

Abstract
Hedgehog signaling plays an essential role in patterning of the vertebrate skeleton. Here we demonstrate that conditional inactivation of the Kif3a subunit of the kinesin-2 intraflagellar transport motor in mesenchymal skeletal progenitor cells results in severe patterning defects in the craniofacial area, the formation of split sternum and the development of polydactyly. These deformities are reminiscent of those previously described in mice with deregulated hedgehog signaling. We show that in Kif3a-deficient mesenchymal tissues both the repressor function of Gli3 transcription factor and the activation of the Shh transcriptional targets Ptch and Gli1 are compromised. Quantitative analysis of gene expression demonstrates that the Gli1 transcript level is dramatically reduced, whereas Gli3 expression is not significantly affected by kinesin-2 depletion. However, the motor appears to be required for the efficient cleavage of the full-length Gli3 transcription factor into a repressor form.
Keywords: Kinesin, Skeletal patterning, Hedgehog signalling, Intraflagellar transport
Introduction
The kinesin superfamily of proteins (KIFs) are microtubule-associated ATP-dependent motors that mediate intracellular transport of organelles, vesicles, proteins and RNA complexes. To date, a total of 45 members have been identified in mice and humans (Miki et al., 2005). Despite some similarity, fairly distant protein species are represented within the kinesin superfamily, strongly suggesting that a certain degree of transport specificity of individual kinesins may be required to support the highly complex system of intracellular trafficking.
Kif3a was first cloned and described as a member of the kinesin superfamily involved in axonal transport of membrane organelles in neurons (Kondo et al., 1994). In the nervous system, the Kif3a subunit has been found associated with Kif3b, a closely related protein, forming a heterodimeric complex, kinesin-2. Kinesin-2 is a microtubule plus end-directed motor essential for the anterograde transport of membrane organelles (Yamazaki et al., 1995). A third component of kinesin-2, the non-motor globular protein KAP3, binds both the Kif3a/Kif3b motor subunits and the membranous organelles. KAP3 was proposed to regulate loading of cargo organelles or protein complexes for transport (Yamazaki et al., 1996).
Early genetic studies of kinesin-2 function in mammals revealed its important role in defining the initial left-right (L-R) asymmetry of the mouse embryo by maintaining the structure of the cilia present on cells in the embryonic node (Marszalek et al., 1999;Nonaka et al., 1998). Kinesin-2 has also been implicated in transporting material into the connecting cilium of photoreceptors and in mediating fast release of vesicles in the ribbon synapses present in multiple cell types of the retina, including rods, cones and bipolar cells (Beech et al., 1996;Muresan et al., 1999;tom et al., 2005). Studies of kinesin-2 activities in non-motile cilia of the sensory neurons in Caenorhabditis elegans and motile cilia of sea urchin embryos suggest that the motor protein is necessary for the structural and functional integrity of various ciliary structures (Cole et al., 1998;Morris and Scholey, 1997). In melanophores of Xenopus lavis, cytoplasmic dynein and kinesin-2 have been implicated in regulating aggregation and dispersion of the cytoplasmic pigment granules, respectively (Rogers et al., 1997;Tuma et al., 1998).
The initial functional studies of kinesin-2 were performed in highly specialised types of cells, such as neurons. However, analysis of various murine organs and tissues demonstrated a rather broad expression pattern of Kif3a suggesting multiple biological functions (Kondo et al., 1994). In this study, we provide comprehensive analysis of the role kinesin-2 plays during skeletal development in mouse. Consistent with the recently proposed roles of ciliary/basal body proteins in regulation of the hedgehog signaling pathway, inactivation of Kif3a leads to patterning defects in the axial and appendicular skeleton. Moreover, we report first experimental evidence suggesting an essential role of kinesin-2 during craniofacial development.
Materials and methods
Mouse strains
Mice harboring floxed Kif3a alleles in C57BL/6 background were a generous gift from Larry Goldstein and were genotyped as previously described (Marszalek et al., 2000). Dermo1-Cre, Prx1-Cre and Wnt1-Cre deleter strains were kindly provided by Drs. David Ornitz, Cliff Tabin and Andy McMahon, respectively. The presence or absence of the Cre transgenes was determined using PCR with primers specific to the Cre-recombinase coding sequence: Cre5′ (TGC TCT GTC CGT TTG CCG) and Cre3′ (ACT GTG TCC AGA CCA GGC). Genomic tail DNA was amplified by PCR; 35 cycles of denaturation at 94°C for 30 seconds, annealing at 58°C for 60 seconds, and elongation for 90 seconds at 72°C in reaction buffer containing 2.5 mM MgCl2, 1 × PCR buffer (Roche Applied Science, Indianapolis, IN) and 0.2 μM each primer. Embryos at desired developmental stages were obtained by timed pregnancies, counting noon of the day of the vaginal plug as E0.5.
Skeletal staining and histology
Skin, muscles and visceral organs were removed from embryos or newborn animals prior to overnight incubation in a solution containing 0.06% Alizarin Red S and 0,02% Alcian Blue (Sigma, St. Louis, MO) in a 1:4 mixture of glacial acetic acid and 95% ethanol. Skeletal samples were cleared by incubation in 1.8% and 0.3% potassium hydroxide and stored in 100% glycerol. The skeletal preparations were analysed and photographed in glycerol/ethanol solution. For histological analysis and in situ hybridization on sections, embryos were fixed overnight in 4% paraformaldehyde in PBS, dehydrated in ethanol and embedded in paraffin. Paraffin blocks were sectioned at 6 μm and sections mounted onto glass slides were dewaxed, stained with hematoxylin and eosin or used for in situ mRNA hybridizations.
Whole mount and paraffin section in situ hybridization
Whole mount and paraffin section in situ hybridization was performed using digoxigenin (DIG)-UTP (Roche Applied Science, Indianapolis, IN) labeled riboprobes. The Bluescript plasmid containing a 1600 base pairs EcoRI fragment of the 3′ untranslated region in exon 8 of mouse Gli1 cDNA was a gift from from Dr. Alex Joyner (Skirball Instutute of Biomolecular Medicine, New York). The Shh and Ptc1 probes were provided by Dr. Andrew McMahon (Harvard University, Cambridge). For whole-mount preparations, embryos were fixed in MEMFA (0.1M MOPS pH 7.4, 2mMEGTA, 1mM MgSO4, 4% formaldehyde for 90 min at RT, dehydrated in methanol, and stored at −20°C. Prior to hybridizations, embryos were rehydrated by 5 min successive incubations in 75%, 50% methanol in water and 25% methanol in PBT (0.1% Tween-20 in PBS). Next, embryos were incubated with proteinase K (10 μg/ml in PBT) for 5 minutes at room temperature. Digestion was stopped by washing with 2 mg/ml glycine in PBT, and embryos were re-fixed for 30 min at RT in 4% paraformaldehyde with 0.2% glutaraldehyde in PBT, washed in PBT, and hybridized overnight at 65°C with 1 μg/ml of digoxigenin-labeled riboprobe (Roche Applied Science, Indianapolis, IN) in hybridization solution (50% formamide, 5× SSC [pH 4.5], 1% SDS, 50 μg/ml yeast tRNA, and 50 μg/ml heparin). Embryos were washed three times in hybridization solution for 30 minutes at 70°C, rinsed three times in TNT (10 mM Tris-HCl [pH 7.5], 0.5 M NaCl, and 0.1% Tween-20) for 5 min each at room temperature, and incubated for 1 hour with 100 μg/ml RNase A in TNT. After three washes in 50% formamide, 2× SSC (pH 4.5), 0.5 M NaCl, and 0.1% Tween-20 for 30 minutes at 65°C, followed by three washes in MAB (100 mM maleic acid [pH 7.5], 150 mM NaCl, 2 mM levamisole, and 0.1% Tween-20) for 5 minutes at room temperature, samples were blocked for 2 hours in 10% sheep serum in MAB/2% blocking agent (Roche Applied Science, Indianapolis, IN). Embryos were incubated overnight at 4°C with anti-digoxigenin alkaline phosphatase-coupled antibody (1:5000 diluted in MAB/2% Roche block/1% sheep serum). After extensive MAB washing, the embryos were washed three times in NTMT (100 mM Tris-HCl pH 9.5, 50 mM MgCl2, 100 mM NaCl, and 0.1% Tween-20) for 5 minutes each, and stained with BM purple (Roche Applied Science, Indianapolis, IN) at room temperature. Genotypes of embryos were determined by PCR before in situ hybridizations.
6 μm tissue sections in on glass slides were deparaffinised, rehydrated and subjected to Proteinase K (10 μg/ml in 10mM Tris pH7.5, 1mM CaCl2) digestion for 15 min at 37°C. Sections were rinsed twice in PBS, fixed in 4% PFA for 5 min at RT and acetylation was performed in 0.25% acetic acid for 15 min at RT. After dehydration in a series of alcohol, sections were air dried for 30 min and incubated overnight at 55° with a appropriate amount of DIG-labelled probe diluted in the hybridisation solution (50% formamide, 10mM Tris-HCl pH7.5, 200mg/ml tRNA, 1x Denhardt's). On the following day, slides were washed for 30 min at 55°C in 5xSSC and in 2xSSC /50% formamide, and were treated with 10μg/ml RNase. Signal was detected using Nuclear Acid Detection kit (Boehringer Mannheim, Pleasanton, CA).
Western blotting
Limb buds from five E11.5-E12.5 embryos were lysed in ice-cold RIPA buffer (50mM Tris pH 7.5, 150mM NaCl, 2mM EDTA, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS) supplemented with proteinase inhibitors. Aliquots of cell lysates (75μg) were separated on 7.5% sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked in PBS with 5% dry fat-free milk for 1 hour and incubated overnight at 4°C with affinity-purified rabbit polyclonal anti-Gli3 antibody (a generous gift from Baolin Wang, used 1:1000) in Tris-buffered saline (TBS) and 0.1% Tween 20. Membranes were washed and incubated with anti-rabbit antibody conjugated to horseradish peroxidase (1:1000). Signal detection was performed using the Super Signal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL).
Real-time PCR
Total RNA was isolated from four E18.5 Prx1-Cre mutant and four control forelimbs using Tri Reagent™ (Sigma, St. Louis, MO) reagent. cDNA was generated using the iScript ™Select cDNA Synthesis Kit (BioRad Laboratories, Hercules, CA). Subsequent amplification of 500ng of cDNA per reaction was performed with RT2Real-Time™ SYBR Green/Fluorescein PCR Master Mix (PA-011, SuperArray Bioscience Corporation, Frederick, MD) under the following conditions: 95°C, 15 min; 40 cycles of (95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s). Gli-1 (PPM41530A)), Gli-3 (PPM25249A) and GAPDH (PPM02946A) primers were purchased from SuperArray Bioscience Corporation (Frederick, MD). RT-PCR analyses were conducted on BioRad iCycler™ machine (BioRad Laboratories, Hercules, CA). The fold-change in gene expression was calculated as 2Λ(−ΔΔCt), where ΔΔCt is the difference in ΔCt value observed between control and mutant mRNA samples (ΔCt-expression of the gene of interest normalized for the expression of the housekeeping GAPDH gene).
Results
Conditional inactivation of Kif3a in neural crest cells results in severe craniofacial defects
Neural crest lineage-specific Kif3a conditional knockout mutants were generated by crossing the previously described Wnt1-Cre deleter and floxed Kif3a mouse strains (Chai et al., 2000;Marszalek et al., 2000). Wnt1-Cre; Kif3a mutant pups were born in the expected Mendelian ratio and displayed a severe form of frontonasal dysplasia and shortening of the lower jaw (Fig. 1A, top panel). They invariably died shortly after birth due to respiratory failure. The most likely cause of the respiratory defect is a profound cleft secondary palate (Fig. 1A, bottom panel). In order to characterize the craniofacial defects observed in the mutants in more detail, we analysed skeletal skull preparations from newborn animals stained with Alizarin Red and Alcian Blue.
Fig. 1.
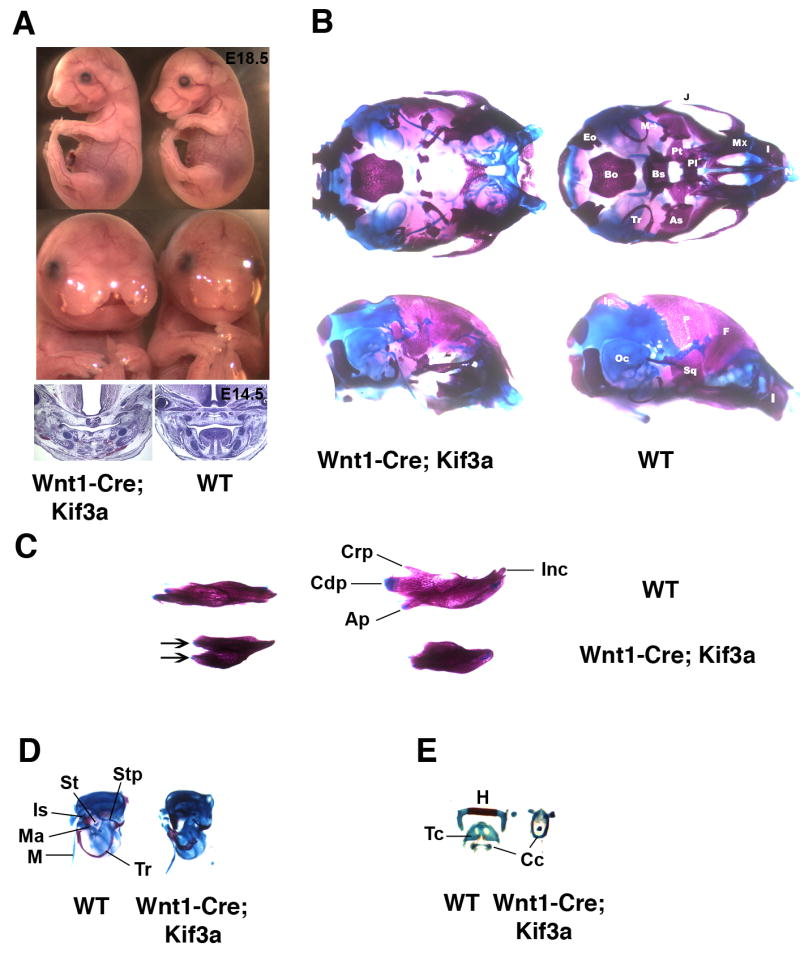
Craniofacial abnormalities induced by Wnt1-Cre-mediated Kif3a inactivation in neural crest cells. Lateral and frontal view of E18.5 Wnt1-Cre; Kif3a embryos (A, top); H&E staining of frontal sections of E14.5 Wnt1-Cre; Kif3a embryos (A, bottom). Ventral (B, top) and lateral (B, bottom) view of the skull from newborn Wnt1-Cre; Kif3a animal and control littermate stained with Alizarin Red and Alcian Blue. Dorsal (C, left) and medial (C, right) view of mandibles from newborn Wnt1-Cre; Kif3a animal and control littermate. Mutant dentaries are truncated and reveal duplicated lamina (arrows). Otic capsule and middle ear ossicles (D). Tympanic rings in Wnt1-Cre; Kif3a mutants are shortened and display thickening of the lamina. Middle ear ossicles malleus, incus and stapes are absent. Hyoid and laryngeal skeletal elements (E).
(Ap) Angular process; (As) alisphenoid; (Bo) basioccipital; (Bs) basisphenoid; (Cc) cricoid cartilage; (Cdp) condylar process; (Crp) coronoid process; (Eo) exoccipital; (F) frontal; (H) hyoid; (I) incisive; (Inc) incisor; (Ip) interparietal; (Is) incus; (J) jugal; (M) Meckel's cartilage; (Ma) malleus; (Mx) maxilla; (Oc) otic capsule; (P) parietal; (Pl) palatine; (Pt) pterygoid; (Sq) squamosal; (St) stapes; (Stp) styloid process; (Tc) thyroid cartilage; (Tr) tympanic ring.
In control Wnt1-Cre; Kif3a+/− embryos, the palate was intact and all the craniofacial skeletal elements well preserved (Fig. 1B). In mutant skulls, the cartilaginous primordium of the nasal capsule was formed but displayed a wide cleft in the midline region. In the rostral part of the cranial base, neural crest-derived skeletal structures that are normally formed by endochondral ossification, were absent (Fig. 1B). In the same area, a subset of intramembranous bones was only mildly affected. These included the maxillary, frontal, incisive, nasal and tympanic bones. In contrast, the squamous part of the temporal bone, vomer, palatine, and jugal bones, also formed by intramembranous ossification, were completely absent. Thus, genetic ablation of Kif3a in neural crest cells has a dramatic effect on the formation of both endochondral and intramembranous skeletal structures of neural crest origin. In the lower jaw, the mandibular lamina appeared duplicated and truncated rostrally, whereas Meckel's cartilage had relatively normal length and morphology (Figs. 1B,C). Endochondral skeletal structures of the middle ear, malleus, incus and stapes, were missing (Fig. 1D). The second through fourth branchial arch derivatives, the lesser horns and the body of the hyoid bone, the styloid process of the tympanic bone and the thyroid cartilage were not formed (Figs. 1D,E). In agreement with the neural crest-specific nature of Wnt1-Cre expression, cephalic mesoderm-derived skeletal elements of the skull, such as basioccipital, exoccipital and supraoccipital bones were not affected. Additional defects in non-skeletal tissues of the Kif3a-deficient animals included absence of tongue and both sets of incisors (Figs. 1A,C).
Wnt1-Cre; Kif3a mutants displayed striking phenotypic similarities to previously described Wnt1-Cre; Smo mice, in which responsiveness of neural crest cells to Sonic Hedgehog (Shh) was abolished by conditional removal of the hedgehog receptor Smoothened (Smo) (Jeong et al., 2004). These similarities prompted us to investigate the status of hedgehog signalling in the craniofacial area of the Wnt1-Cre; Kif3a embryos during the period when patterning of the mid-face occurs. In situ hybridization of E12.5 control embryos revealed Shh expression in the oral epithelium (Fig. 2, top right panel). Up-regulation of the downstream effector of hedgehog signalling, the transcription factor Gli1, was observed both in epithelial cells and the underlying mesenchyme (Fig. 2, bottom right panel). In Wnt1-Cre; Kif3a embryos, transcription of Shh was normal in the oral epithelium, whereas Gli1 expression was limited to epithelial cells and not detectable in the mesenchymal cells (Fig. 2, bottom left panel). This observation suggested that hedgehog signalling was impaired in Kif3a-deficient neural crest-derived mesenchyme.
Fig. 2.
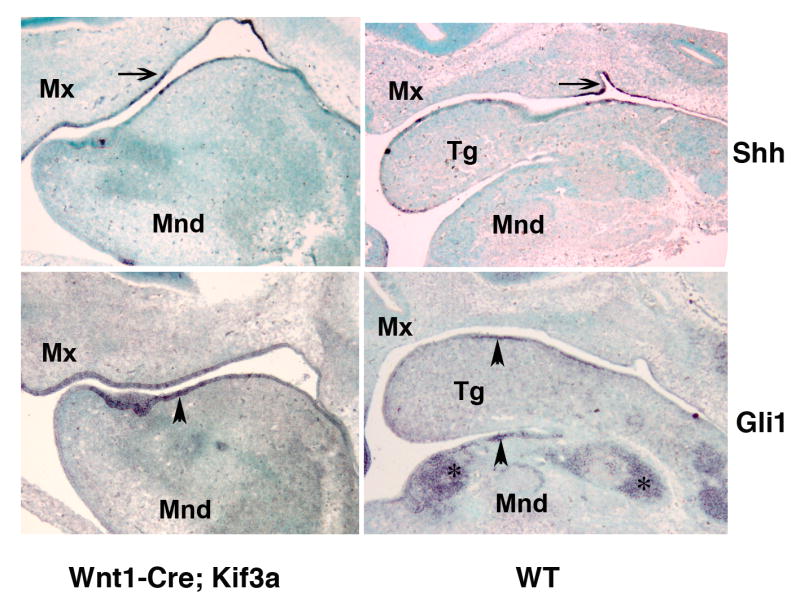
Effect of Kif3a ablation on hedgehog signalling in oral epithelium and craniofacial mesenchyme. In situ hybridization of sagittal sections of E12.5 head visualizing Shh (top) and Gli1 (bottom) expression pattern in Wnt1-Cre; Kif3a and control embryos. The gene expression patterns are representative of 5 -7 sagittal sections obtained from E12.5 embryos of each genotype. The expression of Shh (arrow) and its down-stream effector Gli1 (arrowhead) is preserved in Wnt1-Cre; Kif3a mutant and control oral epithelium. In contrast, activation of Gli1 can only be detected in the skeletogenic mesenchyme (asterisks) in the control embryo, whereas it is not evident in the Kif3a-deficient mesenchymal tissue. (Mnd) mandible; (Mx) maxilla; (Tg) tongue.
Trunk and limb skeletal patterning defects in the Kif3a-deficient mice are due to impaired formation of the Gli3 repressor
To investigate whether kinesin-2 is required for development of the axial and appendicular skeleton, we inactivated Kif3a in the trunk and limb skeletal progenitor cells. For this purpose we employed the previously described mesoderm-specific deleter mouse strain Dermo1-Cre (Yu et al., 2003). Dermo1-specific Kif3a conditional knockout animals were born in the expected Mendelian ratio but died immediately after birth because of respiratory distress. Visual examination of skeletal preparations of the newborn mutant pups revealed multiple deformities of the limbs and rib cage (Fig. 3A). In addition to a split sternum phenotype, the length and thickness of the ribs were considerably reduced relative to those of control littermates (Fig. 3B). These findings lead us to conclude that rib cage defects represented the most likely cause of neonatal lethality in Dermo1-Cre; Kif3a animals.
Fig. 3.

Axial and appendicular skeletal abnormalities induced by genetic ablation of Kif3a in Dermo1-expressing skeletogenic mesoderm. Alizarin Red and Alcian Blue stained skeletal preparations of newborn Dermo1-Cre-mediated Kif3a conditional knockout and control mice. (A-E) Whole mount Dermo1-Cre; Kif3a skeletal preparations demonstrate defects both in axial and appendicular skeleton. The rib cage and long bones of mutant mice are severely reduced in size (A). The Dermo1-Cre; Kif3a mice are characterised by split sternum, thin and shortened ribs (B). The Dermo1-Cre; Kif3a forelimbs display pre-axial polydactyly with duplication of digit I (arrow) and radial hemimelia (C). The Dermo1-Cre; Kif3a hindlimbs develop pre-axial polydactily and digits with missing phalangal elements (arrow). Failure to form joints between embryonic tarsal bones results in multiple bone fusions in newborn limbs (arrowhead) (D). The knee joint display abnormal morphology due to developmental patterning defects of the articular cartilage surfaces of patella, tibia, fibula and femur (E).
In both forelimbs and hindlimbs of the Dermo1-Cre; Kif3a animals, the anterior skeletal elements of zeugopods were shortened, whereas the autopods displayed pre-axial polydactyly with duplication/triplication of the most anterior digit (Figs. 3C,D). The autopods of the hindlimbs revealed poorly defined or missing distal phalanges and fusion of the multiple tarsal bones (Fig. 3D). In addition, the knee joint surfaces were severely misshapen due to early developmental patterning defects of the articular cartilage anlage prior to ossification of long bones (Figs. 3E and 6B).
Fig. 6.

Disorganized growth plate morphology and expression pattern of Ihh in Kif3a-deficient long bones. H&E-stained sections of E18.5 Dermo1-Cre; Kif3a ulna reveals a group of ectopic chondrocytes in the perichondrium (A, left and central panels). In situ hybridization demonstrates abnormal lateral expansion of collagen X-expressing hypertrophic chondrocytes along the affected anterior side of the ulna (A, right panel). H&E staining of paraffin sections of the knee joint area and in situ hybridization with collagen II and collagen-X probes visualize normal epiphyseal distribution of the resting/proliferating and hypertrophic chondrocytes, respectively (B, left panel). In the shortened Kif3a-deficient tibia, an abnormal perichondral distribution of the immature collagen II-positive chondrocytes and hypertrophic chondrocytes is evident (B, right panel). The joint area with articular cartilage defects is marked by arrow. Ihh expression visualized by in situ hybridization is detectable in both control and the Prx1-Cre; Kif3a long bones. Note ectopic expression of Ihh outside the pre-hypertrophic chondrocyte zone of the growth plate in the mutant tibia (C).
In order to prevent rib cage malformation and the perinatal lethality induced by Dermo1-Cre-mediated ablation of Kif3a, we restricted the domain of Kif3a inactivation to the lateral plate mesoderm using the Prx1-Cre deleter mouse strain (Logan et al., 2002). The Prx1-driven Cre recombinase activity is limb/sternum-specific and does not affect the somitic mesoderm skeletal derivatives, such as ribs and vertebrae. Prx1-Cre; Kif3a mutants were viable and had a rib cage of normal volume (Fig. 4A). Although the overall size and morphology of the ribs were preserved, allowing normal respiratory function, the sites of rib attachment to the sternum displayed asymmetry and resulted in abnormal rib pairing (Fig. 4B). Moreover, the sternum appeared wider and shorter in comparison to control animals. Both limbs and sternum are derived from lateral plate mesoderm. Their common origin explains the finding that both skeletal domains are affected in the Prx1-Cre; Kif3a conditional knockout mice.
Fig. 4.

Sternum and limb defects in newborn mice following lateral plate mesoderm-specific Kif3a inactivation. Whole mount preparations of Prx1-Cre; Kif3a mutants and control newborn mice (A). Mutant fore- and hindlimbs are reduced in length, whereas the rib cage has normal volume (A). The sternal bands in Prx1-Cre; Kif3a mice are fused but slightly shortened, the ribs display normal overall morphology but their attachment sites to sternum are asymmetric (B). The forelimbs of Prx1-Cre; Kif3a mutants develop syndactylous autopods with short dysmorphic digits and ill-defined phalanges (arrows) (C).
Prx1-Cre; Kif3a pups displayed forelimb polydactyly and profound shortening of limb bones (Fig. 4A). However, unlike a clearly pre-axial type of polydactyly observed in Dermo1-Cre; Kif3a mice, the forelimbs of the Prx1-Cre; Kif3a mutants displayed complete loss of the anterior-posterior polarity (Fig. 4C). Detailed examination of skeletal preparations of the polydactylous forelimbs revealed formation of six to nine digits, all of which were short and dysmorphic, with ill-defined phalanges.
Multiple polydactylous phenotypes previously reported in chicken and mice, have been attributed to abnormal expression patterns of Shh. During limb bud development, Shh expression is limited to the posterior domain of the growing limb bud, where it generates the zone of polarizing activity (ZPA). Ectopic expression of Shh in the anterior limb bud causes digit duplication in the same region. Furthermore, over-expression of Shh in chondrocytes causes a split sternum phenotype (Tavella et al., 2004).
To investigate whether the pattern of Shh expression was altered in the mutant embryos, we performed in situ hybridization. Examination of E11.5 old embryos showed that Shh expression was not affected and appeared restricted to the posterior limb bud region both in Prx1-Cre; Kif3a and control embryos (Fig. 5 A). We next analysed the expression of a Shh transcriptional target, the transmembrane receptor Patched1 (Ptch1). Robust activation of Ptch1 was detected in the posterior limb bud mesenchyme in control embryos (Fig. 5B). The hindlimbs of the Prx1-Cre; Kif3a mutants, which develop normal number of digits, displayed an expression of Ptch1 comparable to control. However, very low levels of the transcripts were observed in mutant forelimbs, which develop polydactyly later during embryonic development (Fig. 5B). Thus, a difference in hedgehog signalling activity (as reflected in Ptch1 expression) in early forelimb and hindlimb buds correlates with the final autopod phenotype. The phenotypic differences between the hind- and forelimbs in the Prx1-Cre; Kif3a mutant animals can be explained by previously reported differences in the onset of Prx1 expression (Logan et al., 2002). These data suggest that kinesin-2 is an essential positive mediator of hedgehog signal transduction. However, compromised hedgehog signalling cannot explain the polydactyly observed in Kif3a-deficient mice. The development of extra digits suggests that loss of Kif3a results in impaired function of the Gli3 repressor. In fact, several recent reports demonstrate that deficiency for various intraflagellar transport (IFT) proteins in mice results in attenuated activation of downstream targets of Shh and inhibition of the proteolytical processing of full-length Gli3 activator into a truncated repressor form (Haycraft et al., 2005;Huangfu and Anderson, 2005;Liu et al., 2005;May et al., 2005). In order to assess the efficiency of Gli3 processing in the absence of Kif3a, cell lysates from E 11.5-E12.5 Kif3a-deficient forelimb buds were analysed by western blotting. In the control limb buds, the full-length Gli3 was efficiently cleaved into the truncated repressor form, whereas the processing was impaired in the Kif3a−/− limb mesenchyme (Fig. 5C).
Fig. 5.
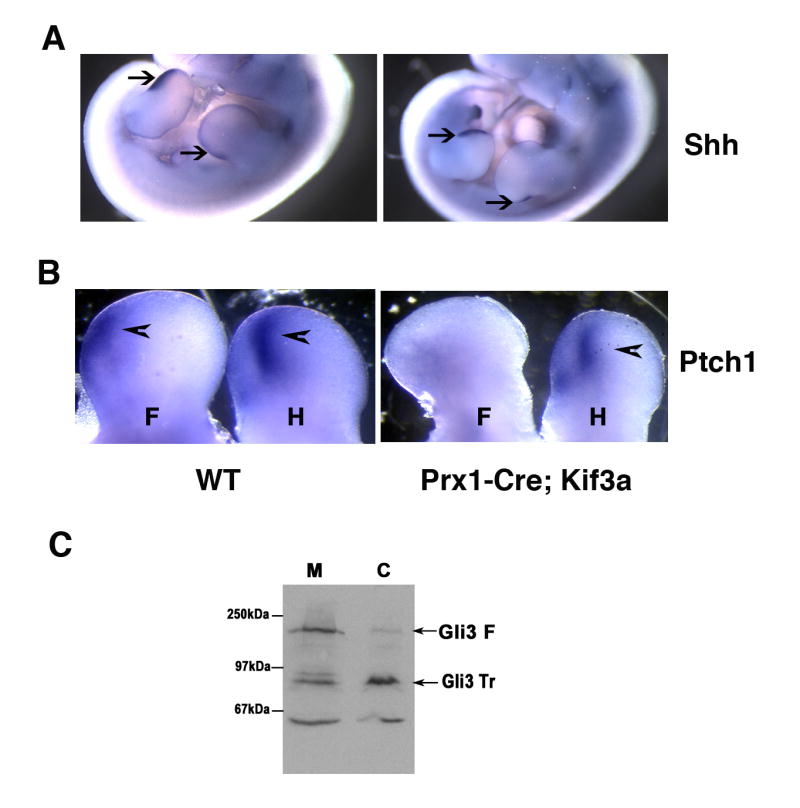
Effect of kinesin-2 inactivation on the expression of Shh ligand, downstream hedgehog target Ptch and generation of the hedgehog signaling repressor Gli3. At E11.5, expression of Shh transcript is restricted to posterior portion of limb bud in both control and Prx1-Cre; Kif3a embryos (arrows) (A). In response to Shh, expression of Ptch1 gene is up-regulated in posterior domain of both control limb buds and in the mutant hindlimb buds (arrowheads). The transcription level of Ptch1 in the Prx1-Cre; Kif3a forelimb remains low (B).
Western blotting of cell lysate from control and Kif3a−/− limb buds demonstrates impaired processing of the full-length Gli3 (Gli3 F) in Kif3a-deficient limbs and decreased amount of the truncated Gli3 repressor (Gli3 Tr) (C).
Kif3a inactivation results in disrupted Ihh signaling and retarded long bone growth
The longitudinal growth of all long bones of the appendicular skeleton was affected by Kif3a inactivation, but the severity of the phenotype varied greatly from slight shortening and lateral bending to profound reduction in length (Figs. 3A,C and 4A,C). Histological and in situ hybridization analysis of Dermo1-Cre; Kif3a ulna demonstrated that its bended appearance correlated with the side-specific presence of ectopic chondrocytes in the perichondrium and abnormal asymmetric pattern of distribution of the hypertrophic chondrocytes in the growth plates (Fig. 6A). Using in situ hybridization analysis with collagen II and collagen X-specific probes, we investigated the distribution of proliferating and hypertrophic chondrocytes in the Dermo1-Cre; Kif3a tibia. Growth plates, composed of proliferating, prehypertrophic and hypertrophic chondrocytes, serve as the growth centers of long bones. In control long bones, the growth plates were restricted to the growing ends (Fig 6B left panels). In contrast, the mutant long bones did not form properly defined growth plates and the immature proliferating chondrocytes could be found along the entire contour of the tibia. The central area of the long bone, normally filled with bone marrow was occupied with collagen X-secreting hypertrophic chondrocytes (Fig. 6B right panels).
Indian hedgehog (Ihh), secreted by prehypertrophic chondrocytes in the growth plate, is a key regulator of the longitudinal growth of long bones. In order to evaluate the effect of Kif3a inactivation of Ihh expression, we performed comparative in situ hybridization analysis of the Prx1-Cre; Kif3a and control tibia using Ihh-specific probe. Ihh-positive cells were detected both in the mutant and control long bones (Fig. 6C). However, Prx1-Cre; Kif3a tibia displayed an abnormal spatial distribution of the Ihh-secreting chondrocytes. Similar to other growth plate markers, such as collagen II and collagen X, Ihh expression was not restricted to the tibial epiphysis, but was also found in the perichondrium.
Since the expression of Ihh was maintained in Kif3a knockouts, albeit spatially disorganized, we evaluated the expression of the Ihh downstream target, the transcriptional activator Gli1 and transcriptional repressor Gli3 using real time PCR. In agreement with the situ hybridization data, the Kif3a-deficient limbs revealed a five-fold reduction in Gli1 expression relative to the control limbs (Fig. 7). Importantly, the Gli3 expression level was not affected by kinesin-2 inactivation. Thus, signaling downstream of Ihh appears to be inhibited in the long bones of Kif3a knockout embryos.
Fig. 7.
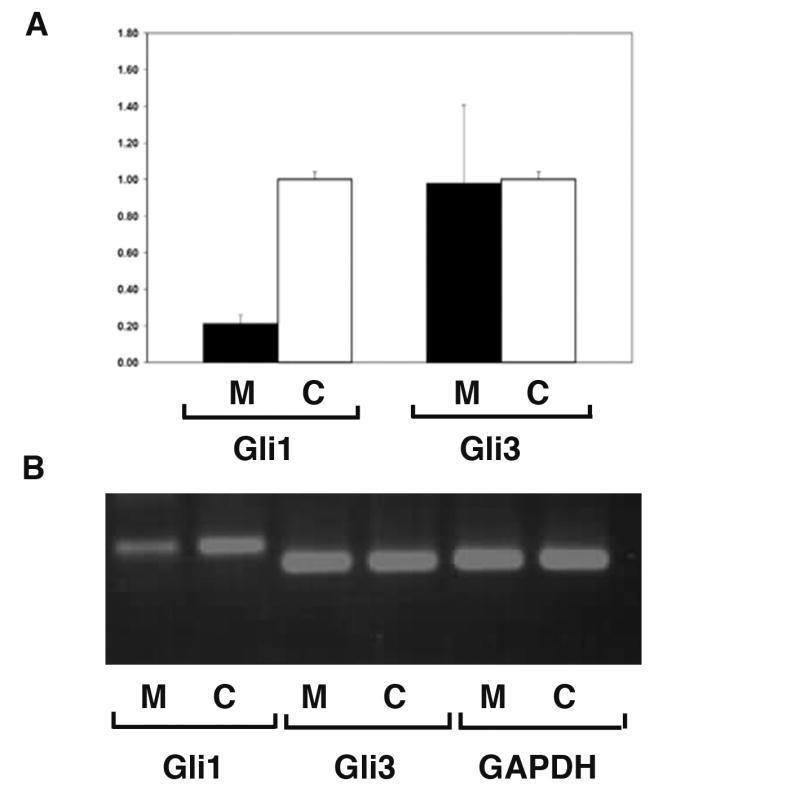
Kif3 inactivation does not affect Gli3 expression level but prevents up-regulation of Gli1 in long bones. Real-time PCR analysis of total RNA isolated from E18.5 Prx1-Cre; Kif3a and wild type long bones was conducted using Gli1- and Gli3- specific primers. The levels of Gli1 and Gli3 mRNA are calculated relative to GAPDH expression. The mean and SD of the ratios of the Gli1 and Gli3 expression in mutant (M, n=6) vs. control (C, n=6)) are presented (A). All error bars=SD, P<0.001 for Gli1 and P>0.9 for Gli3. The probability values were calculated using two-sample equal variance, two-tailed distribution Student's t-Test.
The size and the amount of the Gli1, Gli3 and GAPDH products obtained by RT-PCR were analysed by agarose gel electrophoresis. Gli3 and GAPDH are expressed at comparable level in control and mutant samples, whereas Gli1 expression is markedly reduced in the mutant long bones relative to control (B).
Discussion
In the present study we employ three independent tissue-specific Cre recombinase-expressing mice to inactivate the Kif3a gene in various skeletal progenitor cells. We demonstrate that, similar to other IFT proteins, kinesin-2 controls the patterning and growth of the appendicular and axial skeletal structures. Moreover, we provide the first experimental evidence for an essential role of kinesin-2 in the patterning of the mammalian craniofacial skeleton. This process also appears to require both intact IFT and hedgehog signal transduction.
The roles of intraflagellar transport proteins, including kinesin-2, have been previously studied primarily in the context of ciliary function in non-skeletal tissues (Marszalek et al., 2000;Teng et al., 2005). Until recently, the importance of ciliary proteins for the development and function of skeletal tissues has not been addressed directly, but circumstantial evidence from human genetics and studies of murine models of polycystic kidney disease, suggests that primary cilia and/or associated cell polarity processes are required for normal bone formation. Thus, in addition to polycystic kidney and loss of left-right asymmetry of internal organs, skeletal abnormalities are seen in cilia-associated disorders, such as several types of Bardet-Biedl syndrome (BBS) (Mykytyn and Sheffield, 2004). Moreover, inactivation of the ciliary IFT gene polaris in mice results in multiple skeletal abnormalities, including long bone defects and polydactyly in mice (Haycraft et al., 2007;Haycraft et al., 2005;Zhang et al., 2003). Importantly, a recent study implicates mutations in the IFT80 protein in causing a subset of Jeune syndrome cases. Jeune syndrome, or asphyxiating thoraic dystrophy is a human disorder characterised by a constricted thoracic cage and respirotory insufficiency, whereas polycystic kidney disease, polydactily and retinal degeneration have also been reported (Beales et al., 2007).
The data presented here clearly demonstrate that the IFT motor kinesin-2 regulates many aspects of skeletal development. First, inactivation of Kif3a in neural crest-derived cells severely affects morphogenesis of craniofacial skeletal structures of the mid- and upper face. Second, conditional knockout animals exhibit aplasia of middle ear ossicles and multiple skeletal elements forming the cranial base. The defects appear to be due to impaired hedgehog signal transduction in the mesenchymal cells. In agreement with this hypothesis, the defects in Wnt1-Cre; Kif3a mice are strikingly similar to the craniofacial abnormalities observed upon neural crest-specific inactivation of the transmembrane G-protein coupled hedgehog receptor Smoothened (Smo) (Jeong et al., 2004). A thickened mandible, lack of incisors, middle ear ossicles and multiple skeletal elements of the cranial base have been observed in both knockout mice. Similarly, both Wnt1-Cre; Kif3a and Wnt1-Cre; Smo mutant embryos display complete absence of the tongue.
The profound morphological similarities between the two knockouts along with the impaired activation of downstream Shh effectors, such as Gli1 and Ptch1, strongly suggest a genetic interaction between Kif3a and Smoothened. However, the mid-facial cleft characteristic of Kif3a-deficient mutants has not been described in Wnt1-Cre; Smo embryos. This difference may be attributed to the fact that the hedgehog-independent repressor function of Gli3 would remain intact while the positive hedgehog signalling would be reduced in the Smo-deficient animals. As suggested in this and recent studies of several IFT proteins (Liu et al., 2005;May et al., 2005), the absence of Kif3a would affect both the positive and negative aspects of hedgehog signalling. In agreement with this hypothesis, genetic inactivation of Ski, a transcriptional regulator of Gli3, results in neural tube abnormalities, craniofacial defects and polydactyly (Berk et al., 1997;Dai et al., 2002). Clefting of the mid-face, with or without cleft palate, and broadened forehead in Ski−/− mice develop in a strain-dependent manner and are phenotypically similar to the defects observed in Wnt1-Cre; Kif3a animals (Colmenares et al., 2002).
The profound developmental defects in the axial and appendicular skeleton caused by genetic inactivation of Kif3a in the trunk and limb mesoderm are reminiscent of the defects previously described in mice with altered hedgehog signalling.
Dermo1-Cre driven inactivation of Kif3a leads to a development of small rib cage causing immediate perinatal death by suffocation. It also results in shortening (hemimelia) of the anterior zeugopod, preaxial polydactyly and split sternum. Incomplete fusion of the sternal bands during embryogenesis, polydactyly and tibial hemimelia have been previously described in extra toes mouse strains Gli3 Xt and Gli3 XtJ, with a deletion in 5′ and 3′ regions of the Gli3 gene, respectively (Hui and Joyner, 1993;Johnson, 1967). Interestingly, in the Gli3 XtJ /+ heterozygous animals a mild pre-axial form of polydactyly with duplication of the first digit has been reported (Buscher and Ruther, 1998;Hui and Joyner, 1993). This phenotype is reminiscent of the type of polydactyly seen in the Dermo1-Cre; Kif3a mice. On the other hand, the morphology of Prx1-Cre; Kif3a autopods represents a close phenocopy of the abnormal patterning caused by a complete genetic inactivation of Gli3.
The posterior portion of the limb bud, designated as the zone of polarizing activity (ZPA), expresses high levels of Shh and is thought to play a key role in determination of digit number and digit identity (Riddle et al., 1993). Accordingly, ectopic expression of Shh in the anterior limb bud has been implicated in the development of polydactyly (Capdevila and Izpisua Belmonte, 2001;Johnson and Tabin, 1997). Here we demonstrate that neither pattern nor level of Shh expression is altered upon Kif3a inactivation in the mesenchymal tissues. However, Shh-dependant signal transduction appears to be attenuated as activation of the downstream Shh transcriptional target Ptch1 and Gli1 is strongly reduced. Previously described Shh-deficient mutant embryos exhibit complete lack of autopods in addition to fused skeletal elements in zeugopods (Shh KO). Of interest, double mutants lacking both Gli1 and Gli2 downstream effectors of Shh display no discernable limb patterning defects (Park et al., 2000). In contrast, mice lacking both Shh and Gli3 develop polydactyly, suggesting a crucial role for Gli3 gene in digit number determination (Hui and Joyner, 1993;Litingtung et al., 2002;Schimmang et al., 1992).
Gli3 protein is a member of the Gli-family of transcription factors, which functions as a repressor of multiple Sonic hedgehog targets. Unlike Gli1 and Gli2, Gli3 expression precedes the onset of the Shh transcription and is normally restricted to the posterior early limb bud where it plays an important role in the pre-patterning of the skeletogenic mesenchyme (te et al., 2002). Proteolytic processing of the full-length Gli3 into a truncated form is required for its activity as a repressor and occurs in the absence of hedgehog signaling. Nevertheless, at later stages, hedgehog signalling appears to negatively regulate the availability of the Gli3 repressor by preventing its cleavage by proteasomes.
Similarly to the IFT component, polaris, kinesin-2 is essential for embryonic and postnatal long bone growth (Haycraft et al., 2007;Song et al., 2007). Histological analysis of Kif3a-deficient long bones demonstrated that their bent and shortened morphology correlated with the side-specific or bilateral growth plate abnormalities and the presence of ectopic chondrocytes in the perichondrium. Normal growth plates are composed of proliferating and differentiating chondrocytes and are limited to the extremities of the long bones and thus facilitates the longitudinal growth. In the Kif3a knockouts, the long bone growth plates have lost their normal structure and spatial localization. In addition to the epiphyseal areas, the proliferating, prehypertrophic and hypertrophic chondrocytes were found in the perichondrium.
Several recent reports demonstrate that deficiency for various IFT proteins in mice results in formation of extra digits and attenuated activation of downstream targets of Shh (Haycraft et al., 2007;Haycraft et al., 2005;Huangfu et al., 2003;Liu et al., 2005;May et al., 2005). Moreover, biochemical studies indicate that the proteasomal processing of full-length Gli3 activator into a truncated repressor form is inhibited in the affected limbs (Haycraft et al., 2005;Huangfu and Anderson, 2005;Liu et al., 2005;May et al., 2005). The nature of the phenotypic defects Kif3a-deficient mice described in our study as well as the gene expression data, are in agreement with the notion that kinesin-2 is required for the formation of the hedgehog signaling repressor Gli3 and activation of the downstream targets of Sonic and Indian hedgehog signaling. The biochemical analysis of Gli3 repressor level confirms that its formation is impaired in the Kif3a-deficient tissue. Furthermore, the comparative quantitative analysis of the gene expression reveals that Gli3 transcription is not affected by Kif3a inactivation. In contrast, we detected a five-fold decrease in Gli1 expression in Kif3a-deficient tissues. The list of novel proteins involved in regulation of hedgehog signalling in skeletal tissues is growing rapidly. Elimination of the talpid3 and oral-facial-digital type1 (Ofd1) genes in chicken and mouse, respectively, results in deregulated hedgehog signalling and skeletal abnormalities similar to the defects we describe in Kif3a conditional knockout animals (Davey M 2006, Ferrante M 2006). Although not implicated in IFT, Ofd1 was found to be located in the centrioles/basal bodies (Romio L 2004, Keller 2005). The protein contains the Lis homology (LisH) domain, a novel sequence motif involved in regulating dynein/dynactin binding to microtubules and microtubule dynamics (Emes 2001, Coquelle F 2002). The talpid protein does not share sequence similarity with any protein of known function and was only detected in the cytoplasm, when overexpressed in cultured limb bud cells (Davey 2006). Intriguingly, a kinesin-like protein Costal-2 controls both positive and negative functions of hedgehog signalling in Drosophila (Kalderon, 2004). Costal-2 has been proposed to serve as a scaffold regulating the assembly and activity of two types of hedgehog signalling complexes (HSC), HSC-activator and HSC-repressor (Ogden et al., 2006). Both functions require direct binding of Costal-2 to the Gli homologue Cubitus interruptus (Ci) and Smoothened receptor and are regulated by multiple kinases. KIF7/KIF27 members of the kinesin-4 subfamily of KIFs have been proposed as vertebrate orthologues of Costal-2 (Katoh and Katoh, 2004;Tay et al., 2005). However, despite the fact that the phylogenetic relationship between Costal-2 and kinesin-2 appears to be more remote, the results of our study raise the possibility that kinesin-2 serves as a functional counterpart to Costal-2 in mammalian cells.
A body of experimental evidence suggests that intact IFT is essential for the formation of the primary cilium. Several studies have proposed an essential role of the primary cilium in hedgehog signal transduction (Corbit et al., 2005;Haycraft et al., 2005;May et al., 2005). However, conclusive evidence in support of such a role is not available. As expected, mesenchymal Kif3a-deficient cells are devoid of ciliary axonemes (our unpublished data) and Kif3a knockout mice displayed deregulated hedgehog signaling. However, our data does not allow the conclusion that cilia are required for hedgehog signaling. Heterotrimeric kinesin-2 appears to acquire additional cargo-transporting functions beyond the transport along axonemal microtubules (IFT). In neurons, this motor has been implicated in cytoplasmic transport of organelles, vesicles and soluble proteins (Kondo et al., 1994;Ray et al., 1999). Moreover, transport of late endosomes (Bananis et al., 2004) and lysosomes (Brown et al., 2005) and ER-to-Golgi anterograde traffic of vesicles (Le et al., 1998;Stauber et al., 2006) have been attributed to kinesin-2 activity in mammalian cells. Accordingly, we cannot exclude the possibility that the patterning abnormalities we observe in Kif3a knockout mice are caused by an impaired non-ciliary motor function of kinesin-2.
Acknowledgments
We would like to thank Dr. Larry Goldstein and Eileen Westerman for providing the floxed Kif3a mice. Gli3-specific antibody was a kind gift from Drs. Baolin Wang and Susan Mackem. Drs. Andy McMahon, David Ornitz and Cliff Tabin provided the Wnt1-Cre, Dermo1-Cre and the Prx1-Cre mice, respectively. We thank Sofiya Plotkina and Dr. Donald Glotzer for technical assistance and help with performing RT-PCR experiments, respectively. We acknowledge Michael Hart and Yulia Pittel for help with the manuscript. This work was supported by grants R01 AR036819 and R21 AR053143 (to B.R.O.) from the National Institutes of Health. E.K.H was supported by postdoctoral fellowships from the Norwegian Research Council (155362/310 and 169861/D15).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Bananis E, Nath S, Gordon K, Satir P, Stockert RJ, Murray JW, Wolkoff AW. Microtubule-dependent movement of late endocytic vesicles in vitro: requirements for Dynein and Kinesin. Mol Biol Cell. 2004;15:3688–3697. doi: 10.1091/mbc.E04-04-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007 doi: 10.1038/ng2038. In Press. [DOI] [PubMed] [Google Scholar]
- Beech PL, Pagh-Roehl K, Noda Y, Hirokawa N, Burnside B, Rosenbaum JL. Localization of kinesin superfamily proteins to the connecting cilium of fish photoreceptors. J Cell Sci. 1996;109(Pt 4):889–897. doi: 10.1242/jcs.109.4.889. [DOI] [PubMed] [Google Scholar]
- Berk M, Desai SY, Heyman HC, Colmenares C. Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes Dev. 1997;11:2029–2039. doi: 10.1101/gad.11.16.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CL, Maier KC, Stauber T, Ginkel LM, Wordeman L, Vernos I, Schroer TA. Kinesin-2 is a motor for late endosomes and lysosomes. Traffic. 2005;6:1114–1124. doi: 10.1111/j.1600-0854.2005.00347.x. [DOI] [PubMed] [Google Scholar]
- Buscher D, Ruther U. Expression profile of Gli family members and Shh in normal and mutant mouse limb development. Dev Dyn. 1998;211:88–96. doi: 10.1002/(SICI)1097-0177(199801)211:1<88::AID-AJA8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Izpisua Belmonte JC. Patterning mechanisms controlling vertebrate limb development. Annu Rev Cell Dev Biol. 2001;17:87–132. doi: 10.1146/annurev.cellbio.17.1.87. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141:993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenares C, Heilstedt HA, Shaffer LG, Schwartz S, Berk M, Murray JC, Stavnezer E. Loss of the SKI proto-oncogene in individuals affected with 1p36 deletion syndrome is predicted by strain-dependent defects in Ski−/− mice. Nat Genet. 2002;30:106–109. doi: 10.1038/ng770. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Dai P, Shinagawa T, Nomura T, Harada J, Kaul SC, Wadhwa R, Khan MM, Akimaru H, Sasaki H, Colmenares C, Ishii S. Ski is involved in transcriptional regulation by the repressor and full-length forms of Gli3. Genes Dev. 2002;16:2843–2848. doi: 10.1101/gad.1017302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, ydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR. Extra-toes: anew mutant gene causing multiple abnormalities in the mouse. J Embryol Exp Morphol. 1967;17:543–581. [PubMed] [Google Scholar]
- Johnson RL, Tabin CJ. Molecular models for vertebrate limb development. Cell. 1997;90:979–990. doi: 10.1016/s0092-8674(00)80364-5. [DOI] [PubMed] [Google Scholar]
- Kalderon D. Hedgehog signaling: Costal-2 bridges the transduction gap. Curr Biol. 2004;14:R67–R69. doi: 10.1016/j.cub.2003.12.047. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. KIF27 is one of orthologs for Drosophila Costal-2. Int J Oncol. 2004;25:1875–1880. [PubMed] [Google Scholar]
- Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol. 1994;125:1095–1107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bot N, Antony C, White J, Karsenti E, Vernos I. Role of xklp3, a subunit of the Xenopus kinesin II heterotrimeric complex, in membrane transport between the endoplasmic reticulum and the Golgi apparatus. J Cell Biol. 1998;143:1559–1573. doi: 10.1083/jcb.143.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–983. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005 doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Miki H, Okada Y, Hirokawa N. Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 2005 doi: 10.1016/j.tcb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Morris RL, Scholey JM. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J Cell Biol. 1997;138:1009–1022. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci. 1999;19:1027–1037. doi: 10.1523/JNEUROSCI.19-03-01027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykytyn K, Sheffield VC. Establishing a connection between cilia and Bardet-Biedl Syndrome. Trends Mol Med. 2004;10:106–109. doi: 10.1016/j.molmed.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Ogden SK, Casso DJ, Ascano M, Jr, Yore MM, Kornberg TB, Robbins DJ. Smoothened regulates activator and repressor functions of Hedgehog signaling via two distinct mechanisms. J Biol Chem. 2006;281:7237–7243. doi: 10.1074/jbc.M510169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, Nakashima M, Joyner AL. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Ray K, Perez SE, Yang Z, Xu J, Ritchings BW, Steller H, Goldstein LS. Kinesin-II is required for axonal transport of choline acetyltransferase in Drosophila. J Cell Biol. 1999;147:507–518. doi: 10.1083/jcb.147.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Tint IS, Fanapour PC, Gelfand VI. Regulated bidirectional motility of melanophore pigment granules along microtubules in vitro. Proc Natl Acad Sci U S A. 1997;94:3720–3725. doi: 10.1073/pnas.94.8.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmang T, Lemaistre M, Vortkamp A, Ruther U. Expression of the zinc finger gene Gli3 is affected in the morphogenetic mouse mutant extra-toes (Xt) Development. 1992;116:799–804. doi: 10.1242/dev.116.3.799. [DOI] [PubMed] [Google Scholar]
- Song B, Haycraft CJ, Seo HS, Yoder BK, Serra R. Development of the post-natal growth plate requires intraflagellar transport proteins. Dev Biol. 2007;305(1):202–16. doi: 10.1016/j.ydbio.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber T, Simpson JC, Pepperkok R, Vernos I. A role for kinesin-2 in COPI-dependent recycling between the ER and the Golgi complex. Curr Biol. 2006;16:2245–2251. doi: 10.1016/j.cub.2006.09.060. [DOI] [PubMed] [Google Scholar]
- Tavella S, Biticchi R, Schito A, Minina E, Di MD, Pagano A, Vortkamp A, Horton WA, Cancedda R, Garofalo S. Targeted expression of SHH affects chondrocyte differentiation, growth plate organization, and Sox9 expression. J Bone Miner Res. 2004;19:1678–1688. doi: 10.1359/JBMR.040706. [DOI] [PubMed] [Google Scholar]
- Tay SY, Ingham PW, Roy S. A homologue of the Drosophila kinesin-like protein Costal2 regulates Hedgehog signal transduction in the vertebrate embryo. Development. 2005;132:625–634. doi: 10.1242/dev.01606. [DOI] [PubMed] [Google Scholar]
- te Welscher P, Fernandez-Teran M, Ros MA, Zeller R. Mutual genetic antagonism involving GLI3 and dHAND prepatterns the vertebrate limb bud mesenchyme prior to SHH signaling. Genes Dev. 2002;16:421–426. doi: 10.1101/gad.219202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Rai T, Tanaka Y, Takei Y, Nakata T, Hirasawa M, Kulkarni AB, Hirokawa N. The KIF3 motor transports N-cadherin and organizes the developing neuroepithelium. Nat Cell Biol. 2005;7:474–482. doi: 10.1038/ncb1249. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Altrock WD, Kessels MM, Qualmann B, Regus H, Brauner D, Fejtova A, Bracko O, Gundelfinger ED, Brandstatter JH. Molecular dissection of the photoreceptor ribbon synapse: physical interaction of Bassoon and RIBEYE is essential for the assembly of the ribbon complex. J Cell Biol. 2005;168:825–836. doi: 10.1083/jcb.200408157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma MC, Zill A, Le BN, Vernos I, Gelfand V. Heterotrimeric kinesin II is the microtubule motor protein responsible for pigment dispersion in Xenopus melanophores. J Cell Biol. 1998;143:1547–1558. doi: 10.1083/jcb.143.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Nakata T, Okada Y, Hirokawa N. Cloning and characterization of KAP3: a novel kinesin superfamily-associated protein of KIF3A/3B. Proc Natl Acad Sci U S A. 1996;93:8443–8448. doi: 10.1073/pnas.93.16.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Nakata T, Okada Y, Hirokawa N. KIF3A/B: a heterodimeric kinesin superfamily protein that works as a microtubule plus end-directed motor for membrane organelle transport. J Cell Biol. 1995;130:1387–1399. doi: 10.1083/jcb.130.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn. 2003;227:78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]