

Abstract
Recent studies have sought to identify the genes involved in excitotoxic neurodegeneration. Here we report that certain strains of mice, including strains that are used for gene targeting studies, do not exhibit excitotoxic cell death after kainic acid seizures. Kainic acid produced excitotoxic cell death in the CA3 and CA1 subfields of the hippocampus in 129/SvEMS and FVB/N mice, in the same pattern as described in rats. C57BL/6 and BALB/c mice exhibited excitotoxic cell death only at very high doses of kainate, and then only in a very restricted area, although they exhibited comparable seizures. Hybrids of 129/SvEMS × C57BL/6 mice created using embryonic stem cells from 129/SvEMS mice also did not exhibit excitotoxic cell death. These results demonstrate that C57BL/6 and BALB/c strains carry gene(s) that convey protection from glutamate-induced excitotoxicity. This differential susceptibility to excitotoxicity represents a potential complication for gene targeting studies.
Excitatory amino acid (EAA) neurotoxicity is thought to play a key role in secondary degeneration after central nervous system injury, stroke, and ischemia (1), and also in many neuropathological disorders, including Alzheimer disease, Huntington disease, and epilepsy (2). EAA neurotoxicity is triggered by a massive release of glutamate, which activates glutamate receptors leading to dramatic increases in intracellular Ca2+ (3). The high Ca2+ levels initiate signaling cascades within susceptible neurons that cause neuronal death through an as-yet-undefined sequence of events (4, 5).
Excitotoxic cell death is most often induced experimentally by the administration of kainic acid (KA), a potent agonist of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate class of glutamate receptors. In rodents, peripheral injections of KA result in recurrent seizures and the subsequent degeneration of select populations of neurons in the hippocampus (6–8). Thus, KA administration has been widely used as a model to study EAA neurotoxicity and seizure-related neurologic diseases (9, 10). More recently, KA neurotoxicity has been used as a model to begin to explore the genes that are involved in EAA neurotoxicity using gene targeting techniques to create null mutant mice (11–13).
In the course of studies carried out for other reasons, we have discovered that certain commonly used strains of mice do not exhibit cell death after KA seizures, whereas others exhibit neurotoxicity similar to rats. Significantly, the strains involved are ones that are used for gene targeting studies, raising the possibility that some of the effects reported in gene targeting studies (11–13) may, in fact, be due to the genetic background of the hybrids. The present manuscript characterizes this genetic difference.
MATERIALS AND METHODS
Animals.
Male BALB/c and C57BL/6 mice, purchased from Hilltop Labs (Philadelphia), and male FVB/N and 129/SvEMS mice, purchased from The Jackson Laboratory, served as subjects. Additionally, mice deficient in the p53 tumor suppressor gene were obtained from The Jackson Laboratory and hybrid mice (129/SvEMS × C57BL/6), created using embryonic stem cell technology were obtained from S. Pearson-White (University of Virginia). All mice were 60–90 days old and were housed individually on a 12-h light/dark schedule. Water and food were available ad libitum.
Drug Administration.
KA was dissolved in isotonic saline (pH 7.3) and administered subcutaneously. Dose–response studies, in which a range of 20–45 mg/kg of KA was administered to either FVB/N or C57BL/6 mice, defined seizure thresholds and mortality rate. Mice were monitored continuously for 4 h for the onset and extent of seizure activity. Seizures were rated according to a previously defined scale (14): Stage 1: immobility; stage 2: forelimb and/or tail extension, rigid posture; stage 3: repetitive movements, head bobbing; stage 4: rearing and falling; stage 5: continuous rearing and falling; stage 6: severe tonic-clonic seizures. These studies revealed consistent seizures in both strains with a mortality rate of less than 25% at a dose of 30 mg/kg. All experiments were performed in accordance with approved institutional animal care guidelines.
Neuropathological Analysis.
At 2, 4, 7, 12, or 20 days postinjection (n = 4–10), mice were anesthetized with Nembutal (sodium pentobarbital) and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Horizontal sections were cut on a Vibratome at a thickness of 40 μm. Every sixth section was stained with cresyl violet to determine neuronal cell loss, and a representative series of sections was stained with a modification of the Fink-Heimer technique for degenerating neurons, fibers, and terminals (15).
Neuron Counts.
Neuron counts were made in areas CA3, CA1, the dentate hilus, and the dentate gyrus. Only neurons with a visible nucleus and in which the entire outline of the cell was apparent were counted. Every sixth section (240 μm separation distance) was evaluated with a 63× oil immersion objective on a Zeiss microscope using a video camera and monitor. A counting frame was printed on an acetate sheet and placed over the monitor, and the number of neurons contained within the frame were counted. Cell counts at all levels were averaged, and mean numbers were used for statistical analysis. Data are expressed as percent of control (n = 3–4 control mice of each strain were evaluated).
Pattern and Extent of Neuronal Activation During KA Seizures as Revealed by 2-Deoxy[14C]glucose (2DG) Uptake.
Adult male C57BL/6 and FVB/N mice were injected with 5 μCi of 2DG (New England Nuclear; 323 mCi/mmol) 60 min after administration of 5, 10, 20, or 30 mg/kg of KA (n = 4 mice per dose) or saline (n = 4 per strain). Mice were euthanized with halothane 45 min after the 2DG administration. The brains were removed, snap-frozen in isopentane (−15°C) and stored at −80°C. Horizontal sections (20 μm) throughout the entire rostro-caudal extent of the hippocampus were cut on a cryostat at −20°C and were mounted on gelatin-coated coverslips. Every 10th section was thaw-mounted along with 14C standards of known radioactivity on cardboard and exposed to Kodak Biomax film.
Regional autoradiograph optical density was measured using the MCID (microcomputer imaging device) image analysis system (Imaging Research Inc., St. Catherine’s, Ontario, Canada). An average of six measurements per structure were used on each side, and optical density values were calculated after subtraction of the film background density. Values given represent the mean of four mice for each dose and each strain. Results were assessed statistically by one-way ANOVA, and intergroup differences were analyzed by Newman–Keuls posthoc test.
Production of Hybrid Mice.
129/SvEMS × C57BL/6 hybrid mice were obtained from S. Pearson White. These mice were generated as part of other gene targeting experiments and were homozygous wild-type (i.e. control) with respect to the targeted gene. To produce the hybrids, blastocysts were isolated from C57BL/6 mice on day 3.5 of pregnancy, and 20–25 embryonic stem cells from 129/SvEMS mice were injected into each blastocoele cavity. The blastocysts were reimplanted into pseudopregnant 129/SvEMS mice. Chimeric mice were identified by agouti contribution to the coat color and were backcrossed to C57BL/6 mice. Germ-line transmission was determined by the presence of agouti mice in the offspring.
Homozygous null mutants for the p53 tumor suppressor gene were obtained from The Jackson Laboratory. They were generated on a 129/Sv background as described previously (16). The genotypes of the mating pairs had been confirmed previously using PCR analysis of DNA extracted from mouse tails (16). Mice homozygous for the targeted p53 mutation have previously been determined to be null (17–21), and previous studies have confirmed that these mice do not express any p53 protein (20, 21).
RESULTS
After systemic administration of 30 mg/kg KA, all four strains displayed comparable seizures. Within 15 min of the injection, mice assumed a catatonic posture accompanied by staring behavior. This behavior was followed by myoclonic twitching and often frequent rearing and falling. All strains exhibited continuous tonic-clonic seizures within 1 h of KA administration, with seizures continuing for 2–3 h. Within 4–5 h after administration, animals assumed a hunched posture and were immobile for the next 2–4 h. In all strains, about 75% of the mice showed generalized clonic seizures within 40–60 min after administration of 30 mg/kg of KA. Mice not displaying any type of seizure activity (n = 1) were not included in the study, leaving a total of 66 mice that were included in the analysis at this dose. Table 1 summarizes seizure type and duration in each strain. Mice did not differ significantly on any of the measures of seizure severity (F = 0.541; P = 0.7118).
Table 1.
Classification of seizure parameters in inbred strains of mice after systemic administration of KA
| Mouse strain | Seizure parameters, % of mice
|
Duration of seizures, h | |||||
|---|---|---|---|---|---|---|---|
| Staring | Rigid posture | Repetitive movements | Rearing and falling | Severe seizures | Survival | ||
| BALB/c (n = 11) | 100 | 100 | 100 | 100 | 36.4 | 72.7 | 1.27 |
| C57BL/6 (n = 24) | 100 | 100 | 100 | 87.5 | 37.5 | 83.3 | 1.35 |
| FVB/N (n = 13) | 100 | 100 | 92.3 | 92.3 | 84.6 | 76.9 | 1.42 |
| 129/SvEMS (n = 10) | 100 | 100 | 100 | 100 | 20.0 | 100 | 1.40 |
KA induced a similar level of seizure duration and stage irrespective of mouse strain (F = 0.541; P = 0.7118).
To determine whether the LD50 of KA is strain dependent, we administered KA (35, 40, and 45 mg/kg) to C57BL/6 and FVB/N mice (n = 10 per group) and assessed seizure severity and survival rate. In both strains, the LD50 for KA was 35 mg/kg, when administered subcutaneously. At doses greater than or equal to 35 mg/kg, all mice in both strains (n = 60) exhibited class 5 seizures within 10–30 min postinjection. The duration of seizures increased with the dose of KA administered, such that at doses greater than 35 mg/kg, mice exhibited class 5 seizures for 4–6 h. When 40 mg/kg of KA was administered less than 30% of both strains survived, and at a dose of 45 mg/kg, only one mouse in each strain survived. At both of these doses, mice appeared debilitated 7 days after administration, and some exhibited spontaneous seizures. At all doses of KA administered, no significant differences were found between the duration of seizures, the number of seizures, or the survivability between C57BL/6 and FVB/N mice.
Despite the fact that KA-induced seizures were comparable across strains, some strains exhibited excitotoxic cell loss while other strains did not. Both FVB/N and 129/SvEMS inbred mouse strains exhibited selective excitotoxic cell death comparable to what has been described in rats (14, 22, 23). This can be seen in sections stained to reveal Nissl substance (RNA) within intact neuronal cell bodies (Fig. 1). For example, the CA3 region, which normally contains a many-cell-thick layer of neuronal cell bodies (Fig. 1 A and C) contained few intact neurons in KA-treated FVB/N and 129/SvEMS mice (Fig. 1B). Approximately, 50% of the remaining neurons within the CA3 subfield exhibited signs of degeneration (shrunken nuclei and pale staining, see Fig. 1D). Cell loss was also evident in the hilus of the dentate gyrus and in some cases in the CA1 region. Cell loss was evident throughout the entire rostro-caudal extent of the hippocampus. In striking contrast, there was no cell loss or evidence of cellular degeneration in C57BL/6 and BALB/c mice at doses less than 40 mg/kg (Fig. 1 C and F) at any time points examined (4–20 days postinjection). These conclusions were confirmed by quantitative analyses of neuron numbers (Fig. 2).
Figure 1.

C57BL/6 mice are resistant to excitotoxin-induced neuronal cell loss and degeneration in the hippocampus. (A) Low magnification (10×) cresyl-violet staining of horizontal sections through the hippocampus in a control mouse, (B) in a FVB/N mouse 4 days post-KA injection, and (C) in a C57BL/6 mouse 4 days after KA administration (30 mg/kg). (D–F) Higher magnification (50×) of the CA3 subfield and the dentate hilar neurons in an uninjected control mouse, FVB/N, and C57BL/6 mouse, respectively. Note the destruction of neurons in the CA3 subfield and the dentate hilus (arrows) in FVB/N mice compared with protection against cell loss and degeneration in C57BL/6 mice. (G–I) High magnification (50×) of the CA3 subfield showing the extent of degeneration in an uninjected control mouse, FVB/N mouse, and C57BL/6 mouse 7 days after KA administration, respectively. Extensive degenerative debris is present throughout the CA3 subfield in FVB/N mice only. CA1 and CA3 denote the hippocampal subfields; DG, dentate gyrus. [Scale bars, 750 μm (A–C); 300 μm (D–I).]
Figure 2.

Quantification of excitotoxin-induced neuronal loss in four inbred mouse strains. Differential cell loss in areas CA3, CA1, and the dentate hilus was observed among inbred mouse strains. Cell loss was most dramatic in area CA3, the dentate hilus, and area CA1 of FVB/N and 129/SvEMS inbred mouse strains, whereas no significant cell loss was observed in any areas examined in BALB/c or C57BL/6 mice (F = 6.012; P < 0.005, Tukey test).
Selective silver stains are especially reliable indicators of excitotoxic cell damage because they stain the debris from degenerating axons and synaptic terminals and degenerating neuronal cell bodies. Silver staining of FVB/N and 129/SvEMS mice revealed degenerating neuronal cell bodies within the cell layers in CA3 and terminal degeneration within the neuropil of CA1 and CA3—the principal site of termination of the projections from neurons in CA3, throughout the entire rostro-caudal extent of the hippocampus. In about half of the cases, degeneration debris was also present in the inner molecular layer of the dentate gyrus—the site of termination of projections from neurons in the hilus (24) (Fig. 1H). Degeneration debris was never seen in C57BL/6 or BALB/c mice at doses less than 40 mg/kg at any of the time points examined (4–20 days postinjection).
A very small amount of cell death was observed in two of three C57BL/6 mice that survived after a 40 mg/kg dose of kainate. A small area of cell loss was observed within area CA3b (Fig. 3 A and B), but cell loss was restricted to ventral portions (approximately 200 μm) of the hippocampus. This localized response was confirmed by selective silver staining, which revealed a discrete patch of degeneration that was restricted to the ventral portion of the hippocampus (Fig. 3C). However, the remaining C57BL/6 mouse that survived administration of KA at 40 mg/kg did not show any cell loss or degeneration, and neither did the C57BL/6 mouse that survived administration of KA at 45 mg/kg.
Figure 3.

In a subset of C57BL/6 mice, select cell loss and degeneration is observed in the ventral hippocampus when the dose of administered KA exceeds the LD50. (A) Low magnification (10×) cresyl-violet staining of a horizontal section through the hippocampus in a C57BL/6 mouse 7 days post-KA injection (40 mg/kg). (B) Higher magnification (66×) of the CA3 subfield showing selective cell loss in area CA3b (arrows). (C) High magnification (66×) of the CA3 subfield showing discrete degeneration within a subpopulation of cells in CA3b (arrow). CA1 and CA3 denote the hippocampal subfields; DG, dentate gyrus; H, hilus. [Scale bars, 600 μm (A) and 150 μm (B and C).]
To ensure that the pattern and extent of neuronal activation during KA seizures was comparable across strains, we evaluated 2DG uptake after administration of low (5 mg/kg), intermediate (10–20 mg/kg), and high (30 mg/kg) doses of KA. At low doses of KA (5 mg/kg), which did not elicit behavioral seizures, there was no apparent increase in 2DG uptake within the hippocampus as compared with control mice (Fig. 4 E and F). In contrast, at higher doses (10, 20, or 30 mg/kg) of KA, increased 2DG uptake was observed throughout the entire rostro-caudal extent of the hippocampus (Fig. 4), although the ventral hippocampus in both strains consistently showed greater 2DG metabolism than the dorsal hippocampus, similar to what has been described in rats (22). Surprisingly, a greater metabolic response was observed throughout the hippocampus in C57BL/6 mice than FVB/N mice at higher doses (10–30 mg/kg; Fig. 5).
Figure 4.
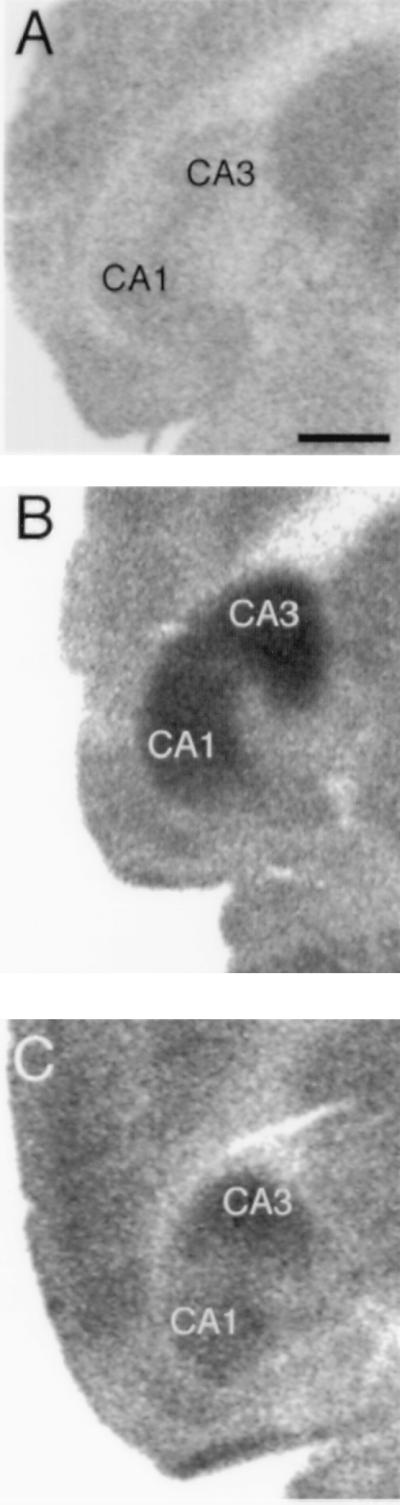
Comparison of 2DG response to administration of KA in C57BL/6 and FVB/N mice. (A) High magnification (5×) of a horizontal section showing 2DG uptake in the hippocampus of a control animal. (B and C) High magnification of a horizontal section displaying 2DG uptake 60 min after injection of 20 mg/kg KA in a C57BL/6 and FVB/N mouse, respectively. Metabolism is greatest in the CA3 and CA1 subfields of the hippocampus, and hippocampal 2DG metabolism is greater in the C57BL/6 mouse (B) as compared with the FVB/N (C). [Scale bar, 750 μm.]
Figure 5.

Quantitation of 2DG uptake in the hippocampus in C57BL/6 and FVB/N mice after KA administration. 2DG uptake was significantly increased in C57BL/6 mice at intermediate and high doses of KA as compared with FVB/N mice (F = 50.702; P < 0.001).
Recent studies have used null mutations to identify genes that may regulate or be associated with the cycle of excitotoxic cell death. Specifically, it has been reported that a null mutation of the p53 tumor suppressor gene protects against neurotoxic cell death after KA administration (12). However, the null mutant mice were hybrids (129/Sv × C57BL/6) in which there is a recombination of 129/Sv and C57BL/6 chromosomes. Because one of the parent strains (C57BL/6) does not exhibit excitotoxic cell death at doses less than or equal to the LD50, it is possible that C57BL/6 chromosomes contain genes other than the null mutation that confer protection from cell death. To evaluate this possibility, we evaluated KA-induced cell death in progeny of 129/SvEMS × C57BL/6 created using embryonic stem cells from 129/SvEMS mice. These mice had a mixed 129/SvEMS × C57BL/6 genetic background and were the homozygous wild-type mice (control) with respect to a null mutation created as part of an unrelated study. After a 35 mg/kg dose of KA, all eight mice exhibited seizures, but no evidence of degeneration or cell loss (Fig. 6A), indicating that protection against EAA neurotoxicity is due to the genetic background of the hybrids (specifically, the presence of C57BL/6 chromosomes).
Figure 6.

Susceptibility to excitotoxic cell death is dependent on genetic background. (A) Silver-stained section from a homozygous wild-type hybrid mouse (129/SvEMS × C57BL/6) showing a lack of degenerative debris after KA administration. (B) Selective silver-stained horizontal section from a p53−/− mouse generated in a 129/Sv background illustrating neuronal and terminal degeneration throughout the CA3 (arrows) and CA1 subfields as well as the dentate hilus 7 days after KA administration. (C) Higher magnification of area CA3 and the dentate hilus (arrows). CA3 denotes the hippocampal subfield. [Scale bars, 750 μm (A and B); 200 μm (C).]
The protection from excitotoxicity in the p53 null mutant animals reported in a previous study (12) could have been due either to the null mutation or the presence of particular C57BL/6 genes. Thus, we evaluated whether a null mutation of p53 in a susceptible genotype (129/SvEMS) would convey protection against cell death. We administered KA to mice deficient in the p53 tumor suppressor gene that had been generated in a 129/SvEMS background as described previously (16–21). As shown in Fig. 6B, silver staining revealed extensive degeneration (Fig. 6C), and cell loss was apparent in Nissl-stained sections. After KA administration, extensive (70–80%) cell loss was observed in the dentate hilus and area CA3 and was also evident (50% cell loss) in area CA1 (F = 3.93; P < 0.001). Thus, null mutations of the p53 gene are not sufficient to confer protection against KA-induced cell death.
DISCUSSION
Four commonly used inbred mouse strains demonstrated a dramatic difference in susceptibility to KA-induced excitotoxic cell death. C57BL/6 and BALB/c mice were virtually invulnerable to KA-induced cell death (except at doses well above the LD50), whereas FVB/N and 129/SvEMS mice exhibited excitotoxic cell death similar to what is seen in rats. These results demonstrate that C57BL/6 and BALB/c strains carry gene(s) that confer protection from glutamate-mediated excitotoxicity.
The virtual invulnerability in the two strains cannot be explained by decreased alterations in the extent of seizure activity, because all mice exhibited the same class of behavioral seizures. Moreover, the pattern and extent of neuronal activity was comparable as revealed by 2DG autoradiography. Thus, differences in cell death are not due to differences in blood brain barrier permeability or differences in the pattern of neuronal activity during the seizures. The absence of cell death could, however, be due to differences in EAA receptor function based on the selective cell loss and degeneration that was observed in several C57BL/6 mice after injection of KA at doses exceeding the LD50. It previously has been reported that at high doses, KA has effects that do not seem to be mediated via conventional glutamate receptors (25, 26). This might explain why the pattern of cell loss and degeneration observed in the select C57BL/6 mice is not qualitatively similar to what has been observed in other mouse strains or in rats.
The finding that two commonly used inbred mouse strains are more resistant to KA-induced excitotoxicity has important implications for studies using these strains to examine neuronal death. It may be that these strains, and any others with similar resistance, will show very different responses in any situation in which excitotoxic cell death plays a role (for example, after brain or spinal cord injury, transient ischemia, or epileptogenesis). In this regard, it is of considerable interest that C57BL/6 mice exhibit a substantially different response to spinal cord injury than rats (27, 28)—a situation in which excitotoxicity is thought to play an important role (29).
Our results documenting strain differences in susceptibility to excitotoxic cell death also bear on recent data examining genes involved in excitotoxicity using null mutations. An example of this can be seen in the case of null mutations of the p53 tumor suppressor gene. It has been reported that deletion of the p53 gene in animals of a mixed genetic background (129/Sv × C57BL/6) conferred protection against KA-induced degeneration (12). Moreover, our findings in hybrids created with embryonic stem cell technology indicate that some subset of genes in the 129/SvEMS × C57BL/6 hybrids is sufficient to confer protection from excitotoxicity. These results indicate that the presence of some subset of C57BL/6 genes in the 129/SvEMS × C57BL/6 hybrids is sufficient to convey protection from excitotoxicity. It is important to note that Morrison et al. (12) did report that hybrid animals that were wild type with respect to the null mutation did exhibit excitotoxic cell death. The differences in susceptibility to KA in animals with mixed C57BL/6 and 129/SvEMS genes may be due to different recombination patterns due to chromosome sorting in the hybrids. In addition, it is possible that modifying elements present within the hybrids may affect processes that involve the null gene. These results substantiate previous concerns regarding the evaluation of the effects of null mutations in animals with a mixed genetic background (30).
A number of recent gene targeting studies have reported effects of null mutations on processes that are triggered by or related to glutamate-mediated excitotoxic cell death. Importantly, all of these have used animals with mixed genetic backgrounds in which there may be a contribution of C57BL/6 genes. For example, null mutations of nitric oxide synthase in 129/SvEMS × C57BL/6 hybrids have been reported to eliminate excitotoxic cell death of cortical neurons in culture after exposure to N-methyl-d-aspartate—an in vitro model of excitotoxicity (12). Another study has evaluated the consequences of a null mutation of c-fos on synaptic reorganization after seizure-induced excitotoxic death of hippocampal neurons—again in C57BL/6 × 129/SvEMS hybrids (14). Although the mechanism of elicitation of excitotoxicity differs from the paradigm used in our study, our data suggests that the use of the hybrids with C57BL/6 genes might influence the resultant phenotype. These findings will have to be reevaluated in light of the present study. To confirm the role that these genes play in excitotoxicity or synaptic reorganization, it will be necessary to evaluate the consequences of null mutations in mice with a pure genetic background.
The fact that C57BL/6 and BALB/c inbred strains possess gene(s) that confer protection against KA-induced excitotoxicity raises the question of whether similar genes exist in other species, especially humans. Such genes could account for differences in susceptibility to neurodegenerative disorders, seizure disorders, and the response to trauma. It obviously will be of considerable interest to identify the gene(s) in mice that are responsible for resistance.
Acknowledgments
We thank S. Pearson-White for supplying the homozygous wild-type mice used in this study and H. Scrable for helpful discussions. This work was supported by National Institutes of Health Grant NS29875 to O.S. and P.E.S. was the recipient of National Research Service Award Fellowship NS09927.
ABBREVIATIONS
- EAA
excitatory amino acid
- KA
kainic acid
- 2DG
2-deoxy[14C]glucose
References
- 1.A. A, Horrocks L A. Brain Res Rev. 1991;16:171–191. doi: 10.1016/0165-0173(91)90004-r. [DOI] [PubMed] [Google Scholar]
- 2.Rothman S M, Olney J W. Trends Neurosci. 1987;10:299–302. [Google Scholar]
- 3.Choi D W. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 4.Hori N, French-Mullen J H M, Carpenter D O. Brain Res. 1985;358:380–384. doi: 10.1016/0006-8993(85)90989-8. [DOI] [PubMed] [Google Scholar]
- 5.Choi D W. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 6.Nadler J V, Perry B W, Gentry C, Cotman C W. J Comp Neurol. 1980;192:333–359. doi: 10.1002/cne.901920209. [DOI] [PubMed] [Google Scholar]
- 7.Nadler J V, Cuthbertson G J. Brain Res. 1980;195:47–56. doi: 10.1016/0006-8993(80)90865-3. [DOI] [PubMed] [Google Scholar]
- 8.Sperk G, Lasmann H, Baran H, Kish S J, Seitelberger F, Hornykiewicz O. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Ari Y. Neuroscience. 1985;14:35–43. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- 10.Nadler J V. Life Sci. 1981;29:2031–2042. doi: 10.1016/0024-3205(81)90659-7. [DOI] [PubMed] [Google Scholar]
- 11.Dawson V L, Kizushi V M, Huang P L, Snyder S H, Dawson T M. J Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison R S, Wenzel H J, Kinoshita Y, Robbins C A, Donehower L A, Schwartzkroin P A. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe Y, Johnson R S, Butler L S, Binder D K, Speigelman B M, Papaioannou V E, McNamara J O. J Neurosci. 1996;16:3827–3836. doi: 10.1523/JNEUROSCI.16-12-03827.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Racine R J. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 15.Fink R P, Heimer L. Brain Res. 1967;4:369–374. doi: 10.1016/0006-8993(67)90166-7. [DOI] [PubMed] [Google Scholar]
- 16.Livingstone L R, White A, Sprouse J, Livanos E, Jacks T, Tisty T D. Cell. 1992;70:923. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 17.Jacks T, Remington L, Williams B O, Schmitt E M, Halachmi S, Bronson R T, Weinberg R A. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 18.Kastan M B, Zhan Q, el-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J., Jr Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 19.Lowe S W, Schmitt E M, Smith S W, Osborne B A, Jacks T. Nature (London) 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 20.Strasser A, Harris A W, Jacks T, Cory S. Cell. 1994;79:329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 21.Ziegler A, Jonason A S, Leffell D J, Simon J A, Sharma H W, Kimmelman J, Remington L, Jacks T, Brash D E. Nature (London) 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 22.Lothman E W, Collins R C. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- 23.Sloviter R S, Dempster D W. Brain Res Bull. 1985;15:39–60. doi: 10.1016/0361-9230(85)90059-0. [DOI] [PubMed] [Google Scholar]
- 24.Swanson L W, Wyss J M, Cowan W M. J Comp Neurol. 1978;181:681–716. doi: 10.1002/cne.901810402. [DOI] [PubMed] [Google Scholar]
- 25.Balchen T, Berg M, Diemer N H. Neurosci Lett. 1993;163:151–154. doi: 10.1016/0304-3940(93)90369-v. [DOI] [PubMed] [Google Scholar]
- 26.Robinson J H, Deadwyler S A. Brain Res. 1981;221:117–127. doi: 10.1016/0006-8993(81)91067-2. [DOI] [PubMed] [Google Scholar]
- 27.Fujuki M, Zhang Z, Guth L, Steward O. J Comp Neurol. 1996;317:469–484. doi: 10.1002/(SICI)1096-9861(19960729)371:3<469::AID-CNE9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Fujuki M, Guth L, Steward O. J Comp Neurol. 1996;371:485–495. doi: 10.1002/(SICI)1096-9861(19960729)371:3<485::AID-CNE10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 29.Liu D, Thangnipon W, McAdoo D J. Brain Res. 1991;547:344–348. doi: 10.1016/0006-8993(91)90984-4. [DOI] [PubMed] [Google Scholar]
- 30.Gerlai R. Trends Neurosci. 1996;19:177. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]