

Abstract
In Streptococcus pneumoniae, competence and bacteriocin genes are controlled by two two-component systems, ComED and BlpRH, respectively. In Streptococcus mutans, both functions are controlled by the ComED system. Recent studies in S. mutans revealed a potential ComE binding site characterized by two 11 bp direct repeats shared by each of the bacteriocin genes responsive to the competence-stimulating peptide (CSP). Interestingly, this sequence was not found in the upstream region of the CSP structural gene comC. Since comC is suggested to be part of a CSP-responsive and ComE-dependent autoregulatory loop, it was of interest to determine how it was possible that the ComED system could simultaneously regulate bacteriocin expression and natural competence. Using the intergenic region IGS1499, shared by the CSP-responsive bacteriocin nlmC and comC, it was demonstrated that both genes are likely to be regulated by a bifunctional ComE. In a comE null mutant, comC gene expression was increased similarly to a fully induced wild-type. In contrast, nlmC gene expression was nearly abolished. Deletion of ComD exerted a similar effect on both genes to that observed with the comE null mutation. Electrophoretic mobility shift assays (EMSAs) with purified ComE revealed specific shift patterns dependent on the presence of one or both direct repeats in the nlmC–comC promoter region. The two direct repeats were also required for the promoter activity of both nlmC and comC. These results suggest that gene regulation of comC in S. mutans is fundamentally different from that reported for S. pneumoniae, which implicates a unique regulatory mechanism that allows the coordination of bacteriocin production with competence development.
INTRODUCTION
Streptococcus mutans is considered to be the primary pathogen causing dental caries (tooth decay). In addition to the known virulence factors of S. mutans, such as biofilm formation, acid production/tolerance and competence development, the ability to produce bacteriocin (mutacin) is thought to play an important role in the establishment of S. mutans in the dental biofilm community (Kreth et al., 2005; Qi et al., 2001; Yonezawa & Kuramitsu, 2005). Currently, two types of mutacins have been characterized, the lantibiotics and the non-lantibiotics (Balakrishnan et al., 2000; Chen et al., 1998, 1999; Hillman et al., 1998; Mota-Meira et al., 1997; Qi et al., 1999a, b, 2000, 2001; Yonezawa & Kuramitsu, 2005). Lantibiotics are ribosomally synthesized and post-translationally modified peptides that contain lanthionine or β-methyllanthionine as well as dehydrated amino acids (McAuliffe et al., 2001; Twomey et al., 2002), whereas non-lantibiotics consist of either one or two small unmodified peptides (Balakrishnan et al., 2000; Qi et al., 2001).
Production of mutacin is initiated at the onset of stationary phase or under conditions of high cell density, allowing the mutacin producers to kill other competitive bacterial species when nutrients become limited. As with other stress responses, mutacin production is stringently regulated. Recent work in our laboratory has demonstrated that production of the lantibiotic mutacin I is regulated by multiple inputs (Tsang et al., 2005, 2006), while production of the non-lantibiotic mutacin IV is controlled by the ComED two-component system in response to the competence-stimulating peptide (CSP) (Kreth et al., 2005). Further studies demonstrated that mutacin IV production and competence development are temporally coordinated, with a 2 h delay in competence development following activation of mutacin IV gene expression by CSP (Kreth et al., 2005).
During our studies of ComED regulation of mutacin IV, we found that in addition to mutacin IV (nlmA), a group of mutacin or mutacin-like genes are also regulated by the ComED system (Kreth et al., 2006). Similar findings have also been reported previously (van der Ploeg, 2005). These CSP-responsive genes include nlmC (Smu1738/SMU.1914C), encoding the newly identified mutacin V (Hale et al., 2005), Smu1731/SMU.1906C and Smu0384/SMU.423, encoding non-lantibiotic mutacin-like peptides, and Smu0839/SMU.925, encoding a protein homologous to bacteriocin immunity proteins. A common feature of these genes is that their promoters each contain a putative ComE binding site sequence (Kreth et al., 2006; van der Ploeg, 2005). More interestingly, the nlmC gene shares the intergenic region with the divergently transcribed comC. This finding raised a puzzling question: since the putative ComE binding sequence resides only on the DNA strand encoding the nlmC promoter, could ComE also regulate comC gene expression? In this study we demonstrate that ComE does indeed bind to the putative ComE binding site. However, unlike in Streptococcus pneumoniae, in S. mutans, ComE can function simultaneously as an activator of nlmC and repressor of comC.
METHODS
Bacterial strains and culture conditions
Bacterial strains used in this study and their relevant characteristics and genotypes are listed in Table 1. All S. mutans derivatives were grown in Brain Heart Infusion (BHI, Difco) or on BHI agar plates. For the selection of antibiotic resistant clones, BHI plates were supplemented with either spectinomycin (Spc; 800 μg ml–1) or erythromycin (Erm; 15 μg ml–1). All S. mutans strains were grown anaerobically (90% N2/5% CO2/5% H2) at 37 °C. For cloning purposes and plasmid proliferation Escherichia coli cells were used. Cells were grown in LB medium supplemented with spectinomycin (100 μg ml–1) or ampicillin (100 μg ml–1) aerobically at 37 °C.
Table 1.
Bacterial strains used in this study
| Strain | Relevant characteristics | Reference(s) |
|---|---|---|
| E. coli | ||
| BL21(DE3)pLys | F- ompT gal hsdSB dcm lon λDE3 pLysS | Studier et al. (1990) |
| S. mutans | ||
| UA159 | Wild-type SpcS ErmS | Ajdic et al. (2002) |
| UA159E | UA159 comE ErmR | This work |
| UA159D | UA159 comD ErmR | This work |
| SMCD1 | comD ErmR | Li et al. (2001) |
| SMCE-L1 | comE ErmR | Li et al. (2001) |
| JK1738−159 | UA159 : : Φ(nlmCp—luc) SpcR | This work |
| JK1738−159D | JK1738−159 comD ErmR | This work |
| JKC159 | UA159 : : Φ(comCp—luc) SpcR | This work |
| JK-DR1738 | UA159 : : Φ(nlmCpΔDRI+II—luc) SpcR deletion of DRI+II in nlmCp | This work |
| JK-DRC | UA159 : : Φ(comCpΔDRI+II—luc) SpcR deletion of DRI+II in comCp | This work |
DNA manipulation
Standard procedures were used for plasmid isolation, DNA digestion and analysis and cloning. PCR was performed with a MyCycler thermocycler (Bio-Rad) using a protocol from the supplier.
Construction of nlmC–luc reporter gene fusion
The backbone vector for the construction of all reporter gene fusions was pFW5–luc, which contains a spectinomycin resistance marker (aad9) that works in both Gram-negative and Gram-positive bacteria (Podbielski et al., 1996). The promoter region of nlmC was amplified by PCR from the chromosomal DNA of strain UA159. Primers used for the amplification incorporated restriction enzyme sites for BamHI as well as SalI and are listed in Table 2. The promoter region was then cloned into pFW5–luc to generate the plasmid pFW5 : : Φ(nlmCp–luc). The plasmid was then transformed into S. mutans strain UA159 and integrated into the chromosome via single crossover recombination. Transformants were selected on BHI+Spc plates and confirmed by PCR and reporter gene activity.
Table 2.
Primers used in this study
| Primer name | Sequence | Gene targeted |
|---|---|---|
| 1738BamHI | 5′-GGATCCCACCTTCAACAGCTGAAAGTGC-3′ | nlmC promoter region |
| 1738SalI | 5′-GTCGACTAAAACTTCTGTTAAACAGCCGG-3′ | nlmC promoter region |
| 1738-RT-F | 5′-ATGGATAATGAAGCACTTTCAGC-3′ | RT-PCR nlmC |
| 1738-RT-R | 5′-ATAACCTTGCCCAGCACCTA-3′ | RT-PCR nlmC |
| comC-RT-F | 5′-GACTGATGAATTAGAGATTATCATTGG-3′ | RT-PCR comC |
| comC-RT-R | 5′-TTTCCCAAAGCTTGTGTAAAACT-3′ | RT-PCR comC |
| ComE inv | 5′-TGGTGATTGCCCTTTTCAG-3′ | comE primer |
| Delta-BS-F | 5′-ATATTTTGCTCCATTTTGAAAATAAATTG-3′ | DR deletion |
| Delta-BS-R | 5′-CTATTTTGTCCTAAACGGTCATTTTTG-3′ | DR deletion |
| Delta-BS-R1 | 5′-GATGTTCTGAAACTATCATTACCAGC-3′ | DR deletion |
| 1738-comC-B | 5′-GGATCCTAAAACTTCTGTTAAACAGCCGG-3′ | promoter flip nlmC to comC |
| 1738-comC-S | 5′-GTCGACCACCTTCAACAGCTGAAAGTGC-3′ | promoter flip nlmC to comC |
Construction of comE and comD mutants
The comE and comD defective strains were constructed by transferring chromosomal DNA from previously constructed mutant strains [SMCE1 ComE– ErmR and SMCD1 ComD– ErmR (Li et al., 2001)] into the nlmC–luc reporter strain. Transformants were confirmed by PCR.
Construction of direct repeat I (DR I) and direct repeat II (DR II) deletions
Deletion of the DR I and DR II regions of the nlmC promoter was performed by inverse PCR using pFW5 : : Φ(nlmCp–luc) as the template and primers Delta-BS-F, Delta-BS-R and Delta-BS-R1 (Table 2). Inverse PCR with Delta-BS-F and Delta-BS-R would create a deletion of DR II, and with Delta-BS-F and Delta-BS-R1 would create a deletion of both DR I and DR II (Fig. 1a). The PCR fragments were then religated and transformed into E. coli. The correct plasmid constructs [pFW5 : : Φ(nlmCpΔDRII–luc)159 and pFW5 : : Φ(nlmCpΔDRI+II–luc)159] were confirmed by restriction enzyme digestion followed by DNA sequencing. The plasmids were then transformed into UA159 and integrated into the nlmC promoter region via single crossover recombination.
Fig. 1.

Intergenic region IGS1499. (a) Nucleotide sequence of the intergenic region (IGS1499) between nlmC (Smu1738) and comC, including the ATG codon for each gene. The top strand is the coding strand for comC, and the bottom strand is the coding strand for nlmC. The putative extended –10 and –35 regions for nlmC are highlighted with boxes. The two direct repeats (DR I and DR II) and their directions are indicated by arrows. Brackets denote the regions that were deleted in the DR II and the DR I+II deletion mutants. (b) Putative ComE binding site in the promoter region of comED (S. mutans) and putative ComE and BlpR binding sites in the promoter regions of comCDE and blpS (S. pneumoniae) and their relative arrangement on the chromosome [modified after (Martin et al., 2006)]. The imperfect direct repeats are underlined in all three sequences and separated by a 10 bp spacer (11 bp in comCDE in S. pneumoniae). The light grey box indicates the location of the putative ComE or BlpR binding sites. Sm, S. mutans; Sp, S. pneumoniae.
To create comC–luc fusion lacking the same direct repeats, the promoter fragment in pFW5 : : Φ(nlmCpΔDRI+II–luc)159 was amplified with the primers 1738comC-B and 1738comC-S, which generated the BamHI and SalI restriction sites for reversal of the promoter region, thus creating the plasmid pFW5 : : Φ(comCpΔDRI+II–luc)159. Plasmid pFW5 : : Φ(comCpΔDRI+II–luc)159 was subsequently integrated into the chromosome of UA159 to create a comC–luc reporter strain lacking the two direct repeats in the upstream region.
Luciferase assay
Luciferase assays were performed using a method similar to one described previously (Loimaranta et al., 1998). Briefly, 25 μl 1 mM d-luciferin (Sigma) suspended in 100 mM citrate buffer, pH 6, was added to 100 μl cell culture. To ensure sufficient levels of intracellular ATP, cells were recharged with 1 % glucose for 10 min prior to luciferin addition. Luciferase activity was measured using a TD 20/20 luminometer (Turner Biosystems). Usually, three parallel cultures were measured at each time point and the mean value was taken. Each experiment was repeated at least two times.
RNA extraction and real-time RT-PCR
Overnight cultures of UA159 and derivatives were diluted 1 : 40 in 200 ml BHI (no antibiotics) and grown for 2–3 h until OD600 reached approx. 0.2. If CSP induction was required, synthetic CSP (1μg ml–1) was added to the culture (Kreth et al., 2005). The cells were further incubated for 2 h until OD600 was between 0.5 and 0.6, a time point where comC induction is at the maximum. Cells were harvested by centrifugation and stored at –80 °C until needed. For RNA extraction, cDNA synthesis and real-time RT-PCR a procedure described previously (Merritt et al., 2005) was followed, using the iCycler(Bio-Rad) real-time PCR System. Total cDNA abundance between test samples was normalized using the 16S RNA gene as a housekeeping control.
ComE purification and electrophoretic mobility shift assays (EMSAs)
The comE coding region was PCR amplified from S. mutans strain UA159, and cloned into pCT T7/NT Topo vector (Invitrogen) with a 6-histidine tag at the N terminus. The vector was transformed into E. coli strain BL21(DE3)pLys. ComE protein was overexpressed by induction with IPTG at a final concentration of 1 mM. Cells were harvested and lysed with lysozyme (8 mg ml–1), followed by sonication (Branson sonifier 450, VWR). Cell lysates were centrifuged at 7800 g at 4 °C for 30 min. For ComE protein purification, cleared cell lysate was mixed with Pro-Bond resin (Invitrogen) and shaken at 4 °C for 1 h. The resin was allowed to settle by gravity to separate the bound ComE and the non-specific proteins in the buffer. The non-specific proteins were secured for later analysis by PAGE. The column was washed four times with 8 ml native washing buffer [native binding buffer (50 mM NaH2PO4, 500 nM NaCl, 10mM Tris/HCl, pH8, 10 %,v/v, glycerol and 5 mM β-mercaptoethanol)+20 mM imidazole]. Purified protein was eluted with 10 ml native elution buffer (native binding buffer+250 mM imidazole). Eluted fractions (1 ml each) were collected. Each eluted fraction had glycerol added to 20 % (v/v) and was quick-frozen in dry ice/methanol before storage at –80 °C. All the fractions were saved and analysed, by running 4–20% Tris-glycine gel (Invitrogen) at 150 V, 1.5 h, for ComE purity. The ComE concentration was determined from a standard curve with BSA standards. Each gel was stained with colloidal blue staining kit (Invitrogen).
For radioactive probe labelling, 1 μM oligonucleotide probe oSG316 (CCCATTTTTAGTTTTTTGTCTG) was labelled at the 5′-end with 0.85 μM [γ-32P]ATP (10 mCi ml–1, 370 MBq ml–1; New Life Science Products) by using T4 polynucleotide kinase (Promega). The labelled primer was used with primer oSG317 (GAAAAAATCATGGATTTCTTG) in a PCR to amplify promoter regions of comC wild-type (204 bp), comCpΔDRII (192 bp) and comCpΔDRI+II (166 bp). For EMSA, ComE (10 nM) was incubated at room temperature for 30 min in ComE dilution buffer (50 mM HEPES, pH 6.5, 1 mM EDTA, 10 % glycerol), 5 × EMSA buffer (45 % glycerol, 250 mM HEPES, pH 6.5, 250 μg BSA ml–1), with varying concentrations of salmon sperm DNA and 1 nM isotopically labelled DNA substrate. Following incubation, EMSA reactions were analysed on 6% non-denaturing polyacrylamide gels in electrophoresis buffer (0.5 × TBE) that was pre-run for at least 2 h at 150 V constant voltage. Nucleoprotein complexes were separated by PAGE at 150 V for 3 h and subsequently dried and scanned with the Bio-Rad imaging system.
RESULTS
ComE is required for nlmC transcription but represses comC gene expression
As a first step towards understanding how the ComED system co-ordinates the temporal expression of nlmC with competence development, we analysed the nlmC–comC intergenic region, IGS1499, and found that it contained two 11 bp direct repeats (DR) separated by a 10 bp spacer (Fig. 1a). This region is localized immediately upstream of the predicted –35 region of nlmC and, like the ComE binding site for S. pneumoniae, is a strong match to the AlgR/AgrA/LytR family binding sites (Fig. 1b) (Nikolskaya & Galperin, 2002). Since this sequence feature exists only on the DNA strand encoding the promoter of nlmC and not that of comC, we were curious as to whether ComED also regulated comC expression. To test this, real-time RT-PCR was performed in wild-type UA159. As shown in Fig. 2(a), comC gene expression increased ∼5-fold in response to CSP. Similar results were also obtained with two other wild-type strains NG8 and UA140 (data not shown). These results were surprising given that the putative ComE binding sequence was not located in the comC promoter.
Fig. 2.
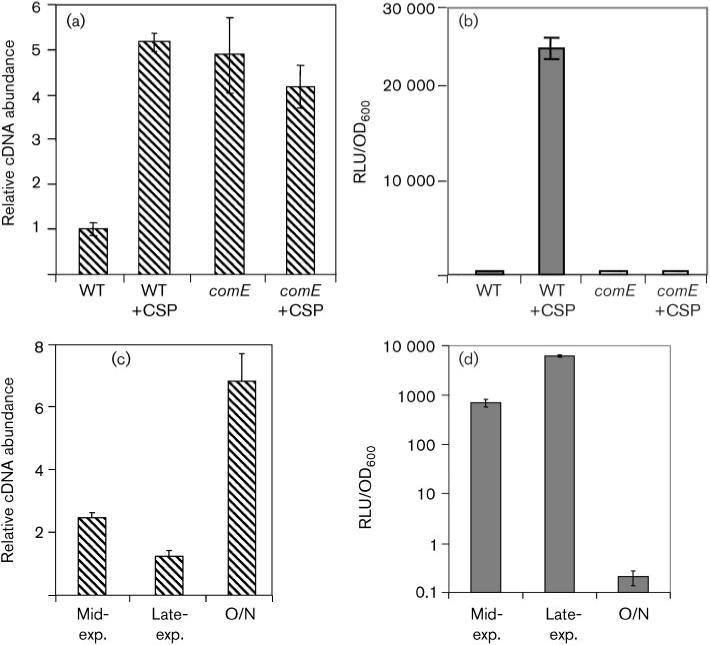
Role of ComE in nlmC and comC gene expression. (a) Expression of comC in wild-type and comE mutant backgrounds with and without addition of CSP. Cells were harvested at late exponential phase. Transcript levels of comC were measured by real-time RT-PCR using 16S RNA as a housekeeping control. cDNA abundance was normalized against the 16S cDNA and expressed as relative cDNA abundance. The wild-type level without CSP was arbitrarily set as 1. (b) nlmC expression in wild-type and comE mutant backgrounds with and without CSP. Cells were harvested at late exponential phase. Transcript levels were measured using the nlmC–luc reporter and normalized against cell density. RLU, relative light units. (c) comC expression over time. comC transcription was measured at mid-exponential phase (2 h after initial dilution from overnight culture), late exponential phase (3 h after initial culture dilution), and after overnight (O/N) incubation in competence-developing medium without CSP. The fold difference in expression with time point 0 h set to 1 is presented. (d) nlmC–luc gene expression in the reporter strain JK1738−159 at mid-exponential phase, late exponential phase and after overnight growth. To measure luciferase activity in the overnight (O/N) culture, cells were recharged with pre-warmed fresh medium (1 : 3 dilution) for 30 min. Data presented in (a), (b), (c) and (d) were collected from at least two independent experiments done in triplicate.
To further investigate, we performed a similar experiment to measure comC gene expression in a comE mutant background in the presence and absence of CSP. To our surprise, comC expression increased approximately fivefold in the comE mutant background as compared to the uninduced wild-type (Fig. 2a). This was the same level of comC induction observed in response to CSP addition in the wild-type. In contrast, the expression of nlmC was nearly abolished in the comE mutant background (Fig. 2b). This result suggested that in the absence of CSP induction ComE acts as a repressor of comC gene expression, whereas ComE is required for nlmC transcription.
As a further verification of the repressor function of comC, we measured nlmC and comC expression at growth stages in which comE expression was either high or low. The expression pattern of comC was measured by real-time RT-PCR in competence development medium in the absence of exogenously added CSP. As shown in Fig. 2(c), comC gene expression was higher in mid-exponential phase than in late exponential phase, whereas nlmC gene expression was much higher in late exponential phase than in mid-exponential phase. The most dramatic difference between the two promoters was observed in overnight cultures. At this stage, comC gene expression was three times that achieved in mid-exponential phase; in contrast, nlmC gene expression dropped to nearly undetectable levels (Fig. 2d). These results further supported a bifunctional role for ComE that could simultaneously activate nlmC and repress comC.
Deletion of comD also has an opposing effect on nlmC and comC gene expression
According to the model of how the ComED two-component system operates, binding of CSP to ComD triggers a phosphorylation cascade comprising autophosphorylation of ComD then phosphorylation of ComE. Phosphorylated ComE then activates or represses its target genes (Alloing et al., 1998; Morrison & Lee, 2000). Based on this model, we predicted that in a comD mutant background comC expression would be unresponsive to addition of CSP, while the wild-type would exhibit the expected increase in comC expression. A similar expression pattern was predicted for nlmC. As shown in Fig. 3(a), comC expression in the comD mutant was similar to that in the wild-type in the absence of CSP, but was reduced >2-fold in the presence of CSP. We speculate that the slightly higher comC expression in the comD background is probably due to reduced comE expression (i.e. less ComE repressor) resulting from autoregulation through the ComED circuit (J. Merritt & F. Qi, unpublished).
Fig. 3.

Effect of a comD mutation on comC and nlmC gene expression. (a) Expression of comC in wild-type and comD mutant backgrounds with and without CSP. Transcript levels were measured by real-time RT-PCR using 16S RNA as a housekeeping control. cDNA abundance was normalized against the 16S cDNA and expressed as relative cDNA abundance. The wild-type level without CSP was arbitrarily set as 1. (b) nlmC expression in wild-type and comD mutant backgrounds with and without CSP. Transcript levels were measured using the nlmC–luc reporter and normalized against cell density. Values and standard deviation are calculated from at least two independent experiments performed in triplicate.
The effect of the comD mutation on nlmC expression was measured with a luciferase reporter gene fusion. As shown in Fig. 3(b), nlmC expression in the comD mutant was reduced >40-fold compared with the wild-type in the presence of CSP. In the comD mutant, the >4-fold reduction of nlmC gene expression relative to the uninduced wild-type was probably caused by the same decrease in comE gene expression mentioned above (Fig. 3b). Taken together, these results further support the notion that ComE is a repressor of comC expression but is required for nlmC gene expression.
The direct repeats in the nlmC–comC promoter region are required for ComE binding
Based on our initial results, we hypothesized that the two direct repeats, DR I and DRII (Fig. 1), in the nlmC–comC intergenic region would be required for ComE binding in S. mutans. To test this, the ComE protein was purified from E. coli and tested for binding to the nlmC–comC intergenic region in an EMSA. A 204 bp DNA fragment encompassing a region 17 bp upstream of the nlmC and 46 bp upstream of the comC initiation codons was used as the substrate for ComE binding. As shown in Fig. 4, ComE bound to the wild-type DNA in a concentration-dependent manner. At a molar ratio of 50 : 1 ComE/DNA (5 nM : 0.1 nM), there were two shifted bands, with the lower band more abundant than the upper band (lane 2). When the ComE concentration was increased to 15 nM, there was a conspicuous increase in the abundance of the upper band (lane 3). These results suggested that the two direct repeats may serve as two binding sites for ComE. To further test this, we constructed two deletion mutations of the direct repeat region; one contained a complete deletion of DR II (ΔDRII, see Fig. 1), and the other contained a complete deletion of both DRI and DRII (ΔDRI+II, see Fig. 1). DNA was PCR amplified from both mutants, and EMSA was performed under the same conditions as for the wild-type. As shown in Fig. 4, deletion of DRII resulted in the generation of only the lower shifted band with both concentrations of ComE (lanes 5 and 6). This result suggested that this deletion mutant provided only one binding site. As expected, deletion of both direct repeats abolished ComE binding completely, as shown by the unshifted bands (lanes 7–9). These results demonstrate that the two direct repeats in the nlmC–comC promoter region are required for ComE binding, although determining the molecular mechanism of how ComE binds to the two direct repeats will require further experiments.
Fig. 4.
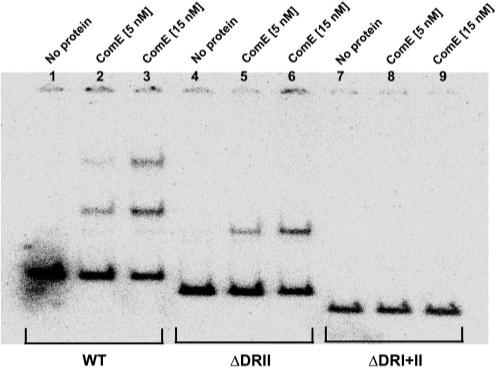
EMSA of the ComE protein with comC wild-type, comCΔDRII and comCΔDRI+II DNA fragments. EMSA analysis was carried out using purified ComE protein with 1 nM radiolabelled DNA probe. The reactions were run on a 6 % non-denaturing polyacrylamide gel and the image was scanned with a Bio-Rad imaging machine. The experiments were repeated three times with similar results. A representative gel image is shown.
The direct repeats in the intergenic region are also required for nlmC and comC gene expression
The comE mutation data presented in Fig. 2 demonstrated that an intact ComE is a repressor for comC, but an activator for nlmC expression. The data presented in Fig. 4 demonstrated that the two direct repeats in the nlmC–comC promoter region are required for ComE binding. Based on these findings, we predicted that deletion of the two direct repeats would diminish nlmC gene expression but derepress comC expression. To test this, luciferase reporter fusions lacking the two direct repeats were constructed for both nlmC and comC. Luciferase activity was measured using the wild-type as a control. Cells were grown to late exponential phase, when expression levels of both promoters can be measured and compared in the absence of CSP. As shown in Fig. 5, deletion of DR I+II reduced nlmC gene expression ∼20-fold as was expected (Fig. 5a). Surprisingly, deletion of the two direct repeats nearly completely abolished comC gene expression (Fig. 5b). These results were rather puzzling since the two direct repeats are located 130 bp upstream of the initiation codon for comC. To find a possible explanation for this unexpected result, we tried to localize the comC transcriptional start site by RACE PCR and RT-PCR. Despite multiple attempts, these experiments have so far yielded ambiguous results. It appeared that none of the predicted promoter sequences served as classical promoters for comC; rather, the comC transcript appeared to start further upstream, even beyond the promoter region for nlmC (J. Merritt & F. Qi, unpublished results). Therefore, a mechanistic explanation of how ComE regulates comC gene expression would require additional research.
Fig. 5.

Effects of the DRI+II deletion on nlmC and comC gene expression. (a) Luciferase gene expression driven by the wild-type and the DR I+II-deleted nlmC promoter. Cells were harvested at late exponential phase. Note the log scale of the y-axis. (b) Luciferase gene expression driven by the wild-type and the DR I+II-deleted comC promoter. Cells were harvested at the late exponential phase. All values are the mean of at least two independent experiments done in triplicate.
DISCUSSION
The objective of this study was to determine how ComE co-ordinates the temporal gene expression of both nlmC and comC. Both luciferase gene fusion and real-time RT-PCR were used to characterize promoter activities of nlmC and comC in different mutant backgrounds. EMSAs were used to analyse ComE binding to the wild-type and DR deletion mutants of the nlmC–comC intergenic region. We demonstrated that ComE is required for both activation of nlmC transcription and repression of comC gene expression. Furthermore, we showed that the two 11 bp direct repeats in the upstream regions of nlmC and comC are required for ComE binding to the promoter region, suggesting that the two direct repeats may serve as the binding sites for ComE.
The strongest evidence supporting the bifunctionality of ComE in regulating nlmC and comC gene expression came from the mutagenesis studies of comE. In a comE null mutant background, nlmC gene expression was nearly abolished, whilst comC gene expression was increased to the same level as in CSP-induced wild-type cells (Fig. 2). Further support for ComE bifunctionality came from the expression pattern analyses of nlmC and comC. In overnight cultures, when ComE was presumably absent, comC gene expression displayed its strongest induction; in contrast, nlmC gene expression was reduced to below detectable levels. In addition, the differential response of nlmC and comC to CSP induction could also add further support to the opposing regulatory mechanisms for nlmC and comC gene expression via ComE. For example, the maximal induction for nlmC is usually between 50- and 100-fold (Fig. 2b), whereas the maximal induction for comC is usually between 3- and 5-fold (Fig. 2a). In addition to the potentially dissimilar promoter strengths, an additional factor in the different levels of CSP induction could result from the positive feedback loop regulation of nlmC gene expression vs the derepressive regulation of comC gene expression. Unlike nlmC induction, the derepression of comC in response to CSP could not be further enhanced by autoinduction of the comED operon.
As a further support for the direct role of ComE in nlmC and comC gene regulation, purified ComE was shown to bind to the nlmC–comC intergenic region. This binding was also dependent upon the presence of two direct repeats, which are reminiscent of the S. pneumoniae ComE binding site. In the presence of both direct repeats, there are two shifted bands, presumably reflecting binding of ComE to either one or both direct repeats (Fig. 4). Deletion of one direct repeat resulted in only one shifted band, whilst deletion of both direct repeats abolished ComE binding completely. These results suggest that ComE can bind to either direct repeat; however, whether the presence of both direct repeats promotes cooperative binding is not known.
The direct involvement of the two direct repeats in gene expression of nlmC and comC was tested by deletion mutagenesis. Deletion of the two direct repeats diminished nlmC gene expression (Fig. 5). This is consistent with previous findings (van der Ploeg, 2005), which showed that point mutations made in either of the direct repeats severely impaired nlmA gene expression [nlmA has a near-identical promoter region to nlmC (Kreth et al., 2006)]. Unexpectedly, the same direct repeat deletion also abolished comC gene expression. This result is rather puzzling as the two direct repeats exist only on the DNA strand encoding the promoter of nlmC, not that encoding the comC promoter. Two explanations may be entertained at this point. (1) The promoter region of comC may overlap the direct repeats region; thus deletion of the direct repeats may have abolished RNA polymerase binding to the comC promoter. (2) The comC transcript may start further upstream of the direct repeat region and the direct repeat region is required for RNA stability of the comC transcript. Since real-time RT-PCR measures RNA at steady state, rapid degradation of the comC transcript as a result of the DR deletion may have resulted in the nearly undetectable levels of comC–luc gene expression as observed in Fig. 5. Although the detailed mechanisms of how ComE regulates comC gene expression await further research, our data appear to suggest that the mechanism of comC transcription is more complicated than is currently accepted.
Taken together, this study demonstrates that ComE plays a very different regulatory role in S. mutans from that of its homologue in S. pneumoniae. Interestingly, both of the C-termini from the ComE proteins of S. pneumoniae and S. mutans share high homology to the DNA binding domains of AlgR/AgrA/LytR family members. In addition, both proteins bind to direct repeat sequences that match extremely well to the predicted binding sites for the AlgR/AgrA/LytR family members (Nikolskaya & Galperin, 2002). However, despite this similarity, S. mutans has evolved a repressive mode of regulation for comC and a delayed competence response to the addition of CSP. This suggests that the mechanism of competence regulation in S. mutans may be quite distinct from that in S. pneumoniae.
ACKNOWLEDGEMENTS
This work was supported in part by NIH grant R01-DE014757 to F. Q., NIDCR T32 training grant DE007296 to J. M. and a Delta Dental grant WDS78956 to W. S.
Glossary
Abbreviations
- CSP
competence-stimulating peptide
- DR
direct repeat
- EMSA
electrophoretic mobility shift assay
REFERENCES
- Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A. 2002;99:14434–14439. doi: 10.1073/pnas.172501299. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloing G, Martin B, Granadel C, Claverys JP. Development of competence in Streptococcus pneumoniae: pheromone autoinduction and control of quorum sensing by the oligopeptide permease. Mol Microbiol. 1998;29:75–83. doi: 10.1046/j.1365-2958.1998.00904.x. [DOI] [PubMed] [Google Scholar]
- Balakrishnan M, Simmonds RS, Carne A, Tagg JR. Streptococcus mutans strain N produces a novel low molecular mass non-lantibiotic bacteriocin. FEMS Microbiol Lett. 2000;183:165–169. doi: 10.1111/j.1574-6968.2000.tb08952.x. [DOI] [PubMed] [Google Scholar]
- Chen P, Novak J, Kirk M, Barnes S, Qi F, Caufield PW. Structure-activity study of the lantibiotic mutacin II from Streptococcus mutans T8 by a gene replacement strategy. Appl Environ Microbiol. 1998;64:2335–2340. doi: 10.1128/aem.64.7.2335-2340.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Qi F, Novak J, Caufield PW. The specific genes for lantibiotic mutacin II biosynthesis in Streptococcus mutans T8 are clustered and can be transferred en bloc. Appl Environ Microbiol. 1999;65:1356–1360. doi: 10.1128/aem.65.3.1356-1360.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale JD, Ting YT, Jack RW, Tagg JR, Heng NC. Bacteriocin (mutacin) production by Streptococcus mutans genome sequence reference strain UA159: elucidation of the antimicrobial repertoire by genetic dissection. Appl Environ Microbiol. 2005;71:7613–7617. doi: 10.1128/AEM.71.11.7613-7617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman JD, Novak J, Sagura E, Gutierrez JA, Brooks TA, Crowley PJ, Hess M, Azizi A, Leung K. Genetic and biochemical analysis of mutacin 1140, a lantibiotic from Streptococcus mutans. Infect Immun. 1998;66:2743–2749. doi: 10.1128/iai.66.6.2743-2749.1998. other authors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Merritt J, Shi W, Qi F. Co-ordinated bacteriocin production and competence development: a possible mechanism for taking up DNA from neighbouring species. Mol Microbiol. 2005;57:392–404. doi: 10.1111/j.1365-2958.2005.04695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Merritt J, Zhu L, Shi W, Qi F. Cell density- and ComE-dependent expression of a group of mutacin-like genes in Streptococcus mutans. FEMS Microbiol Lett. 2006;265:11–17. doi: 10.1111/j.1574-6968.2006.00459.x. [DOI] [PubMed] [Google Scholar]
- Li YH, Lau PC, Lee JH, Ellen RP, Cvitkovitch DG. Natural genetic transformation of Streptococcus mutans growing in biofilms. J Bacteriol. 2001;183:897–908. doi: 10.1128/JB.183.3.897-908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loimaranta V, Tenovuo J, Koivisto L, Karp M. Generation of bioluminescent Streptococcus mutans and its usage in rapid analysis of the efficacy of antimicrobial compounds. Antimicrob Agents Chemother. 1998;42:1906–1910. doi: 10.1128/aac.42.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Quentin Y, Fichant G, Claverys JP. Independent evolution of competence regulatory cascades in streptococci? Trends Microbiol. 2006;14:339–345. doi: 10.1016/j.tim.2006.06.007. [DOI] [PubMed] [Google Scholar]
- McAuliffe O, Ross RP, Hill C. Lantibiotics: structure, biosynthesis and mode of action. FEMS Microbiol Rev. 2001;25:285–308. doi: 10.1111/j.1574-6976.2001.tb00579.x. [DOI] [PubMed] [Google Scholar]
- Merritt J, Kreth J, Shi W, Qi F. LuxS controls bacteriocin production in Streptococcus mutans through a novel regulatory component. Mol Microbiol. 2005;57:960–969. doi: 10.1111/j.1365-2958.2005.04733.x. [DOI] [PubMed] [Google Scholar]
- Morrison DA, Lee MS. Regulation of competence for genetic transformation in Streptococcus pneumoniae: a link between quorum sensing and DNA processing genes. Res Microbiol. 2000;151:445–451. doi: 10.1016/s0923-2508(00)00171-6. [DOI] [PubMed] [Google Scholar]
- Mota-Meira M, Lacroix C, LaPointe G, Lavoie MC. Purification and structure of mutacin B-Ny266: a new lantibiotic produced by Streptococcus mutans. FEBS Lett. 1997;410:275–279. doi: 10.1016/s0014-5793(97)00425-0. [DOI] [PubMed] [Google Scholar]
- Nikolskaya AN, Galperin MY. A novel type of conserved DNA-binding domain in the transcriptional regulators of the AlgR/AgrA/LytR family. Nucleic Acids Res. 2002;30:2453–2459. doi: 10.1093/nar/30.11.2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbielski A, Spellerberg B, Woischnik M, Pohl B, Lutticken R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS). Gene. 1996;177:137–147. doi: 10.1016/0378-1119(96)84178-3. [DOI] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. Purification of mutacin III from group III Streptococcus mutans UA787 and genetic analyses of mutacin III biosynthesis genes. Appl Environ Microbiol. 1999a;65:3880–3887. doi: 10.1128/aem.65.9.3880-3887.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. Functional analyses of the promoters in the lantibiotic mutacin II biosynthetic locus in Streptococcus mutans. Appl Environ Microbiol. 1999b;65:652–658. doi: 10.1128/aem.65.2.652-658.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. Purification and biochemical characterization of mutacin I from the group I strain of Streptococcus mutans, CH43, and genetic analysis of mutacin I biosynthesis genes. Appl Environ Microbiol. 2000;66:3221–3229. doi: 10.1128/aem.66.8.3221-3229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Chen P, Caufield PW. The group I strain of Streptococcus mutans, UA140, produces both the lantibiotic mutacin I and a nonlantibiotic bacteriocin, mutacin IV. Appl Environ Microbiol. 2001;67:15–21. doi: 10.1128/AEM.67.1.15-21.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Tsang P, Merritt J, Nguyen T, Shi W, Qi F. Identification of genes associated with mutacin I production in Streptococcus mutans using random insertional mutagenesis. Microbiology. 2005;151:3947–3955. doi: 10.1099/mic.0.28221-0. [DOI] [PubMed] [Google Scholar]
- Tsang P, Merritt J, Shi W, Qi F. IrvA-dependent and IrvA-independent pathways for mutacin gene regulation in Streptococcus mutans. FEMS Microbiol Lett. 2006;261:231–234. doi: 10.1111/j.1574-6968.2006.00351.x. [DOI] [PubMed] [Google Scholar]
- Twomey D, Ross RP, Ryan M, Meaney B, Hill C. Lantibiotics produced by lactic acid bacteria: structure, function and applications. Antonie Van Leeuwenhoek. 2002;82:165–185. [PubMed] [Google Scholar]
- van der Ploeg JR. Regulation of bacteriocin production in Streptococcus mutans by the quorum-sensing system required for development of genetic competence. J Bacteriol. 2005;187:3980–3989. doi: 10.1128/JB.187.12.3980-3989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonezawa H, Kuramitsu HK. Genetic analysis of a unique bacteriocin, Smb, produced by Streptococcus mutans GS5. Antimicrob Agents Chemother. 2005;49:541–548. doi: 10.1128/AAC.49.2.541-548.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]