

Abstract
The WT1 gene, located on chromosome 11p13, is mutated in a low number of Wilms tumors (WTs). Germ-line mutations in the WT1 gene are found in patients with bilateral WT and/or associated genital tract malformations (GU). We have identified 19 hemizygous WT1 gene mutations/deletions in 64 patient samples. The histology of the tumors with mutations was stromal–predominant in 13, triphasic in 3, blastemal–predominant in 1, and unknown in 2 cases. Thirteen of 21 patients with stromal–predominant tumors had WT1 mutations and 10 of these were present in the germ line. Of the patients with germ-line alterations, six had GU and a unilateral tumor, two had a bilateral tumor and normal GU tracts, and two had a unilateral tumor and normal GU. Three mutations were tumor-specific and were found in patients with unilateral tumors without GU. These data demonstrate a correlation of WT1 mutations with stromal–predominant histology, suggesting that a germ-line mutation in WT1 predisposes to the development of tumors with this histology. Twelve mutations are nonsense mutations resulting in truncations at different positions in the WT1 protein and only two are missense mutations. Of the stromal–predominant tumors, 67% showed loss of heterozygosity, and in one tumor a different somatic mutation in addition to the germ-line mutation was identified. These data show that in a large proportion of a histopathologically distinct subset of WTs the classical two-hit inactivation model, with loss of a functional WT1 protein, is the underlying cause of tumor development.
Keywords: histopathology, truncation mutations, tumor suppressor gene, nephroblastoma, SSCP analysis
The WT1 gene was isolated by positional cloning from chromosome 11p13 (1, 2) and encodes a transcription factor of the zinc finger (ZF) family. Loss of heterozygosity (LOH) studies showed that tumors frequently have lost markers from chromosome 11p. Subsequently, it was found that this loss is often limited to the region 11p15, where a second locus, WT2, involved in the development of Wilms tumor (WT) has been assumed to exist. Further LOH studies revealed loss of chromosome 16q in about 20% of WTs, suggesting that a third locus is located at this site. Susceptibility for the rare form of familial WT in several generations does not show linkage to either of these regions; therefore, another WT locus must exist in these cases (3–5). The WT1 gene encodes four transcripts produced by alternative splicing (6, 7) and encodes a protein with a predicted size of 45–49 kDa. DNA binding to a GC-rich motif identical to the early growth response-binding site was demonstrated for the WT1 protein lacking splice II in the ZF (WT/−KTS), whereas the WT1 protein containing these amino acids (WT/+KTS) does not bind to this sequence (8). Recently it was shown that WT1/+KTS can also bind to a similar GC-rich DNA motif (9). More recent studies have established that the proline–glutamine rich amino terminus has transcriptional regulatory properties. It was shown that WT1 containing splice I (WT/+17aa) is a repressor, whereas WT1/−17aa can be either a repressor or activator depending on the architecture of the DNA binding site(s) under study (9–11).
Molecular analysis of WTs revealed that intragenic microdeletions/insertions and point mutations of one allele within the WT1 gene are found in only 10–15% of WTs, suggesting that other alterations must contribute to Wilms tumorigenesis. Constitutional hemizygous deletions and mutations of the WT1 gene have been demonstrated in most patients affected by WAGR (WT, aniridia, genitourinary anomalies, and mental retardation) and Denys–Drash syndrome, both predisposing to WT and urogenital malformations (GU) (12).
The WT1 protein is predominantly expressed in components of the urogenital system (fetal kidneys, the genital ridge, and the gonads), spleen, and mesothelial cells (13–16). The level of WT1 expression in WTs varies greatly and can be correlated with tumor histology, with low levels in stromal–predominant tumors, and high levels in epithelial/blastemal–predominant tumors (13–20). In situ mRNA hybridization of tumors revealed that WT1 is expressed in blastema and in immature epithelial structures but not in stroma or collecting duct derivatives or in more mature epithelial structures (15). A similar pattern is seen during normal nephrogenesis.
Pathogenetic heterogeneity in WTs may be due to the involvement of different genes in the different tumor types. However, WTs seen in Beckwith–Wiedeman syndrome (11p15) and WAGR patients (11p13) can show similar histology. According to Beckwith, two pathogenetic groupings can be classified by their association with nephrogenic rests, which according to their position and cellular components were termed intralobar nephrogenic rests (ILNR) and perilobar nephrogenic rests (PLNR) (21). These authors observed that ILNR-associated tumors have prominent stroma and PLNR-associated tumors are mainly composed of blastemal and epithelial elements, corresponding to later stages of development. ILNR are often found in tumors from patients with WAGR Denys–Drash syndrome and PLNRs in those from Beckwith–Wiedeman syndrome patients. Since ILNRs are found in tumors from patients with a clear 11p13 (WT1) involvement it was postulated that cells lacking WT1 have a defect in epithelial cell differentiation resulting in stromal differentiation of the malignant clone (15, 22).
We analyzed the status of the entire WT1 gene in 64 cases of WTs predominantly from patients with preoperative chemotherapy and ascertained through the German SIOP9/GPO study (46). First we studied 32 consecutive cases, followed by patients selected for GU anomalies and bilateral tumors. When we noticed that most tumors with mutations had stromal–predominant histology we selected 11 more cases of this histological subtype; all patients had a unilateral tumor and normal GU. Of the 21 tumors/patients, 13 had WT1 mutations/deletions and 10 of these were germ line. Six patients carrying germ-line mutations/deletions had GU anomalies and unilateral tumors, two had bilateral tumors, and two had a unilateral tumor and normal GU. Three mutations in patients with a unilateral tumor and normal GU were tumor-specific. A total of 15 WT1 gene mutations and 4 deletions of the entire gene were identified in our series of 64 patients. Of the 15 WT1 gene mutations, 12 were nonsense, 1 was a splice site, and 2 were missense mutations. In 7 of 11 cases where LOH analysis was performed the normal allele was lost.
MATERIALS AND METHODS
Patients.
Of the 64 patients analyzed, 53 were registered in the nephroblastoma study trial SIOP9/GPO. Of the 64 patients, 9 were operated on without pretreatment, 52 patients received cytostatic preoperative treatment with vincristine and actinomycin D (23), and in 3 cases the status of pretreatment was unknown. The histological classification was known for 59 of the 64 cases analyzed. The subclassification of the standard tumors was as follows: 21 stromal–predominant (37%), 16 mixed triphasic (28%), 8 predominant–regressive (14%), 5 blastemal–predominant (9%), 3 epithelial–predominant (5.3%), and 2 without subclassification. In addition, four anaplastic tumors were analyzed. In the SIOP9/GPO study these numbers are: 14%, 38.2%, 36.2%, 7.7%, and 3.9%, respectively (24). The higher percentage of tumors with stromal–predominant histology is based on selection for these tumors. The mutation analysis was done on blood DNA in 8, on tumor metastases in 2, on tumor DNA in 41, and on blood/tumor DNA in 13 cases. All information on the patients, their clinical features, and the material and types of analysis performed is summarized in Table 1.
Table 1.
Summary of clinical and molecular features of all patients analyzed
| Patient | Sex | Clinical features | Tumor | Stage/ histology | Preoperative treatment | DNA | PFGE | SSCP | Sequence analysis |
|---|---|---|---|---|---|---|---|---|---|
| WT patients with associated abnormalities | |||||||||
| ANS1 | m | Hypospadias, maldescended testis, aniridia | ul | c | + | − | − | ||
| HDWT2 | m | Hypospadias, cryptorchidism | ul | II/ s | + | c | + | − | − |
| HDWT9 | m | Hypospadia | ul | − | t | + | + | − | |
| 9139 | f | Duplex kidney | ul | I/ s | + | t | − | + | + |
| 9148 | m | Hypospadias | ul | II/regr | + | t | − | + | − |
| 9184 | m | Unilateral testicular aplasia | ul | I/ s | + | t/c | + | + | ex 1b |
| 9274 | m | Hypospadias, cryptorchidism | ul | I/ s | + | t/c | − | + | ex/in 6 |
| 9391 | m | Hypospadias | ul | IV/ tri | + | t | − | + | − |
| 9595 | m | Hypospadias, proteinuria | ul | I/ s | + | c | − | + | ex/in 6 |
| ANS2-9115 | m | Aniridia, cryptorchidism | ul | II/ tri | − | t/c | + | + | − |
| ANS3-9416 | m | Maldescended testis, aniridia | ul | I/ s | + | t/c | + | + | − |
| HDWT8-9378 | m | Maldescended testis, proteinuria | ul | I/ s | + | c | − | + | ex 9 |
| Bilateral WT | |||||||||
| HDWT7 | f | Normal | bl | V/ tri | + | c | − | + | ex 7 |
| 9200 | f | Normal | bl | V/ l:s, r:tri | + | t/c | − | + | ex 1b |
| 9318 | f | Normal | bl | V/ s | + | t/c | − | + | ex 7 |
| 9464 | m | Normal | bl | V/ tri | + | t/c | − | + | ex 1a |
| 9501 | m | Normal | bl | V/ s | + | t | − | + | + |
| WT patients with no associated abnormalities | |||||||||
| HDWT1 | f | Proteinuria | ul | II/ stan | + | c | − | + | − |
| HDWT3 | m | Normal | ul | c | − | + | − | ||
| HDWT4 | m | Normal | ul | III/ | + | met | + | + | − |
| HDWT5 | f | Normal | ul | I/ e | − | t | + | + | − |
| HDWT6 | f | Normal | ul | III/ stan | + | c | − | + | ex 7 |
| HDWT10 | m | Normal | ul | IV/ b | + | met | + | + | − |
| HDWT11 | f | Normal | ul | t | − | + | + | ||
| 5018 | m | Normal | ul | I/ tri | + | t | + | + | − |
| 5024 | m | Normal | ul | I/ tgri | + | t | + | + | − |
| 5302 | f | Normal | ul | I/ anapl | + | t | − | + | − |
| 9066 | m | Normal | ul | III/ tri | + | t | + | + | − |
| 9073 | m | Normal | ul | II/ anapl | + | t | + | + | − |
| 9074 | m | Normal | ul | IV/ b | − | t | − | + | − |
| 9094 | f | Normal | ul | III/ b | + | t/c | + | + | ex 2 |
| 9098 | m | Normal | ul | I/ tri | + | t | + | + | − |
| 9112 | m | Normal | ul | I/ tri | + | t | + | + | − |
| 9125 | f | Normal | ul | IV/ tri | + | t | + | + | − |
| 9147 | f | Normal | ul | IV/ regr | + | t | + | + | − |
| 9165 | f | Normal | ul | I/ tri | − | t | − | + | − |
| 9168 | f | Normal | ul | I/ b | − | t | + | + | − |
| 9169 | m | Normal | ul | IV/ negr | + | t | + | + | − |
| 9174 | m | Normal | ul | I/ tri | + | t | + | + | − |
| 9177 | m | Normal | ul | IA/ s | + | t/c | + | + | ex 6 |
| 9193 | m | Normal | ul | II/ regr | + | t | − | + | − |
| 9196 | m | Normal | ul | II/anapl | + | t | − | + | − |
| 9201 | m | Normal | ul | I/ e | + | t | − | + | − |
| 9206 | f | Familial WT | ul | IV/ tri | + | t | − | + | − |
| 9210 | m | Normal | ul | II/ regr | + | t | − | + | − |
| 9211 | m | Normal | ul | III/ tri | − | t | + | + | − |
| 9249 | m | Normal | ul | IV/ tri | + | t | − | + | − |
| 9258 | f | Normal | ul | I/ regr | + | t | − | + | − |
| 9288 | m | Normal | ul | I/ s | + | t | − | + | − |
| 9304 | m | Normal | u | III/ regr | + | t | − | + | − |
| 9310 | m | Normal | ul | III/ b | − | t | − | + | − |
| 9343 | m | Normal | ul | IV/anapl | + | t | − | + | − |
| 9358 | m | Normal | ul | I/ s | + | t | − | + | + |
| 9362 | m | Normal | ul | I/ s | + | t | − | + | + |
| 9385 | m | Normal | ul | II/ s | + | t/c | − | + | ex 7 |
| 9394 | f | Normal | ul | III/ s | + | t/c | − | + | ex 7 |
| 9415 | f | Normal | ul | II/ e | − | t | − | + | − |
| 9422 | m | Normal | ul | IV/ s | + | t | − | + | + |
| 9496 | m | Normal | ul | I/ s | + | t | ∓ | + | + |
| 9518 | f | Normal | ul | II/ tri | + | t | − | + | − |
| 9554 | f | Normal | ul | II/ regr | + | t | + | + | − |
| 9561 | m | Normal | ul | II/ s | + | t/c | − | + | ex 7, 8 |
| 9572 | m | Normal | ul | II/ s | + | t | − | + | + |
| 9614 | f | Normal | ul | III/ s | + | t/c | − | + | ex 5 |
This table presents a list of all patients, whether blood or tumor DNA was analyzed, the clinical details, preoperative treatment, and the methods used for analysis. m, male; f, female; ul, unilateral; bl, bilateral tumor; regr, regressive; tri, triphasic; s, stromal–predominant; b, blastemal–predominant; e, epithelial–predominant; anapl, anaplastic; stan, standard histology—no subclassification available; t, tumor; l, left; r, right, c, constitutiona;; PFGE, pulsed-field gel electrophoresis; met, metastasis—no primary tumor available; ex, exon; in, intron; sequence analysis; +, exons 1a, 1b, 3, 6, 7, 8, 9, 10 were sequenced. Patient numbers correspond to the SIOP study numbers.
DNA/RNA Preparation from Tumors.
Simultaneous extraction of high molecular weight DNA and RNA from frozen pulverized tumor tissues was performed as described (25). Constitutional DNA was isolated from the peripheral leukocytes of 21 patients as described (26).
PCR–SSCP Analysis.
All WT1 exonic sequences (exons 1–10) were amplified from genomic DNA by PCR and analyzed by SSCP. Exon 1 was analyzed in two overlapping fragments (1a and 1b) because of its large size. The sequences of the oligonucleotide primers for exons 1a, 1b, 2, 4, 6, 7, 8, 9, and 10 were as published (14, 27–30). New primers were designed for exons 3 and 5 (exon 3, 5′-GCTGTCTTCGGTTCTCTCTG-3′ and 5′-AGGACCCAGACGCAGAGC-3′; exon 5, 5′-GGAATTCGGGGCTTGCAGATCCATG-3′ and 5′-GGAATTCTCCTAACTCCTGCATTGCCC-3′). Each PCR reaction contained 100–300 ng genomic DNA, 1× Taq DNA polymerase buffer [ex2, ex4-ex10: 10 mM Tris·HCl, pH 8.3/50 mM KCl/1.5 mM MgCl2/0.01% gelatine; ex1a: 60 mM Tris·HCl, pH 8.5/50 mM KCl/15 mM (NH4)2SO4/1.5 mM MgCl2/0.02% gelatine plus 10% dimethyl sulfoxide (DMSO); ex1b: 60 mM Tris·HCl, pH 8.5/75 mM (NH4)2SO4/7.5 mM MgCl2 plus 10% DMSO], 25 pmol of each primer, 200 μM nucleotides, and 1 unit Taq polymerase in a total volume of 50 μl. Hot start PCR conditions were: 30 cycles at 94°C for 1 min, annealing for 1–2 min at 57°C–60°C, extension at 72°C for 1–2 min. For SSCP, 1–1.5-μl aliquots of the amplified product were added to 4–4.5 μl of 95% formamide, 10 mM EDTA. The DNA samples were denatured and run on 8% PAGE (29:1 or 49:1 acrylamide:bisacrylamide) containing 2% glycerol. Electrophoresis was performed in 1× TBE running buffer at 200 V and 10°C, 15°C, or 20°C for 4–7 hr and silver stained (Qiagen, Chatsworth, CA). Every sample was analyzed using two sets of electrophoresis conditions to maximize the sensitivity of the technique.
Direct Sequencing of PCR Products.
Sequencing of the biotinylated PCR product was performed on streptavidin-coated magnetic Dynabeads as described by the manufacturer (Dynal). Direct sequencing was performed using the Sequenase version 2.0 sequencing kit (United States Biochemical) with T7 polymerase and [35S]dATP according to the instructions of the manufacturer.
Analysis of Allele Loss.
For studies on loss of heterozygosity, pairs of blood and tumor DNA from the same patient were analyzed by PCR in 13 cases with 4 polymorphic markers: restriction fragment length polymorphisms (RFLPs) in PR1 and PR4 will be described elsewhere, exon 7 (31), and a CA-repeat marker in the 3′ untranslated region (32). All PCR products were analyzed on 8 or 15% PAGE (29:1) containing 2% glycerol.
RESULTS
SSCP Analysis of Exons 1–10 of the WT1 Gene.
WTs were initially analyzed for gross alterations in the WT1 gene by pulsed-field gel electrophoresis, Southern blot analysis, and reverse transcriptase–PCR. One homozygous deletion in a sporadic WT (WT21) of 200 kb spanning WT1, a constitutional deletion of 1300 kb in a patient with WT and GU (HDWT2, previously called KJ) and three hemizygous, constitutional submicroscopic deletions in WAGR patients were identified with pulsed-field gel electrophoresis and have been described (ANS1, ANS2, and ANS3) (33, 34). No alterations were seen in Southern blots and reverse transcription–PCR, where two overlapping fragments corresponding to the Pro/Gln-rich region and the four ZFs were amplified. Because no alterations were found with these methods we used the more sensitive SSCP method to search for single base pair mutations within the WT1 gene (35). With this method all coding exons of the WT1 gene, including exon 1, were analyzed in 54 WT samples, including tumor material from 2 WAGR and 2 metastasis patients and in 6 constitutional DNAs from WT patients. From these samples, 44 were unilateral sporadic, 8 unilateral with genitourinary abnormalities (GU), 5 bilateral, 2 WAGR, 2 metastasis, and 1 familial WT.
WT Patients with Associated Abnormalities.
Ten patients with GU abnormalities were analyzed and two of these (ANS2 and ANS3) were WAGR patients. In the remaining eight patients, mutations were found in four and all were germ line. WTs from patients with WT1 mutations/deletions were of stromal–predominant histology in six cases, triphasic in one, and unknown in one. No mutations were found in the other four patients with SSCP and sequencing of exons 1a, 1b, 3, 6, 7, 8, 9, and 10. The other exons have not been sequenced.
Tumor DNA from patient 9184 showed only altered bands in SSCP of exon 1b and blood DNA was heterozygous for this alteration. Direct sequencing of tumor DNA showed a 7-bp deletion in exon 1b, resulting in a stop codon in exon 3. Blood DNA from patient 9595 showed a SSCP alteration in exon 6 and direct sequencing revealed a GT to GG change at position +2 in the conserved splice donor site in intron 6 (not shown). We have previously described patient 9274 with a germ-line mutation 1 nt 5′ of this mutation, affecting the same splice site in intron 6 and resulting in exon skipping (30). No tumor material from patient 9595 was available for RNA analysis, therefore, we could not determine the consequence of the splice site mutation. Blood DNA from patient HDWT8 showed an altered band in exon 9. Direct sequencing revealed a C → T transition at codon 1546, creating a stop codon at this position. Tumor material was not available in this case. Tumor DNAs from two WAGR patients ANS3 and ANS2 were analyzed with SSCP during the course of this work and no alteration in the second allele could be identified.
Bilateral WTs.
Five patients with bilateral WT were analyzed, the histology was stromal–predominant in three and mixed triphasic in two cases. None of these patients had GU anomalies. Mutations were found in four cases and all were present in the germ line. In three cases the tumor has lost the normal allele and therefore the wild-type WT1 protein should be absent. In one case, HDWT7, no tumor material was available for analysis.
Patient HDWT7 was the twin of HDWT6 who developed bilateral and unilateral WT, respectively. SSCP analysis of blood DNA showed an alteration in exon 7 (Fig. 1). Direct sequencing revealed a C → T transition, resulting in a stop codon in both patients. In patient 9200, an alteration in exon 1b was found in blood DNA. No normal bands were seen in the DNAs isolated from the right and left tumor, demonstrating that both tumors have lost the normal WT1 allele. Direct sequencing of tumor DNA showed a deletion of 26 bp, resulting in a stop codon in exon 2. Blood DNA from patient 9464 showed an alteration in exon 1a by SSCP. The tumor showed only the altered bands and direct sequencing revealed a 29-bp deletion creating a stop codon at position 89 in exon 1b. Analysis of the DNA from one of the two tumors from patient 9318 showed a slight shift in SSCP bands in exon 7 (Fig. 1). Direct sequencing of the tumor DNA revealed a C → A transversion, resulting in creation of a stop codon. The mutation is germ line and the tumor has lost the normal WT1 allele.
Figure 1.
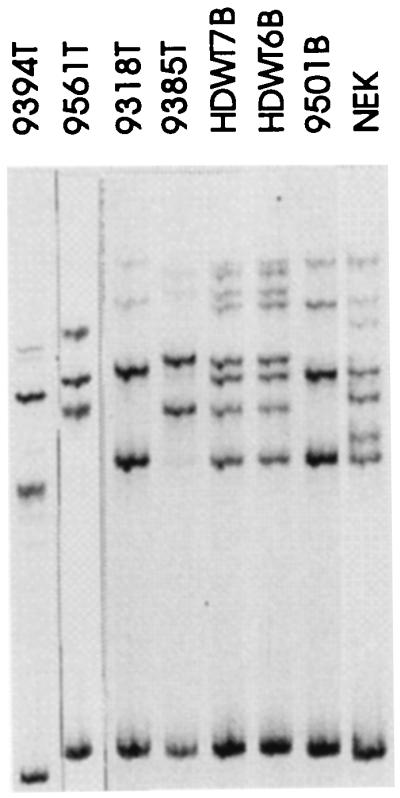
SSCP analysis of exon 7 from patients 9394, 9561, 9318, 9385, HDWT7, and HDWT6. Only a slight shift can be seen in the upper bands in patient 9318 under the conditions used for this gel. NEK control DNA is heterozygous for the RFLP (alleles A and B) and patient 9501 is homozygous for allele A. Patients HDWT7 and HDWT6 are homozygous for allele A and the additional bands correspond to the mutation, which is identical to the mutation in patient 9385. Lanes 9394, 9561, 9318, and 9385 contain tumor DNA; only altered bands are seen.
WT Patients with No Associated Abnormalities.
Of the 47 patients analyzed the histology was known in 44 cases: 11 stromal–predominant, 12 mixed triphasic, 8 epithelial or blastemal–predominant, 7 regressive, 4 anaplastic, and 2 standard with no further subclassification. Seven mutations were found, five in stromal–predominant tumors, one in a blastemal tumor, and in one case with standard histology. Four mutations were germ line and 3 were tumor-specific. In one tumor, a second, different, tumor-specific mutation was identified in addition to the germ-line mutation. Two of the mutations were missense and six nonsense.
In patient 9094, SSCP analysis showed an alteration in blood DNA in exon 2 and the tumor was heterozygous. Direct sequencing of blood and tumor DNA revealed a C → T transition, changing Pro-181 to Ser in the Pro/Gln-rich region of the WT1 gene. This patient had a germ-line missense mutation and the tumor retained one normal allele. SSCP analysis of tumor DNA from patient 9614 revealed an alteration in exon 5. To our knowledge, alterations in this alternative exon have never before been described. Sequence analysis revealed a G → C transversion changing a glycine to an alanine. This mutation is present in the germ line and the tumor has retained the wild-type allele. Tumor DNA from patient 9177 showed an altered band in exon 6. Subcloning of the PCR product and sequencing identified a 4-nt insertion changing the reading frame and resulting in a stop codon in exon 6. This mutation was not present in the germ line and the tumor had retained one normal allele.
SSCP analysis of tumor DNA from patient 9385 showed only altered bands in exon 7 (Fig. 1). Direct sequencing of tumor DNA revealed a C → T transition in exon 7, resulting in a stop codon. This mutation was not found in blood DNA and is therefore a somatic alteration. Using RFLP markers in the promoter region, the LOH of this genomic segment in tumor DNA could be identified, demonstrating that no normal WT1 allele was present in the tumor. This mutation is identical to the germ-line mutation found in the twin patients HDWT6 and HDWT7. SSCP of tumor DNA from patient 9394 showed only altered bands in exon 7 (see also altered sized double-stranded DNA at the bottom of the gel in Fig. 1). Direct sequencing of tumor DNA showed a deletion of 16 bp, resulting in a frameshift and a stop codon at position 380 in exon 9. The alteration was not present in blood DNA and was therefore a tumor-specific alteration with the loss of the normal WT1 allele in the tumor.
SSCP analysis of tumor DNA from patient 9561 revealed an alteration in exon 8. Direct sequencing of tumor DNA showed a GA insertion resulting in a stop in exon 9. This alteration was not found in blood DNA. In addition, the tumor was homozygous for the rare allele B of the RFLP in exon 7 (Fig. 1). Only a very few patients/normal controls are homozygous for this allele (S.S. and B.R.-P., unpublished work); therefore, we confirmed this interpretation with restriction enzyme analysis and sequencing. This analysis revealed that the patient is indeed homozygous for allele B and an additional alteration was found, changing a C → A (Fig. 2), creating a stop codon identical to the mutation found in patient 9318. This mutation was present in blood DNA and the tumor was heterozygous for this alteration. Thus, this tumor has two different alterations in the WT1 gene, one germ-line nonsense mutation in exon 7 and one tumor-specific nonsense mutation in exon 8, fulfilling Knudson’s classical two-hit hypothesis. To our knowledge, this is the second example where two different mutations in the WT1 gene could be identified (36). In addition, several examples of a mutation in the remaining allele in tumors from WAGR patients with a germ-line deletion have been reported.
Figure 2.

BsiEI digests of exon 7 PCR products from blood (B) and tumor (T) DNA from patient 9318 with a bilateral WT and a normal control (NEK). Loss of the recognition sequence by the mutation results in an undigested fragment of 235 bp; the digested products are 80 and 155 bp. M, 1-kb size marker. Schematic shows the mutation with loss of BsiEI site and the stop codon.
A summary of all alterations found in the WT1 gene in our collection of patients is given in Fig. 3. The details of the alterations with the position within the gene, LOH data and patient descriptions are summarized in Table 2.
Figure 3.

Summary of all WT1 gene mutations described here. Exons are boxed, the Pro/Gln-rich region is shaded, and the ZF exons contain wavy lines. The positions of the alternative splice sites are indicated. Patients with constitutional alterations are boxed. The clustering of mutations in exon 7 is evident. In addition, patients 9318 and 9561 have identical germ-line mutations and patients HDWT6, HDWT7 (twins), and 9385 also have identical mutations. The two missense mutations are shown as open triangles and the truncations as closed triangles.
Table 2.
Clinical and molecular features of patients with WT1 alterations
| Patient | sex | Clinical features | Tumor age at diagnosis | Pathology | Tumor LOH | Location of mutations
|
Mutation | Effect on protein (codon) | c/t | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ex/in | nt | Codon | |||||||||
| 9464 | m | Normal | bl/3 mo | stage V, tri | LOH | ex1a | 408–436 | 10–20 | del 29 bp | Stop ex1b (89) | c |
| 9200 | f | Normal | bl/15 mo | st V; l: s, r, tri | LOH | ex1b | 746–771 | 123–131 | del 26 bp | Stop ex2 (178) | c |
| 9184 | m | ul testicular aplasia | ul/12 mo | stage I, s | LOH | ex1b | 787–793 | 137–139 | del 7 bp | Stop ex3 (217) | c |
| 9094 | f | Normal | ul/30 mo | stage III, b | HT | ex2 | 919 | 181 | C → T | Pro → Ser | c |
| 9614 | f | Normal | ul/29 mo | stage III, s | HT | ex5 | 1136 | 253 | G → C | GLY → Ala | c |
| 9177 | m | Normal | ul/30 mo | stage I, s | HT | ex6 | 1192 | 272 | ins TACG | Stop ex6 (276) | t |
| 9274 | m | amb ext | ul/9 mo | stage I, s | LOH | in6 | +1 | — | G → C | Stop ex7 (306 | c |
| 9595 | m | Hypospadias | ul/7 mo | stage I, s | ND* | in6 | +2 | — | T → G | ? | c |
| HDWT6 | f | Normal | ul/19 mo | stage III, stand | ND* | ex7 | 1279 | 301 | C → T | Arg → Stop | c |
| HDWT7 | f | Normal | bl/18 mo | stage V, tri | ND* | ex7 | 1279 | 301 | C → T | Arg → Stop | c |
| 9385 | m | Normal | ul/12 mo | stage II, s | LOH | ex7 | 1279 | 301 | C → T | Arg → Stop | t |
| 9394 | f | Normal | ul/8 mo | stage III, s | LOH | ex7 | 1297–1312 | 307–312 | del 16 bp | Stop ex9 (380) | t |
| 9318 | f | Normal | bl/7 mo | stage V, s | LOH | ex7 | 1316 | 313 | C → A | Ser → Stop | c |
| 9561 | m | Normal | ul/34 mo | stage II, s | HT | ex7 | 1316 | 313 | C → A | Ser → Stop | c |
| ex8 | 1494 | 372 | ins GA | Stop ex9 (380) | t | ||||||
| HDWT8 | m | mald testis, prot | u/24 mo | stage I, s | ND* | ex9 | 1546 | 390 | C → T | Arg → Stop | c |
| HDWT2 | m | Hypospadias, crypt | ul/87 mo | stage II, s | ND* | del WT1 | del 1300 kb | c | |||
| ANS1 | m | WAGR | ul/23 mo | ND* | del WT1 | del 1800 kb | c | ||||
| ANS2 | m | WAGR | ul/21 mo | stage II, tri | HEM | del WT1 | del 1700 kb | c | |||
| ANS3 | m | WAGR | ul/20 mo | stage I, s | HEM | del WT1 | del 11p13 | c | |||
Patient descriptions and details on all alteratons. c, constitutional and t, tumor specific alteration. HT, heterozygous; HEM, hemizygous; amb ext, ambigious external genitalia; mald, maldescended; prot, proteinuria; crypt, cryptorchidism; del, deletion; ins, insertion. All other abbreviations are described in legend of Table 1.
ND, no tumor was available for LOH analysis.
DISCUSSION
We have analyzed 64 WT patients for WT1 mutations. The first analysis was done on consecutive cases and later patients with associated anomalies or bilateral WTs were selected. None of the patients studied in this report had Denys–Drash syndrome. Alterations were found in 19 cases; most of these occurred in tumors with stromal–predominant or mixed triphasic histology and only one was found in a blastemal–predominant tumor. Of the 21 stromal–predominant tumors analyzed, 62% had WT1 gene mutations, including 4 deletions of the entire gene in three WAGR and one WT/GU patient. In cases where no alterations were found with SSCP, we have sequenced several exons (exons 1a, 1b, 3, 6, 7, 8, 9, and 10) and no further mutations have been identified so far. When the frequencies of mutations were determined according to histopathology, the numbers were 13 of 21 (62%) in stromal–predominant, 3 of 16 (19%) in triphasic, and 1 of 8 (12.5%) in epithelial or blastemal–predominant tumors.
Most of the mutations are nonsense mutations leading to protein truncation at different positions of the protein. If a protein were synthesized from the mutant genes the shortest would be 89 aa in length and the longest would lack part of ZF 3 and all of ZF 4; none of these would be able to bind to their normal target sequence. Mutations in most exons were detected, even in the alternatively spliced exon 5. Two missense mutations were found, the first is identical to a previously described mutation in exon 2 (37), changing Pro → Ser and was a germ-line alteration in a patient who developed a blastemal–predominant tumor. The tumor has retained the wild-type allele as previously described (37), indicating that this mutation may act by a dominant-negative or gain-of-function mechanism. The altered amino acid lies at the beginning of the domain that has recently been shown to be responsible for protein oligomerization (38). The second missense mutation changes a Gly → Ala next to the Ser-rich region in exon 5 corresponding to the alternative splice I. A mutation in this exon has to our knowledge never before been described. To our knowledge, this germ-line alteration demonstrates for the first time the importance of this splice variant for the normal function of the WT1 protein.
All other mutations led to protein truncations. No correlation was found between the site of the mutation in the gene and the development of either GU, unilateral, or bilateral tumor. GU was present in patients with protein truncations in exons 3, 7, and 9. Bilateral tumors were found in patients with stops in exons 1b, 2, and 7. Unilateral or bilateral tumors were observed in patients with identical mutations: 9318 and 9561 in exon 7 and HDWT6, HDWT7, and 9385 with another mutation in exon 7. This shows a clear difference in expressivity of identical germ-line mutations. Two of the 4 male patients without GU had germ-line truncation mutations and the other two tumor-specific mutations. Lack of the entire gene as in WAGR patients or in patient HDWT2 almost always led to GU malformations. Truncation in exon 1b in the male patient 9464 should probably lead to a complete loss of the protein as in WAGR patients, however, he has no GU. Therefore, it seems possible that truncated proteins are produced, having a partial function and that there may be subtle differences between the lack of the entire gene and the presence of a truncated protein. A similar observation has been made in familial adenomatous polyposis (FAP) patients with mutations leading to truncation of the APC protein at the amino terminus, resulting in an attenuated form of FAP with fewer polyps and later age of onset (39). It has been postulated that in these cases the short protein may have a protective effect.
The three alterations in exon 1 and one in exon 7 are deletions. It has been suggested that exon 1 could be a hot spot for deletion/insertion mutations due to the presence of multiple tri- and tetranucleotide repeat sequences (40). The CCTG (CAGG) sequence was found within 6 nt of deletion breakpoints in 25% of all human gene deletion mutations (41). Four previously described mutations (40) and the three exon 1 mutations described here are all deletion/insertion mutations occurring within 6–9 nt of such sequences, further supporting the role of these repeat sequences in the creation of the deletions.
The mutations at nucleotide positions 1279, 1316, and 1546 involve a CpG dinucleotide, suggesting that deamination of a methylated cytosine generated the mutation. We found the 1279 mutation three times, the 1316 mutation two times, and the 1546 mutation once. The 1546 mutation was previously found in three other WTs (36, 42, 43). It was observed that the triplet CGN coding for Arg is a frequent site of spontaneous mutations in the p53 gene (44). Here we show that mutations in triplets with a CpG (CGN or TCG) also occur frequently in the WT1 gene.
The WT1 gene is expressed during the normal induction phase of the nephrogenic mesenchyme. For epithelial differentiation a high level of WT1 expression is needed in the cells giving rise to specific structures of the nephron. After completion of the differentiation process, WT1 gene expression is restricted to podocytes. It has been observed that the expression level of WT1 correlates with specific histologic subtypes of WTs, with stromal–predominant tumors showing the lowest level (15, 18–20). This correlates with low expression of WT1 in normal stromal components of the kidney (15, 19, 20).
Patients with germ-line mutations have only one intact copy of the WT1 gene, resulting in a reduced level of the normal protein in the induced mesenchymal cells. Higher levels required for differentiation along the epithelial pathway may not be reached. The development of stromal–predominant WTs could be explained by two mechanisms. In the first, induced cells may be blocked early in their differentiation process due to the low level of WT1, leading to an increase in proliferation of these cells. If in one of these cells the second allele is also lost and no normal WT1 protein is present, this cell eventually develops into a tumor with stromal–predominant phenotype. It has been suggested that the term “stromal component” in a WT may be a misnomer and that the stromal cells have characteristics of nonaggregated-induced nephrogenic mesenchyme. If this is the case, the stromal cells may therefore correspond to the induction phase of normal renal development (22). The second possibility is that the lack of WT1 expression drives cells into a stromal differentiation pathway by default, because epithelial differentiation cannot proceed. In this case, stromal cells in a tumor would correspond to more differentiated cells, e.g., skeletal muscle. The two possibilities can now be distinguished in further studies. In the light of our data that WT1 mutations are found mostly in tumors of stromal–predominant histology, it could be postulated that mutational WT1 inactivation is the cause of stromal–predominant histology. Our data strongly suggest that WTs of stromal–predominant histology and inactivating WT1 mutations constitute a new specific molecular subset, which arises by complete loss of a functional WT1 protein, following a classical two-hit inactivation mechanism of a tumor suppressor gene as proposed by Knudson and Strong (45). In contrast, epithelial/blastemal–predominant tumors, which show a low frequency of WT1 mutations, probably occur by a different mechanism and/or involve different genes. A large proportion of the mutations that we have identified are present in the germ line, increasing the risk in developing a second tumor. Many of the patients with a tumor of this histologic subgroup have either bilateral WT or GU anomalies, suggesting that a germ-line mutation in WT1 predisposes to this subtype of WT. These patients may also transmit this mutation to their offspring. Therefore, it seems warranted that patients with stromal–predominant WT should be analyzed for WT1 mutations.
We are currently analyzing tumors with WT1 mutations for the presence of truncated WT1 proteins. Oligomerization of the WT1 protein has been mapped to the first 180 amino acids at the amino terminus and interestingly truncated proteins, lacking the zinc fingers, have a higher activity in oligomerization (38). This could indicate that these truncated proteins may act in a dominant-negative fashion.
Acknowledgments
We would like to thank all the clinicians participating in the SIOP9/GPO study for providing us with frozen tumor material. We also acknowledge the help of Drs. Schärer and Wühl (Children’s Hospital, Heidelberg) and Dr. Waldherr (Institute of Pathology, Heidelberg) in the evaluation of patient data. This work was supported by Deutsche Krebshilfe Grant M32/92/Ro I and a Deutsche Forschungsgemeinschaft fellowship to V.S.
ABBREVIATIONS
- LOH
loss of heterozygosity
- WT
Wilms tumor
- ZF
zinc finger
- GU
genital tract malformations
- RFLP
restriction fragment length polymorphism
References
- 1.Call K M, Glaser T, Ito C Y, Buckler A J, Pelletier J, Haber D A, Rose E A, Kral A, Yeger H, Lewis W H, Jones C, Housman D E. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 2.Gessler M, Poustka A, Cavenee W, Neve R L, Orkin S H, Bruns G A P. Nature (London) 1990;343:774–778. doi: 10.1038/343774a0. [DOI] [PubMed] [Google Scholar]
- 3.Grundy P, Koufos A, Morgan K, Li F P, Meadows A T, Cavenee W K. Nature (London) 1988;336:374–376. doi: 10.1038/336374a0. [DOI] [PubMed] [Google Scholar]
- 4.Huff V, Compton D A, Chao L Y, Strong L C, Geiser C F, Saunders G F. Nature (London) 1988;336:377–378. doi: 10.1038/336377a0. [DOI] [PubMed] [Google Scholar]
- 5.Huff V, Reeve A E, Leppert M, Strong L C, Douglass E C, Geiser C F, Li F P, Meadows A, Callen D F, Lenoir G, Saunders G F. Cancer Res. 1992;52:6117–6120. [PubMed] [Google Scholar]
- 6.Haber D A, Sohn R L, Buckler A J, Pelletier J, Call K M, Housman D E. Proc Natl Acad Sci USA. 1991;88:9618–9622. doi: 10.1073/pnas.88.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner B, Wildhardt G, Schneider S, Royer-Pokora B. Oncogene. 1992;7:1431–1433. [PubMed] [Google Scholar]
- 8.Rauscher F J, Morris J F, Tournay O E, Cook D M, Curran T. Science. 1990;250:1259–1262. doi: 10.1126/science.2244209. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z Y, Qiu Q Q, Huang J, Gurrieri M, Deuel T F. Oncogene. 1995;10:415–422. [PubMed] [Google Scholar]
- 10.Wang Z Y, Qiu Q Q, Enger K T, Deuel T F. Proc Natl Acad Sci USA. 1993;90:8896–8900. doi: 10.1073/pnas.90.19.8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z Y, Qiu Q Q, Deuel T F. J Biol Chem. 1993;268:9172–9175. [PubMed] [Google Scholar]
- 12.Hastie N D. Annu Rev Genet. 1994;28:523–558. doi: 10.1146/annurev.ge.28.120194.002515. [DOI] [PubMed] [Google Scholar]
- 13.Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Husman D, van Heyningen V, Hastie N. Nature (London) 1990;346:194–197. doi: 10.1038/346194a0. [DOI] [PubMed] [Google Scholar]
- 14.Pelletier J, Bruening W, Li F P, Haber D A, Glaser T, Housman D E. Nature (London) 1991;353:431–434. doi: 10.1038/353431a0. [DOI] [PubMed] [Google Scholar]
- 15.Pritchard-Jones K, Fleming S. Oncogene. 1991;6:2211–2220. [PubMed] [Google Scholar]
- 16.Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpracht A, Zabel B. Development (Cambridge, UK) 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- 17.Huang A, Campbell C E, Bonetta L, McAndrews-Hill M S, Chilton-McNeill S, Coppes M J, Law D J, Feinberg A P, Yeger H, Williams B R G. Science. 1990;250:991–994. doi: 10.1126/science.2173145. [DOI] [PubMed] [Google Scholar]
- 18.Gerald W L, Gramling T S, Sens D A, Garvin A J. Am J Pathol. 1992;140:1031–1037. [PMC free article] [PubMed] [Google Scholar]
- 19.Miwa H, Tomlinson G E, Timmons C F, Huff V, Cohn S L, Strong L C, Saunders G F. J Natl Cancer Inst. 1992;84:181–187. doi: 10.1093/jnci/84.3.181. [DOI] [PubMed] [Google Scholar]
- 20.Yeger H, Culliane C, Flenniken A, Chilton-MacNeill S, Campbell C, Huang A, Bonetta L, Coppes M J, Thorner P, Williams B R G. Cell Growth Differ. 1992;3:855–864. [PubMed] [Google Scholar]
- 21.Beckwith J B, Kiviat N B, Bonadio J F. Pediatr Pathol. 1990;10:1–36. doi: 10.3109/15513819009067094. [DOI] [PubMed] [Google Scholar]
- 22.Mierau G W, Beckwith J B, Weeks D A. Ultrastruct Pathol. 1987;11:313–333. doi: 10.3109/01913128709048329. [DOI] [PubMed] [Google Scholar]
- 23.Ludwig R, Weirich A, Pötter R, Harms D, Bürger D, Michaelis J, Erttmann R, Weinel P, Haas R J, Ritter J M, Jacobi H. Klin Pädiatr. 1992;204:204–213. doi: 10.1055/s-2007-1025350. [DOI] [PubMed] [Google Scholar]
- 24.Weirich A, Schmidt D, Harms D, Ludwig R. Med Pediatr Oncol. 1994;23:217. (abstr.). [Google Scholar]
- 25.Royer-Pokora B, Schneider S. Genes Chromosomes Cancer. 1992;5:132–140. doi: 10.1002/gcc.2870050207. [DOI] [PubMed] [Google Scholar]
- 26.Kunkel L M, Smith K D, Boyer S H, Borgaonkar D S, Wachtel S S, Miller O J, Breg W R, Jones H W, Jr, Rary J M. Proc Natl Acad Sci USA. 1977;74:1245–1249. doi: 10.1073/pnas.74.3.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelletier J, Bruening W, Kashtan C E, Mauer S M, Manivel J C, Striegel J E, Houghton D C, Junien C, Habib R, Fouser L, Fine R N, Silverman B L, Haber D A, Housman D. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 28.Bruening W, Bardeesy N, Silverman B L, Cohn R A, Machin G A, Aronson A J, Housman D, Pelletier J. Nat Genet. 1992;1:144–148. doi: 10.1038/ng0592-144. [DOI] [PubMed] [Google Scholar]
- 29.Bruening W, Gros P, Sato T, Stanimir J, Nakamura Y, Housman D, Pelletier J. Cancer Invest. 1993;11:393–399. doi: 10.3109/07357909309018871. [DOI] [PubMed] [Google Scholar]
- 30.Schneider S, Wildhardt G, Ludwig R, Royer-Pokora B. Hum Genet. 1993;91:599–604. doi: 10.1007/BF00205087. [DOI] [PubMed] [Google Scholar]
- 31.Groves N, Baird P N, Hogg A, Cowell J K. Hum Genet. 1992;90:440–442. doi: 10.1007/BF00220474. [DOI] [PubMed] [Google Scholar]
- 32.Haber D A, Buckler A J, Glaser T, Call K M, Pelletier J, Sohn R L, Douglass E C, Housman D E. Cell. 1990;61:1257–1266. doi: 10.1016/0092-8674(90)90690-g. [DOI] [PubMed] [Google Scholar]
- 33.Royer-Pokora B, Ragg S, Heckl-Östreicher B, Held M, Loos U, Call K, Glaser T, Housman D, Saunders G, Zabel B, Williams B, Poustka A. Genes Chromosomes Cancer. 1991;3:89–100. doi: 10.1002/gcc.2870030203. [DOI] [PubMed] [Google Scholar]
- 34.Drechsler M, Meijers-Heijboer E J, Schneider S, Schurich B, Grond-Ginsbach C, Tariverdian G, Kantner G, Blankenagel A, Kaps D, Schroeder-Kurth T, Royer-Pokora B. Hum Genet. 1994;94:331–338. doi: 10.1007/BF00201588. [DOI] [PubMed] [Google Scholar]
- 35.Orita M, Suzuki Y, Sekiya T, Hayashi K. Genomics. 1989;5:874–879. doi: 10.1016/0888-7543(89)90129-8. [DOI] [PubMed] [Google Scholar]
- 36.Varanasi R, Bardeesy N, Ghahremani M, Petruzzi M J, Nowak N, Adam M A, Grundy P, Shows T B, Pelletier J. Proc Natl Acad Sci USA. 1994;91:3554–3558. doi: 10.1073/pnas.91.9.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gessler M, König A, Arden K, Grundy P, Orkin S, Sallan S, Peters C, Ruyle S, Mandell J, Li F, Cavenee W, Bruns G. Hum Mutat. 1994;3:212–222. doi: 10.1002/humu.1380030307. [DOI] [PubMed] [Google Scholar]
- 38.Moffett P, Bruening W, Nakagama H, Bardeesy N, Housman D E, Pelletier J. Proc Natl Acad Sci USA. 1995;92:11105–11109. doi: 10.1073/pnas.92.24.11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spirio L, Olschwang S, Groden J, Roberston M, Samowitz W, Joslyn G, Gelbert L, Thiliveris A, Carlson M, Otterud B, Lynch H, Watson P, Lynch P, Laureut-Puig P, Burt R, Hughes J P, Thomas G, Leppert M, White R. Cell. 1993;75:951–957. doi: 10.1016/0092-8674(93)90538-2. [DOI] [PubMed] [Google Scholar]
- 40.Huff V, Jaffe N, Saunders G F, Strong L C, Villalba F, Ruteshouser E C. Am J Hum Genet. 1995;56:84–90. [PMC free article] [PubMed] [Google Scholar]
- 41.Krawczak M, Cooper D N. Hum Genet. 1991;86:425–441. doi: 10.1007/BF00194629. [DOI] [PubMed] [Google Scholar]
- 42.Little M H, Prosser J, Condie A, Smith P J, Van Heyningen V, Hastie N D. Proc Natl Acad Sci USA. 1992;89:4791–4795. doi: 10.1073/pnas.89.11.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quek H H, Chow V T, Tock E. Anticancer Res. 1993;13:1575–1580. [PubMed] [Google Scholar]
- 44.Caron de Fromentel C, Soussi T. Genes Chromosomes Cancer. 1992;4:1–15. doi: 10.1002/gcc.2870040102. [DOI] [PubMed] [Google Scholar]
- 45.Knudson A G, Strong L C. J Natl Cancer Inst. 1972;48:313–324. [PubMed] [Google Scholar]
- 46.Ludwig R, Weind A, Bürger D, Graf N, Harms D, Kaatsch P, Pötter R, Rieden K, Tröger J, Zimmermann H. Monatsschr Kinderheilkd. 1997;145:128–135. [Google Scholar]