to peak and peak values are reasonably simulated. There is a discrepancy between the plateau regions. The elevated plateau in the simulation is due to a sustained increase in calcium influx.
To assess the validity of the model, the simulation was compared with a benchmark experiment in HUVECs. Here, a HUVEC monolayer in a square capillary tube was exposed to a sudden onset of flow corresponding to a shear stress of 3 N/m2. The perfusate contained 1 unit per ml (≈9 nM) of α-thrombin and physiologic calcium. Calcium concentrations were measured with fluorescent microscopy. Details of the experimental protocol are given by Helmlinger et al. (24).
The model prediction is compared with the data in Fig. 6. The simulation for τ = 3 N/m2 reasonably predicts the calcium level for most of the time course, particularly in the peak region. The recovery phase is not reproduced as accurately as the peak region. The simulation goes to an elevated plateau. This is due to the sustained increase in qin. The plateau region is examined in more detail in the Discussion.
DISCUSSION
The agreement with the experiment was judged adequate for a first step in the modeling of the intracellular calcium transient, in that the model reproduces many experimental findings. A biphasic response is predicted. The transient peak is due to agonist-stimulated release from intracellular stores, whereas the sustained plateau is attributable to the influx of intracellular calcium. This prediction agrees with the conclusions of Hallam et al. (30). The model reproduces the experimental finding that flow and agonist together elicit a stronger calcium transient than either alone (24). This phenomenon is manifested not only in agonist experiments, but also in the use of serum-supplemented media, which may contain calcium-mobilizing agonists. The simulations clarify the relative contributions of the two pathways to the recognition and transduction of the fluid mechanical signal. This model predicts that shear stress modulates the calcium transient primarily by the influx pathway, and this modulation occurs at shear stress levels above 1 N/m2. Calcium is one of the most exquisite second messengers in a cell. Given the wide variety of flow patterns in the circulation, the influence of calcium-dependent processes can vary widely as well.
This modeling study also offers new insights into the individual mechanisms by which shear stress elicits a calcium transient. Receptor dynamics and development of concentration boundary layer both appear to be rate-limiting processes in comparison to intracellular calcium dynamics. Resistance to diffusion of agonist is the controlling step at shear stresses below 0.1 N/m2. The calcium transient moves from transport-limited to kinetically limited regime in the range of 0.1 N/m2 to 0.5 N/m2. The kinetic limitation here lies in the receptor dynamics.
The mechanosensitive ion channel model predicts a sigmoidal dependence of calcium influx upon shear stress. Many cellular responses to shear stress are sigmoidal in nature. Schwarz et al. (31) found a sigmoidal dependence of the amplitude of calcium transients upon shear stress. Tseng et al. (32) likewise noted that microtubule-associated protein kinase is stimulated in a sigmoidal fashion by fluid shear stress. The sigmoidal dependence of calcium influx arises from the Boltzmann dependence of channel open probability upon membrane strain energy density.
As a first approximation to estimating membrane stresses and membrane strain energy density, a simple uniaxial tension field employed by Fung and Liu (18) has been used. The key to this analysis is the assumption that the stress perpendicular to the flow direction in the plane of the membrane is zero. The endothelial surface, however, is not one which is flat but rather a topography of “hills” and “valleys.” For such a surface it is quite possible that stresses in the endothelial membrane perpendicular to the direction of flow may not be negligible locally. Therefore, it is possible that the membrane is in fact subjected to a biaxial stress field. The open channel probability is then most properly correlated with a strain energy density based upon a two-dimensional stress field. This would be a worthwhile future enhancement to the model.
The magnitude of calcium influx is influenced by the load-bearing contribution of the cytoskeleton. This contribution is embodied in the parameter ɛ, which has not been experimentally determined. It is hypothesized that many direct mechanical effects are initiated by deformation of the membrane. The deformation of the membrane depends upon its load-bearing capacity relative to submembranous components. This suggests that differences in cytoskeletal and plasmalemmal structure could account for heterogeneities in the calcium response and other responses among different cell types. Establishing the value of ɛ would help validate this hypothesis of mechanotransduction.
The simulations raise the question of what is the magnitude of shear stress-induced calcium influx. Schwarz et al. (3) reported a calcium current of 80 picoamps in cells exposed to a shear stress of 1 N/m2 using voltage clamp techniques. This corresponds to a calcium influx of 138 μM/sec, assuming a cytosolic volume of 3 picoliters. Use of such a large influx in conjunction with the published capacities of the plasma membrane efflux mechanisms and cytosolic buffers completely removes the biphasic nature of the transient and yields a plateau level of calcium several orders of magnitude larger than seen in vitro. The current measured by a voltage-clamp procedure may be nonphysiologic, in that the pipette represents a large additional conductance of calcium in parallel to plasmalemmal efflux mechanisms. It is also possible that this current is composed of other ions in addition to calcium. Thus, the magnitude of shear stress-induced calcium influx is not well established and requires further experiments.
The model predicts an elevated plateau in response to shear stress. Helmlinger et al. (24) (whose data are used as the benchmark experiment in this work) indicate that the calcium level returns to the initial baseline when the cells are stimulated with both thrombin and shear stress. In the case of shear stress only, however, they found an elevated plateau in response to shear stress. It is unclear why the plateau level should vary between these two experiments. Several mechanisms that could explain a return to baseline are not included in the model. These include deactivation of thrombin receptors and calcium entry channels. This suggests that further experiments are needed to establish both the behavior and mechanism for the calcium level at asymptotically long times.
The model predicts the modulation of the calcium transient in the physiologically relevant range. Thus, the findings of this study may have profound implications in vivo. Transport of biologically active molecules and membrane permeability depend upon the hemodynamic flow pattern. Events that alter the flow pattern, such as clot lysis and stenosis formation, can thereby influence the initiation and progression of disease. The model can better approximate the in vivo circulation with some enhancements. These enhancements include a pulsatile velocity profile and the use of a geometry similar to a blood vessel.
This model is intended as a first step in the mathematical description of flow effects upon intracellular calcium. Because calcium as a second messenger regulates many intracellular activities, the model can be an important tool in understanding the bridge between shear stress and the multitude of cellular processes it influences.
Acknowledgments
We are grateful to Dr. Gabriel Helmlinger for the experiments in which the response of the calcium transients in HUVECs to thrombin was established. T.F.W. was supported by National Science Foundation Grant BCS-911761 and National Institutes of Health Grant HL52218. R.C.B. is an Established Investigator of the American Heart Association (AHA) and his efforts were also supported by a National Grant-in-Aid from the AHA.
ABBREVIATION
- HUVEC
human umbilical vein endothelial cell
References
- 1.Geiger R V, Berk B C, Alexander R W, Nerem R M. Am J Physiol. 1992;262:C1411–C1417. doi: 10.1152/ajpcell.1992.262.6.C1411. [DOI] [PubMed] [Google Scholar]
- 2.Dull R, Davies P. Am J Physiol. 1991;261:H149–H154. doi: 10.1152/ajpheart.1991.261.1.H149. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz G, Droogmans G, Nilius B. Pflügers Arch. 1992;421:394–396. doi: 10.1007/BF00374230. [DOI] [PubMed] [Google Scholar]
- 4.Kawahara K, Matsuzaki K. Biochem Biophys Res Commun. 1992;184:198–205. doi: 10.1016/0006-291x(92)91178-s. [DOI] [PubMed] [Google Scholar]
- 5.Prasad A R S, Logan S A, Nerem R M, Schwartz C J, Sprague E A. Circ Res. 1993;72:827–836. doi: 10.1161/01.res.72.4.827. [DOI] [PubMed] [Google Scholar]
- 6.Wiesner T F, Berk B C, Nerem R M. Am J Physiol. 1996;270:C1556–C1569. doi: 10.1152/ajpcell.1996.270.5.C1556. [DOI] [PubMed] [Google Scholar]
- 7.Mahama P A, Linderman J J. Biotechnol Prog. 1994;10:45–54. [Google Scholar]
- 8.Nollert M U, McIntire L V. J Biomech Eng. 1992;114:321–326. doi: 10.1115/1.2891390. [DOI] [PubMed] [Google Scholar]
- 9.Winston F K, Thibault L E, Macarak E J. J Biomech Eng. 1993;115:160–168. doi: 10.1115/1.2894116. [DOI] [PubMed] [Google Scholar]
- 10.Fan C. ZAMP. 1965;16:351–360. [Google Scholar]
- 11.Savino J, Siegel R. Int J Heat Mass Transfer. 1964;7:733–741. [Google Scholar]
- 12.Johns A, Lategan T W, Lodge N J, Ryan U S, Van Breemen C, Adams D J. Tissue Cell. 1987;19:733–745. doi: 10.1016/0040-8166(87)90015-2. [DOI] [PubMed] [Google Scholar]
- 13.Schilling W P, Elliott S J. Am J Physiol. 1992;262:H1617–H1630. doi: 10.1152/ajpheart.1992.262.6.H1617. [DOI] [PubMed] [Google Scholar]
- 14.Adams D J, Barakeh J, Laskey R, Van Breeman C. FASEB J. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- 15.Chakravarthy S R, Giorgio T D. Biochim Biophys Acta. 1992;1112:197–204. doi: 10.1016/0005-2736(92)90392-y. [DOI] [PubMed] [Google Scholar]
- 16.Schultz S G. Basic Principles of Membrane Transport. London: Cambridge Univ. Press; 1980. p. 25. , 119. [Google Scholar]
- 17.Sachs F. Membr Biochem. 1986;6:173–195. doi: 10.3109/09687688609065448. [DOI] [PubMed] [Google Scholar]
- 18.Fung Y C, Liu S Q. J Biomech Eng. 1993;115:1–12. doi: 10.1115/1.2895465. [DOI] [PubMed] [Google Scholar]
- 19.Evans E A, Skalak R. Mechanics and Thermodynamics of Biomembranes. Boca Raton, FL: CRC; 1980. p. 99. [Google Scholar]
- 20.Skalak R, Tozern A, Zarda R P, Chien S. Biophys J. 1973;13:245–264. doi: 10.1016/S0006-3495(73)85983-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakache M, Gaub H E. Proc Natl Acad Sci USA. 1988;85:1841–1843. doi: 10.1073/pnas.85.6.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiesner T F. Ph.D. thesis. Atlanta: Georgia Institute of Technology; 1994. [Google Scholar]
- 23.Sato M, Levesque M J, Nerem R M. J Biomech Eng. 1987;107:27–34. doi: 10.1115/1.3138638. [DOI] [PubMed] [Google Scholar]
- 24.Helmlinger G, Berk B C, Nerem R M. J Cell Eng. 1995;1:13–20. [Google Scholar]
- 25.Helmlinger G. Ph.D. thesis. Atlanta: Georgia Institute of Technology; 1994. [Google Scholar]
- 26.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. 2nd Ed. New York: Garland; 1989. p. 301. [Google Scholar]
- 27.Sober H A. Handbook of Biochemistry: Selected Data for Molecular Biology. 2nd Ed. Cleveland: CRC Press; 1970. pp. C–10. [Google Scholar]
- 28.Levesque M, Nerem R M. J Biomech Eng. 1985;107:341–347. doi: 10.1115/1.3138567. [DOI] [PubMed] [Google Scholar]
- 29.Lollar P, Hoak J C, Owen W G. J Biol Chem. 1979;255:10279–10283. [PubMed] [Google Scholar]
- 30.Hallam T J, Jacob R, Merritt J E. Biochem J. 1988;255:179–184. doi: 10.1042/bj2550179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarz G, Callewaert G, Droogmans G, Nilius B. J Physiol (London) 1992;458:527–538. doi: 10.1113/jphysiol.1992.sp019432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng, H., Peterson, T. & Berk, B. C. (1995) Circ. Res., in press. [DOI] [PubMed]