

Abstract
Oxidative stress plays a key role in the neuronal loss exhibited in amyotrophic lateral sclerosis (ALS), an event precipitating irreversible muscle atrophy. By crossing ALS mouse models (SODG93A and SODH46RH48Q) with an antioxidant response element (ARE) reporter mouse, we identified activation characteristics of the ARE system throughout the timecourse of motor neuron disease. Surprisingly, the earliest and most significant activation of this genetic sensor of oxidative stress occurred in the distal muscles of mutant SOD mice. The resultant data supports existing hypotheses that the muscle is somehow implicated during the initial pathology of these mice. Subsequently, Nrf2-ARE activation appears to progress in a retrograde fashion along the motor pathway. These data provide timely information concerning the contributions of the Nrf2-ARE pathway in ALS disease progression.
Keywords: NFE2-related factor 2 (Nrf2), antioxidant response element (ARE), oxidative stress, amyotrophic lateral sclerosis (ALS), superoxide dismutase (SOD), motor neuron disease, muscle, neurodegeneration
Introduction
Motor neuron degeneration is manifest in one of its most severe forms in amyotrophic lateral sclerosis (ALS), a progressively debilitating disease culminating in paralysis and death of the patient. In humans, as well as animal models, it has been clearly shown that oxidative stress plays a central role in the progression of this motor neuron loss, possibly in concert with a chronically enhanced excitotoxin profile [(Rothstein et al. 1990; Gurney et al. 1996; Andrus et al. 1998; Heath and Shaw 2002; Agar and Durham 2003)]. Five to 10% of all human ALS cases have a familial cause (fALS), of which about 20% are caused by a mutation in the gene encoding Cu/Zn superoxide dismutase (SOD1) [(Rosen 1993; Gurney et al. 1994)]. The most commonly used rodent model of ALS exploits the consequence of a glycine to alanine mutation at position 93 of human SOD1 (SODG93A mice) [(Gurney et al. 1994)]. These mice replicate much of the human ALS phenotype. They develop eventual paralysis of their hindlimbs at approximately 120 days of age subsequent to mitochondrial damage, an accumulation of extracellular glutamate and intracellular filamentous inclusions, motor neuron (MN) cell death, and loss of axons innervating the muscle [(Gurney et al. 1994; Klivenyi et al. 1999; Alexander et al. 2000; Fischer et al. 2004)].
Therapeutically, the molecular avenues targeted for treatment of fALS include neurotrophic factors, antioxidants, modifiers of the proteasome system, as well as inhibitors of inflammation or excitotoxicity [(Gurney et al. 1996; Bruijn et al. 1998; Drachman et al. 2002; Guo et al. 2003; Valentine and Hart 2003; Wang et al. 2003; Ascherio et al. 2005; McGeer and McGeer 2005)]. A recent study suggests that reconfiguration of the muscle alone is sufficient for trophic support of endangered spinal cord neurons [(Dobrowolny et al. 2005)]. While motor neurons receive much attention as the indicators of ALS pathology, the muscle provides a uniquely accessible target for human therapies aimed towards maintenance of MNs via neuromuscular synapses.
The antioxidant response element (ARE) is an enhancer element that initiates the transcription of a battery of genes encoding phase II detoxification enzymes and factors essential for neuronal survival [(Rushmore and Pickett 1990; Rushmore et al. 1991)]. The ARE is activated through heterodimeric DNA binding of its transcription factor, NFE2-related factor 2 [Nrf2; (Moi et al. 1994; Venugopal and Jaiswal 1996)]. The mechanism by which this binding is induced is still emerging, but likely includes contributions from the repressor of Nrf2, KEAP1, heterodimeric binding partners, and proteasomal regulation of Nrf2 half-life [(Moi et al. 1994; Itoh et al. 1997; Itoh et al. 1999; Itoh et al. 2003; Katoh et al. 2005; Kobayashi and Yamamoto 2005)]. Importantly, in in vitro and in vivo models, the activation of this system blocks neurotoxicity resulting from glutathione depletion, lipid peroxidation, intracellular calcium overload, excitotoxins, and disruption of the mitochondrial electron transport chain [(Lee et al. 2003; Shih et al. 2003; Kraft et al. 2004; Kraft et al. 2006)]. In addition to the expected induction of detoxification enzymes, Nrf2-ARE activation results in genetic increases to cellular energetics and redox potential, inhibitory neurotransmitter signaling, and metabolic processes [(Li and Jaiswal 1992; Prestera et al. 1995; Thimmulappa et al. 2002; Lee et al. 2003; Shih et al. 2003; Kraft et al. 2004; Nguyen et al. 2004)].
Evaluation of this multipotent signaling system during the generation of pathology in ALS-afflicted animals may prove integral in developing a therapeutically effective weapon against the loss of vulnerable neuron populations. Accordingly, in this study, we evaluated the endogenous activation of the Nrf2-ARE system during the induction of pathology in mutant SOD mouse models. Understanding the temporal-spatial activation of this signaling system during ALS-like disease progression provides useful information regarding the endogenous response of the endangered cell populations and the surrounding, supportive tissue.
Materials and Methods
Animals
Animals were cared for and maintained in accordance with protocols approved by the University of Wisconsin-Madison Animal Care and Use Committee (ACUC). All mice were maintained on a C57BL6/SJL background. Male mice with a glycine to alanine mutation at position 93 of the superoxide dismutase (SODG93A) gene or over-expressing wild-type SOD (SODWT) were purchased from Jackson laboratories. The SODG93A transgenic mouse model was developed and characterized as a model for amyotrophic lateral sclerosis by Gurney et al. [(Gurney et al. 1994)]. These mice lose motor neurons in the ventral horn of the spinal cord and become paralyzed at 4−5 months of age. SODH46RH48Q mice were a generous gift from Dr. David Borchelt at the Johns Hopkins University. The SODH46RH48Q mouse model was created by disrupting the copper binding site of Cu/Zn SOD through mutation of two out of the four histidines in the copper binding site of the enzyme [(Wang et al. 2002)]. Mice which express wild-type human Cu/Zn SOD at levels comparable to those seen in the SODG93A mice (SODWT mice) serve as a control for elevated levels of human SOD [(Dal Canto and Gurney 1995)]. ARE-hPAP mice containing the core ARE sequence from the rat NADPH: quinone oxidoreductase 1 (NQO1) gene that drives a thermostable hPAP were previously developed and characterized [(Johnson et al. 2002)]. Mice were maintained on a heterozygous background so that each litter would generate littermate controls.
ARE-hPAP Activity
ARE activity was measured through calculation of hPAP enzyme activity. Two to five mg frozen tissue was homogenized in 1mL TMNC buffer (50mM Tris pH 7.5, 5mM MgCl2, 100mM NaCl, and 4% CHAPS). The homogenate was freeze-thawed and 25μL was added to a white-walled 96 well plate and mixed with 75μL 200mM diethanolamine (DEA) buffer. Plates were incubated for 20 min at 65°C to inhibit endogenous phosphatases. Following the addition of 100μL of substrate solution [2X CSPD (Applied Biosystems, Bedford, MA), 2X Emerald reagent (Applied Biosystems, Bedford, MA), 5mM MgCl2, and 150mM DEA] and incubation in the dark for 20 min at room temperature, luminescence was read in an Orion microplate luminometer (Berthold Detection Systems, Pforzheim, Germany) with a one second integration. Luminescence values were normalized to protein (bicinchoninic acid protein assay, Pierce, Rockford, IL). ARE-hPAP activity is expressed as relative luminescence units (RLU) per mg protein (minus the remaining background phosphatase activity present in respective ARE-hPAP negative control mouse tissue).
Enzymatic activity staining
Histochemical hPAP activity was determined using 5-bromo-4-chloro-3-indolyl phosphate (BCIP), nitroblue tetrazolium (NBT). During harvesting of hindlimb muscle (medial and lateral gastrocnemius), the tissue was pinned in stretch during fixation. Tissues were sectioned at 20μm,rehydrated, incubated at 65°C in TMN buffer (50mM Tris, pH 10.0; 5mM MgCl2; 100mM NaCl) for 15 min to inactivate endogenous phosphatases, and stained in AP buffer (100mM Tris, pH 9.5; 5mM MgCl2; 100mM NaCl) containing 0.33mg NBT and 0.165mg BCIP (Promega) for 30−45 minutes at 37°C. Slides were counterstained with nuclear fast red (Vector), dehydrated, and mounted with Permount (Fisher).
Myofibril ATPase staining
Tissue was processed as above and evaluated for fiber type as described by Brooke and Kaiser [(Brooke and Kaiser 1970)]. Sections were incubated in acidic buffer (3.5mM barbital and 3.5mM sodium acetate, pH 4.4−4.6) for 15 min at room temperature followed by ATP staining for two hours (3.6mM ATP, 20mM barbital, and 18mM CaCl2, pH 9.4). Next, slides were placed in 1% CaCl2 for 10 min, followed by incubation in 2% CoCl2 for 10 min. A final incubation in 2% (NH4)2S for 1 min brought about the desired color change. Wash steps in ddH2O were performed between each incubation. Sections were dehydrated and mounted with Permount (Fisher). Light or unstained fibers contain MyHC2A, while intermediate-stained fibers contain MyHC2B or MyHC2D (fast fibers). The darkest staining indicates the presence of MyHC1 (slow fibers). ATPase images are shown in grayscale to give clear contrast.
Immunohistochemistry
Slides were stained for glial fibrillary acidic protein (GFAP, 1:1000, DAKO) for astrocytes and SMI-32 for neuronal cells (1:200, Sternberger monoclonals). Fluorescent secondary antibodies were used at a concentration of 1:200 (Vector Labs). Hoechst 33258 (Sigma) was used as a nuclear counterstain at a concentration of 0.1mM in PBS. All slides were imaged on a Zeiss Axiovert 200 inverted fluorescent microscope.
Statistical Analysis
Statistical significance was determined using students' T tests to determine mean ± SE and p-value. Samples with a p ≤ 0.05 are considered significantly different from specified control values. For all figure component images, at least 3 animals were evaluated and representative images are shown.
Results
Activation characteristics of the Nrf2-ARE system in SOD models of ALS
By using a transgenic mouse line developed in the lab expressing a thermostable human placental alkaline phosphatase (hPAP) reporter construct upon Nrf2 binding to the ARE (ARE-hPAP mice) [(Johnson et al. 2002)], we were able to dissect the activation characteristics of the Nrf2-ARE system throughout the timecourse of ALS pathology in SODG93A mice. The presence of the ARE-hPAP transgene had no effect on the timecourse of pathology in the mutant SOD animals, as evaluated by time to paralysis, staining for markers of gliosis such as GFAP, and motor neuron cell loss (data not shown). ARE activation has been characterized as a sensitive index of cellular oxidative stress. Therefore, the presence of elevated hPAP can reflect both an increased level of reactive toxins, as well as the compensatory response within cells in the vicinity of this toxin accumulation. In vivo studies were performed to identify hPAP activity in SODG93A mice crossed with the ARE-hPAP mice, compared to littermate control, ARE-hPAP positive mice (negative for the SOD transgene). The SODG93A mouse model displays an approximate four-fold increase in SOD enzymatic activity [(Gurney et al. 1994)]. As SOD is regulated, in part, through Nrf2-ARE signaling, we controlled for these experiments in three ways. Firstly, we performed crosses of mice expressing an alternative form of mutated human SOD1 (SODH46RH48Q mice) with ARE-hPAP mice and evaluated these animals at 180 days of age (these mice become paralyzed around an age of eight months). SODH46RH48Q mice were previously created by disrupting the copper binding site of Cu/Zn SOD through mutation of two out of the four histidines in the copper binding site of the enzyme [(Wang et al. 2002)]. SODH46RH48Q mice recapitulate human disease over a timecourse approximately twice the duration of SODG93A mouse pathology, but display little or no human SOD enzymatic activity [(Wang et al. 2002)]. Both the SODG93A and SODH46RH48Q mutations have previously been linked to ALS disease in humans [(Aoki et al. 1994), (Enayat et al. 1995), and (Gurney et al. 1994)]. Additionally, as a negative control, mice which express wild-type human Cu/Zn SOD at levels comparable to those seen in the SODG93A mice (SODWT mice) were crossed onto the ARE-hPAP line and evaluated at the maximal timepoint assayed in the SODG93A animals (110 days) to ensure that the presence of elevated human SOD, itself, wasn't the causative factor behind any observed changes [(Dal Canto and Gurney 1995)]. Finally, in all comparisons, transgenic mice expressing mutated SOD1 were assayed in comparison to littermate controls possessing endogenous SOD1 and the ARE-hPAP reporter. It should be noted that no change in hPAP activity was observed in mice lacking the ARE-hPAP transgene and background activity detected in these animals was subtracted from the corresponding values obtained from ARE-hPAP positive mice in the homogenate tissue assays.
Specific stages of disease progression were evaluated in the SODG93A mice, specifically correlating with a presymptomatic timepoint (30 days), the initiation of molecular changes including elevated extracellular glutamate levels (60 days), the onset of muscle weakness and motor neuron loss (90 days), and an end stage timepoint of robust motor neuron pathology, but prior to paralysis (110 days), as complicating factors from moribund mice could mask the activation characteristics of this system. As previously mentioned, SODWT mice and their non-transgenic controls were harvested at 110 days, while SODH46RH48Q mice and littermates, were evaluated at 6 months of age. Interestingly, using a tissue homogenate assay of hPAP activity, we noticed no significant changes in the lumbar/sacral and cervical/thoracic spinal cord, brainstem, motor cortex, or liver between SODG93A and SODH46RH48Q positive and negative animals at any timepoint analyzed (data not shown). In addition, in all of the tissues analyzed (gastrocnemius, triceps, intercostals, spinal cord, brainstem, motor cortex, and liver), no changes were noted in mice expressing the SODWT mutation compared to littermate controls at 110 days of age (data not shown). However, when evaluating muscular tissue over the timecourse of ALS-like disease in SODG93A and SODH46RH48Q animals, we witnessed a robust increase in ARE-hPAP activity (Figure 1).
Fig. 1. ARE-hPAP activation in mutant SOD mice during the timecourse of ALS-like disease.

ARE-hPAP transgenic mice expressing the mutant SOD transgene (+) and their ARE-hPAP positive, mutant SOD negative controls (−) were evaluated for ARE-hPAP enzymatic activity in a tissue homoegenate assay at different timepoints of pathology. Shown are the relative luminescent values corrected for protein of SODG93A mice at 30 (n≥3), 60 (n≥4), 90 (n≥4), and 110 days of age (n≥5), as well as SODH46RH48Q mice at 180 days of age (n≥3). Shown above the bars displaying significant differences between mutant SOD positive mice and negative, littermate controls is the fold change increase in ARE-hPAP activity caused by mutant SOD (* p<0.05; Gastroc. = Gastrocnemius).
The earliest significant increase occurred in the gastrocnemius at 30 days of age. This activation is dramatically elevated with the onset of molecular changes at the 60 day timepoint and continues to increase exponentially throughout the onset and progression of muscle weakness and motor neuron loss. Also at 60 days, ARE activity within the forelimb muscle (triceps brachii) becomes significantly elevated, an effect which is amplified as the mice approach paralysis. ARE activity in the intercostals does not become significantly elevated until 110 days of age. At 180 days in SODH46RH48Q mice, only the gastrocnemius muscle displays any significant elevation in Nrf2-ARE signaling, suggesting that the extent of pathogenesis in these mice at 180 days is roughly equivalent to a timepoint between 30 and 60 days in the SODG93A animals. The liver is shown as an example of a tissue with no noticeable hPAP increase (Figure 1).
Histological evaluation confirmed the activation of the ARE-hPAP reporter within the hindlimb and forelimb muscle of mutant SOD mice compared to their non-transgenic littermate controls (Figure 2). The increase in activity in the gastrocnemius over time is especially clear when comparing 30 to 60 days in the SODG93A mice (Figure 2b and d). As with the homogenate assay, the staining in the SODH46RH48Q gastrocnemius appears closer to the early stages of pathology seen in the SODG93A mice. In the diseased mouse, the activated region of muscle localized specifically to individual fibers and clustered groups of muscle fibers, as observed in longitudinal sections (data not shown).
Fig. 2. Muscular ARE-hPAP staining in mutant SOD mice.
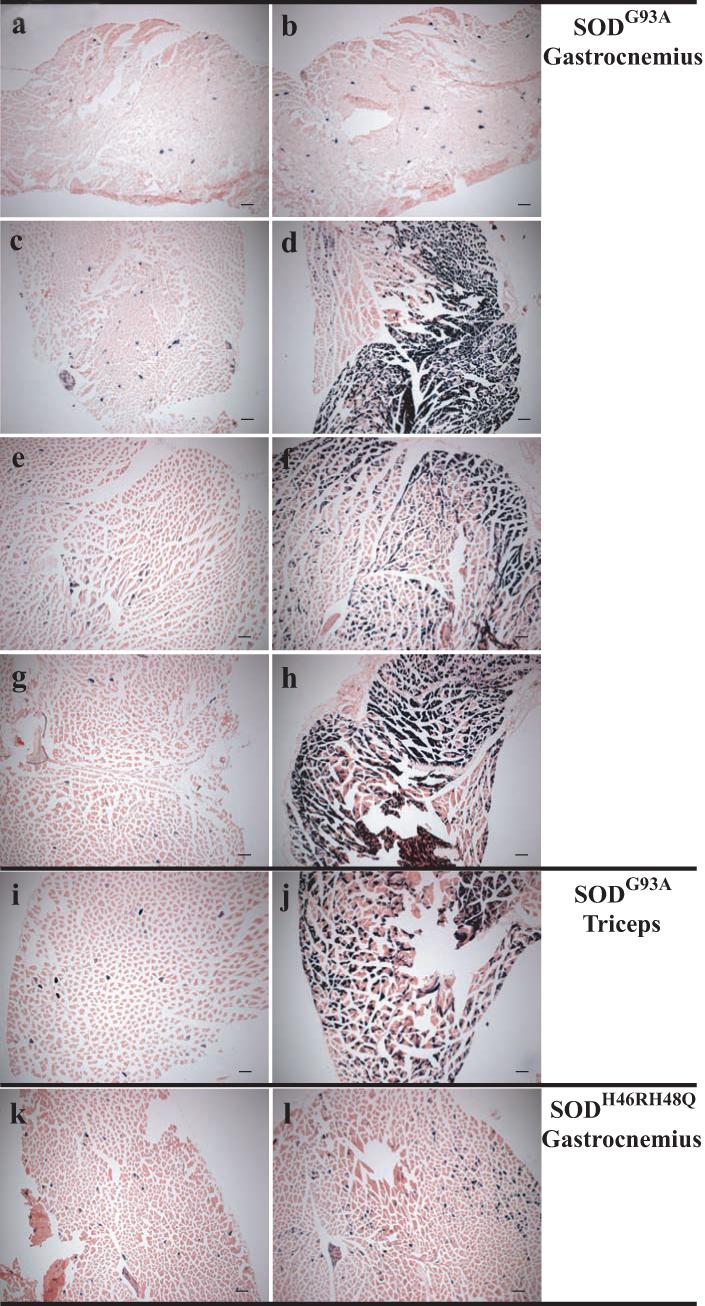
ARE-hPAP positive mice possessing the mutant SOD transgene (b,d,f,h,j,l) were compared to ARE-hPAP positive littermates without the mutant SOD transgene (a,c,e,g,i,k). Gastrocnemius (a-h,k-l) and triceps (i-j) muscle was assayed for hPAP presence at 30 (a-b), 60 (c-d), 90 (e-f), 110 (g-j), and 180 (k-l) days of age in SODG93A (a-j) and SODH46RH48Q (k-l) mice. Magnification= 5x (scale bars= 500μm).
Fiber type specificity of muscular activation of the Nrf2-ARE pathway by mutant SOD
The fiber type of ALS-afflicted muscle may have a strong influence on the timing of muscle fiber denervation, as type I fibers appear to maintain synaptic connections with motor neurons for far longer than type II fibers during ALS pathogenesis [(Frey et al. 2000)]. The fiber types of skeletal muscle are identified in these studies as either type I, IIa, or IIb. Evaluation of serial sections from diseased, mutant SOD mice for fiber type specificity via ATPase staining revealed that the majority of fibers displaying the strongest ARE-hPAP enzymatic activity were type I fibers rather than type II fibers (Figure 3; arrows are type I fibers). This trend was also apparent in SODG93A muscle at 60 days of age and SODH46RH48Q gastrocnemius at 180 days of age (data not shown).
Fig. 3. Fiber-type specificity of ARE-hPAP activation in mutant SOD muscle.

Serial transverse sections of hindlimb muscle were analyzed for ARE-hPAP activity (a,c) and fiber type via ATPase staining (b,d). SODG93A positive mice were analyzed at 90 (a-b) and 110 (c-d) days of age. Type I fibers are highlighted with red arrows corresponding to ARE-hPAP active cells in corresponding sections (b corresponds to a and d to b). Magnification= 20x (scale bars= 20μm).
Spinal cord activation of the ARE-hPAP reporter by mutant SOD-induced disease
Although no significant increase in hPAP activity was detectable in homogenates of spinal cord from mutant SOD positive mice relative to negative animals, evaluation of the spinal cord using an enzymatic stain did reveal some interesting patterns of hPAP activity. First, as observed in muscle, no staining was observed in animals negative for the ARE-hPAP transgene at any timepoint (data not shown). Robust staining in the dorsal horn of ARE-hPAP positive animals was noticeable regardless of the presence or absence of mutant SOD (Figure 4a and b). This basal activity was seen in the vicinity of the substantia gelatinosa and fasciculus gracilis, a collection of fiber bundles conveying ascending information from the lower extremities. This dorsal stain was dimly noticeable at the 30 day timepoint and increased to a dark, maximal staining pattern by 60 days of age in these animals (data not shown). Additionally, at the 30 and 60-day timepoints in SODG93A mice or the 180 timepoint in SODH46R48Q mice, no readily recognizable differences in hPAP staining intensity were seen between mutant SOD positive and negative spinal cords (data not shown).
Fig. 4. ARE activity in the spinal cord of mutant SOD mice.

SODG93A mice were evaluated for ARE-hPAP activity in the lumbar spinal cord at 110 days of age. SODG93A negative mice (a) and SODG93A positive littermates (b) display a basal level of ARE activity in both the dorsal horn and within specific regions of the ventral horn gray matter (boxes in a-b indicate the restricted expression of ARE-hPAP positive cells in the ventral horn of animals lacking the SODG93A transgene; magnification= 5x; scale bar= 500μm). Also shown are several examples of morphologically-distinct, ARE-hPAP positive cells found almost exclusively within the anterior horn of SODG93A positive animals (arrows in c-h; magnification= 40x; scale bar= 20μm).
However, looking specifically at the ventral horn gray matter of the spinal cord, a region containing motor neuron cell bodies, interneurons, and infiltrating glia, there was an observable difference in the ARE-hPAP staining pattern between SODG93A positive and negative animals. In the anterior horn of ARE-hPAP animals negative for the SOD transgene, there was a basal level of activity in the most dorsally oriented lateral motor nuclei (predominantly consisting of MNs associated with flexor muscles, shown boxed in Figure 4a-b). In the lumbar spinal cord, there was a profound induction of the reporter by 110 days in the SODG93A mice (Figure 4a-b). As is shown in Figure 4b, this activation in response to the presence of mutant SOD is reflected by both a more intense staining pattern as well as a wider distribution throughout the lumbar spinal cord. For the most part, increased ARE reporter expression was observed in the region of ventral lateral motor nuclei (particularly in locations of MNs that innervate extensor muscles), in some cases, cells bordering the central canal (most likely ependymal cells), and the posterior funicular gray matter, a region associated with the integration of sensory information. Closer inspection of the diseased anterior horn revealed morphologically distinct staining patterns that we routinely observed in SODG93A mouse spinal cord by 110 days of age that were absent or seldom observed in the ventral horn gray matter of age-matched, nontransgenic controls (Figure 4c-h). The presence of Nrf2-ARE activity was observed in densely stained cell bodies (Figure 4c) with stunted, swollen or broken processes (Figure 4d), wispy clusters of positively-stained processes typically displaying a unidirectional orientation (Figure 4e), small and large spherical cells (Figures 4f and g, respectively) often connected by thin, dimly-stained processes, and extended strands of positively-stained processes exhibiting a beaded morphology (Figure 4h).
This cellular ARE-hPAP staining is seen in motor neuron cell bodies and astrocyte processes. As shown in Figure 5, a slight, but noticeable increase in spinal cord ARE activity is observable by 90 days in SODG93A mice (Figure 5c), becoming a clear and robust expression of the reporter by 110 days (Figure 5e), compared to the maximal basal activity witnessed in control animals (Figure 5a). In control animals, there is a basal level of staining in occasional motor neuron cell bodies and GFAP-positive processes (arrows and arrowheads, respectively, in Figure 5a-b). Similarly, in the SODG93A positive mice at 90 days, there are positively-stained MNs and astrocyte processes, the elevated number of ARE-hPAP positive astroglial properties coinciding with the increased GFAP staining (Figure 5c-d). By 110 days in the SODG93A mice, there is a clear elevation in ARE-hPAP positive motor neurons as well as a plethora of ARE-hPAP positive astrocytes (Figure 5e-f). In addition, in the 110 day old SODG93A mice in particular, there are many cells that do not co-localize well with either GFAP or SMI-32.
Fig. 5. Colocalization of motor neurons and astrocytes with ARE-hPAP staining.

Serial spinal cord sections were stained for hPAP enzyme activity in a,c,e or SMI-32 (green), GFAP (red), and a nuclear counterstain (blue) in b,d,f. Colocalized neurons are highlighted with arrows and GFAP-positive processes, arrowheads (boxed motor neurons in a-b are shown as a reference point). a-b, 110 day mutant SOD negative animals; c-d, 90 day SODG93A positive mice; e-f, 110 day mutant SOD positive animals (magnification= 20x; scale bar= 20μm).
Discussion
Previous research into the pathogenesis of ALS supports the hypothesis that the production of free radicals precipitates, or at least facilitates, motor neuron loss, while multiple antioxidants attenuate this deleterious progression [(Gurney et al. 1996; Henderson et al. 1996)]. By crossing the SODG93A mouse model of ALS with a reporter of ARE activation, we were able to discern that the Nrf2-ARE pathway is indeed active within the hindlimb muscle at a very early timepoint (30 days, Figure 1). This is in accordance with previous studies, which suggest alterations in either the muscle or at the level of the neuromuscular junction (NMJ) at timepoints prior to the onset of observable symptoms such as increases to extracellular glutamate levels, which occurs at approximately 60 days of age [(Frey et al. 2000; Fischer et al. 2004)]. The activation of the ARE reporter within the gastrocnemius persists throughout pathology and increases in concert with disease severity. It is important to note that, in addition to assaying ARE-hPAP littermates expressing endogenous SOD1 at the various timepoints, we controlled for these experiments in two additional ways. We evaluated SOD1 mutants that do not display elevated SOD enzyme activity (SODH46R/H48Q mice) yet exhibit elevated ARE-hPAP activity in the hindlimb muscle at six months of age (Figures 1 and 2), as well as mice that over-express the wild type human SOD enzyme (SODWT), but show no induction of the ARE at 110 days of age. Importantly, muscular activation of the Nrf2-ARE system by mutant SOD at early timepoints likely reflects an early induction of protective mechanisms in response to a low level of oxidative stress, but prior to overt cellular damage. This hypothesis is supported in recent studies identifying markers of oxidative stress such as malondialdehyde, protein carbonyls, and oxidative stress-responsive proteins at timepoints approaching disease onset in the gastrocnemius muscle of mutant SOD mice without corresponding changes in the spinal cord or liver [(Dupuis et al. 2003; Jokic et al. 2003; Mahoney et al. 2006)]. The study by Mahoney et al. (2006) described increases in the ARE-driven genes Cu/Zn SOD, MnSOD, and catalase by 95 days in SODG93A mice, while Jokic et al. (2003) witnessed an elevation in ARE-responsive glutathione peroxidase and gamma-glutamate cysteine ligase, as well as the transcription factor, Nrf2, in the gastrocnemius of SODG86R mice. As ALS disease progresses, so does muscular ARE activation (Figures 1 and 2), presumably because of increasing free radical deposition as the mutant SOD-induced defects are magnified and overrun endogenous defenses. It is plausible that this stress is occurring in response to a decrease in trophic factor secretion to the hindlimb from sub-lethally-damaged motor neurons, as early defects in axonal transport caused by SOD mutations have previously been shown [(Kennel et al. 1996; Williamson and Cleveland 1999)].
Further analysis of mutant SOD-induced alterations in these reporter mice highlights the induction of a massive ARE response within the damaged ventral horn of these animals by end stage (Figure 4). In a study by Hall et al. (1998), an increase in lipid peroxidation was noticeable in lumbar motor neurons of SODG93A mice at 30, 100, and 120 days of age (no significant differences were found at 60 days) [(Hall et al. 1998b)]. In the current study, however, we did not witness an increase in ARE activity within the lumbar spinal cord until approximately 90 days of age, possibly owing to robust basal levels of ARE activity in the dorsal horn of the spinal cord masking any slight changes in the ventral horn motor neurons at early timepoints. Looking more closely at the specific cell types involved, it is clear that both motor neurons and astrocytes upregulate the activity of the Nrf2-ARE system as muscle weakness progresses towards paralysis in SODG93A mice (Figure 5). However, it is also clear that these two cell types are not the only ones displaying ARE-activity within the diseased ventral spinal cord (Figures 4 and 5). The cell type-specific activation of this system in the spinal cord is of particular interest, as the motor neurons of the lateral division of the anterior horn have elevated ARE activity. Previous research has implicated the astrocyte as being the dominant ARE-responsive cell type, having higher levels of Nrf2 and ARE-inducible genes, while neurons remain relatively unresponsive [(Shih et al. 2003; Kraft et al. 2004; Vargas et al. 2005)]. In support of this elevation in the spinal cord, a recent manuscript identifies symptomatic increases in Nrf2 and ARE-driven heme oxygenase-1 in the spinal cord of SODG93A rats [(Vargas et al. 2005)]. There is known to be a strong cellular infiltration toward the sites of motor neuron degeneration in the mutant SOD models as damaged MNs accumulate aggregates and become surrounded by activated astrocytes [(Ince et al. 1996; Hall et al. 1998a; Howland et al. 2002; Wang et al. 2002)]. Importantly, as is shown in Figure 4, the ARE-hPAP positive cells closely resemble multiple-processed reactive astrocytes (4e), phagocytic infiltrating cells (4f-g), and MNs undergoing degeneration (4c-d) with swollen axons (4d) and malformed, possibly debris-filled, processes (4h). Thus, the coordinated glial activation and MN death, due in part to intracellular aggregates within axons, may have direct linkages with activation of the Nrf2-ARE system.
The early activation of the ARE genetic system occurs specifically within the muscle fibers themselves (Figure 2). Interestingly, the predominant ARE-active fibers are type I fibers (Figure 3). Type I, or “slow twitch” fibers, are rich in mitochondria, highly perfused, and contain a large amount of oxygen for maintaining aerobic activity, while type II fibers typically exhibit greater force but fatigue more quickly. More applicable to these studies, previous research has described a decline in motor unit function as early as 47 days of age, as well as a significant muscle denervation prior to postnatal day 50 (P50) in mutant SOD mice [(Kennel et al. 1996; Frey et al. 2000; Fischer et al. 2004)]. This denervation occurs preferentially in type II fibers (mostly IIb) following a thinning of neurofilament-positive terminal branches between P30-P40 and, beginning around P60, a local reinnervation of vacated synapses by type I fibers, which appear to increase in number and resist denervation until paralysis [(Frey et al. 2000)]. Interestingly, in support of this “dying back” model of disease progression in ALS, despite these early muscular defects, no ventral root pathology is seen until 80 days of age, coincident with the appearance of motor neuron vacuolization [(Fischer et al. 2004)]. These data, alongside of the observation of early muscular activation of an oxidative stress-responsive system, make it tempting to speculate that early alterations in trophic factor communication affect the functioning capacity of sensitive muscle fibers, eventually leading to axonal “dying back” and motor neuron loss.
Also of interest is the observed ARE activation within the triceps and intercostal muscles beginning at 60 and 110 days of age in SODG93A mice, respectively (Figures 1 and 2). The triceps brachii are extensor forelimb muscles, which become paralyzed in mutant SOD mice following asymmetrical hindlimb paralysis. This symptomatic activation may reflect oxidative stress events in the forelimb muscle resulting from molecular changes in the lateral motor nuclei of the spinal cord. The external intercostals are inspiratory muscles found between the ribs. Both patients and mutant SOD mice experience a progressive weakness and eventual loss of function in breathing muscles. Thus, this late stage activation likely reflects a final oxidative burst as this tissue begins to atrophy. The selective activation of distal muscles prior to respiratory muscle ARE activation correlates well with the clinical progression of ALS disease.
These studies add support to the growing body of evidence highlighting the initiation of ALS pathology in distal regions. Interestingly, the muscular ARE activation occurs most robustly in type I fibers, possibly because these fibers are more resistant to neuromuscular pathology. A recent study by Miller et al. shows that specific knockdown of mutant SOD1 in the muscle alone has no effect on ALS pathology, whereas knockdown in the muscle and MNs together is sufficient to delay disease, suggesting that mutant SOD in the muscle is not a causative factor in ALS onset and progression [(Miller et al. 2006)]. Taking these observations into consideration when evaluating our Nrf2-ARE activation in muscle leads us to hypothesize that early distal stress and muscle activation of the Nrf2-ARE pathway are independent of mutant SOD expression in muscle. This implies that the changes in muscle described herein may occur in sporadic ALS patients as well as those carrying mutations in SOD1. Hopefully, this information can be used to aid in the early diagnosis of ALS and other motor neuron diseases as well as foster the development of ALS therapies focused on relieving early symptoms at targeted distal sites to maintain the essential, two-way trophic support at the neuromuscular junction.
Acknowledgements
These studies were funded by grants from the ALS Association and the Robert Packard Center for ALS Research at Johns Hopkins as well as ES08089 and ES10042 from the National Institute of Environmental Health Sciences (NIEHS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agar J, Durham H. Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:232–242. doi: 10.1080/14660820310011278. [DOI] [PubMed] [Google Scholar]
- Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Patterson TD. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) Cu/Zn superoxide dismutase. J Neurochem. 2000;74:1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x. [DOI] [PubMed] [Google Scholar]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- Aoki M, Ogasawara M, Matsubara Y, Narisawa K, Nakamura S, Itoyama Y, Abe K. Familial amyotrophic lateral sclerosis (ALS) in Japan associated with H46R mutation in Cu/Zn superoxide dismutase gene: a possible new subtype of familial ALS. J Neurol Sci. 1994;126:77–83. doi: 10.1016/0022-510x(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Ascherio A, Weisskopf MG, O'Reilly EJ, Jacobs EJ, McCullough ML, Calle EE, Cudkowicz M, Thun MJ. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann Neurol. 2005;57:104–110. doi: 10.1002/ana.20316. [DOI] [PubMed] [Google Scholar]
- Brooke MH, Kaiser KK. Three “myosin adenosine triphosphatase” systems: the nature of their pH lability and sulfhydryl dependence. J Histochem Cytochem. 1970;18:670–672. doi: 10.1177/18.9.670. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu,Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musaro A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168:193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF, Meininger V, Loeffler JP. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. Faseb J. 2003;17:2091–2093. doi: 10.1096/fj.02-1182fje. [DOI] [PubMed] [Google Scholar]
- Enayat ZE, Orrell RW, Claus A, Ludolph A, Bachus R, Brockmuller J, Ray-Chaudhuri K, Radunovic A, Shaw C, Wilkinson J, et al. Two novel mutations in the gene for copper zinc superoxide dismutase in UK families with amyotrophic lateral sclerosis. Hum Mol Genet. 1995;4:1239–1240. doi: 10.1093/hmg/4.7.1239. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet. 2003;12:2519–2532. doi: 10.1093/hmg/ddg267. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998a;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res. 1998b;53:66–77. doi: 10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Heath PR, Shaw PJ. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:438–458. doi: 10.1002/mus.10186. [DOI] [PubMed] [Google Scholar]
- Henderson JT, Javaheri M, Kopko S, Roder JC. Reduction of lower motor neuron degeneration in wobbler mice by N-acetyl-L-cysteine. J Neurosci. 1996;16:7574–7582. doi: 10.1523/JNEUROSCI.16-23-07574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince PG, Shaw PJ, Slade JY, Jones C, Hudgson P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: pathological and immunocytochemical changes. Acta Neuropathol (Berl) 1996;92:395–403. doi: 10.1007/s004010050535. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- Jokic N, Di Scala F, Dupuis L, Rene F, Muller A, Gonzalez De Aguilar JL, Loeffler JP. Early activation of antioxidant mechanisms in muscle of mutant Cu/Zn-superoxide dismutase-linked amyotrophic lateral sclerosis mice. Ann N Y Acad Sci. 2003;1010:552–556. doi: 10.1196/annals.1299.102. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Iida K, Kang MI, Kobayashi A, Mizukami M, Tong KI, McMahon M, Hayes JD, Itoh K, Yamamoto M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Ferrante RJ, Matthews RT, Bogdanov MB, Klein AM, Andreassen OA, Mueller G, Wermer M, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5:347–350. doi: 10.1038/6568. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem. 2006;98:1852–1865. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- Li Y, Jaiswal AK. Regulation of human NAD(P)H:quinone oxidoreductase gene. Role of AP1 binding site contained within human antioxidant response element. J Biol Chem. 1992;267:15097–15104. [PubMed] [Google Scholar]
- Mahoney DJ, Kaczor JJ, Bourgeois J, Yasuda N, Tarnopolsky MA. Oxidative stress and antioxidant enzyme upregulation in SOD1-G93A mouse skeletal muscle. Muscle Nerve. 2006;33:809–816. doi: 10.1002/mus.20542. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. BioDrugs. 2005;19:31–37. doi: 10.2165/00063030-200519010-00004. [DOI] [PubMed] [Google Scholar]
- Miller TM, Kim SH, Yamanaka K, Hester M, Umapathi P, Arnson H, Rizo L, Mendell JR, Gage FH, Cleveland DW, Kaspar BK. Gene transfer demonstrates that muscle is not a primary target for non-cell-autonomous toxicity in familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:19546–19551. doi: 10.1073/pnas.0609411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Yang CS, Pickett CB. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic Biol Med. 2004;37:433–441. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol Med. 1995;1:827–837. [PMC free article] [PubMed] [Google Scholar]
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:362. doi: 10.1038/364362c0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 1990;265:14648–14653. [PubMed] [Google Scholar]
- Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- Valentine JS, Hart PJ. Misfolded CuZnSOD and amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, Beckman JS, Barbeito L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: Consequences for motor neuron survival. J Biol Chem. 2005 doi: 10.1074/jbc.M501920200. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]