

Abstract
Nucleoside reverse transcriptase inhibitors (NRTIs) are known to produce painful neuropathies and to enhance states of pain hypersensitivity produced by HIV-1 infection. It has also been observed that in some neuropathic pain models, chemokines and their receptors are upregulated, perhaps contributing to the pain state. In order to understand if chemokines are involved in NRTI-mediated sensory neuropathies, we treated rats with the anti-retroviral drug, 2′,3′- dideoxycytidine (ddC), which is known to produce an extended period of hyperalgesia and allodynia. Using in situ hybridization, we observed that under normal conditions, CXCR4 chemokine receptors were widely expressed by satellite glia in the dorsal root ganglia (DRG) and Schwann cells in the sciatic nerve. A limited number of DRG neurons also expressed CXCR4 receptors. The chemokine SDF-1/CXCL12 was similarly expressed in glial cells in the DRG and peripheral nerve. Following a single administration of ddC, expression levels of CXCR4 mRNA in glia and neurons and SDF-1 mRNA in glia increased considerably. The functional nature of increased CXCR4 mRNA expression was confirmed by measuring SDF-1 induced [Ca2+]i increases in acutely isolated DRG neurons and glia. In contrast, the expression of the chemokine receptors CCR2 and CCR5 did not change following ddC treatment. Pain hypersensitivity produced by ddC could be inhibited by treatment with the CXCR4 antagonist, AMD3100. Hence, we postulate that NRTIs produce pain hypersensitivity through the upregulation of CXCR4 signaling in the DRG. Increased numbers of CXCR4 receptors would also explain the synergism observed between NRTI treatment and the proalgesic effects of HIV-1 infection.
INTRODUCTION
HIV-1 infection is associated with several types of sensory and motor neuropathies (Kolson et al, 2001;Luciano et al., 2003;Verma et al., 2005). However, as HIV-1 does not replicate effectively in neurons, the molecular and cellular basis of its effects on the nervous system are unclear. It is thought that viral proteins, cytokines and neurotoxins may be released by infected macrophages or microglia and that these may ultimately mediate the effects of the virus on neuronal function (White et al., 2005a). HIV-1 infection has been successfully treated with nucleoside reverse transcriptase inhibitors (NRTIs). However, treatment with many of these drugs produces serious side effects. In particular, NRTI treatment has been frequently shown to produce painful neuropathies that are indistinguishable from those resulting from HIV-1 infection (Dalakas, 2001). NRTI-induced pain hypersensitivity can be severely limiting in individuals who are taking these drugs for their beneficial effects.
The interaction of HIV-1 with target cells is mediated by its coat protein gp120, which binds to the cellular co-receptors, hCD4 and either the CCR5 or CXCR4 chemokine receptors (Kaul et al., 2005). Chemokines are small, secreted proteins responsible for orchestrating the migration of leukocytes during inflammatory responses. However, recent studies have revealed that chemokines also regulate aspects of neural development and modulate glial and neuronal responses to injury and disease (Lu et al., 2002; Tran and Miller, 2003a, b; Belmadani et al., 2006).
Chemokines and their receptors are expressed by sensory neurons and glia in the DRG and their expression is upregulated following peripheral injury (Abbadie et al., 2003; Tanaka et al., 2004; White et al., 2005c; Bhangoo et al., 2006). Activation of these chemokine receptors can produce excitation of DRG neurons, which is thought to be mediated by transactivation of the TRPV1 channel (Zhang et al., 2005). Moreover, gp120 can also activate neuronal chemokine receptors, including those expressed by DRG neurons, producing similar excitatory effects (Meucci et al., 1998; Kaul and Lipton, 1999; Oh et al.). Predictably, injection of several chemokines or gp120 into the inflamed rat paw produces pain (Oh et al., 2001).
These results suggest that chemokine signaling in the DRG may play a role in the development and/or maintenance of chronic pain syndromes. Indeed, we have demonstrated that the chemokine receptor CCR2 and its primary ligand the chemokine, MCP-1/CCL2, are upregulated in the DRG in association with a spinal stenosis model of neuropathic pain (White et al., 2005c). Moreover, Abbadie et al (Abbadie et al., 2003) have demonstrated that mice deficient in the CCR2 chemokine receptor are resistant to the induction of several types of neuropathic pain.
Recently, Joseph and colleagues (Joseph et al., 2004) described a model of neuropathic pain associated with the administration of NRTIs used in the treatment of HIV-1 infection/AIDS. Although it is widely believed that NRTIs initiate their effects on peripheral neurons through interactions with mitochondria (Keswani et al., 2003; Bodner et al., 2004), exactly how this produces pain is not known. We have now investigated the effects of NRTI treatment on chemokine signaling in the DRG. We report that NRTIs specifically upregulate signaling via the CXCR4 chemokine receptor in the DRG and that SDF-1/CXCR4 signaling is central to ddC-induced tactile allodynia.
METHODS
Animals
Pathogen-free adult female Sprague-Dawley rats (150-200 g; Harlan Laboratories, Madison, WI) were used in all experiments (n=45). Rats were housed in temperature (23 ± 3 °C) and light (12-h light:12-h dark cycle; lights on at 07:00 h) controlled rooms with standard rodent chow and water available ad libitum. Experiments were performed during the light cycle. These experiments were approved by the Institutional Animal Care and Use Committee of Loyola University, Chicago. All procedures were conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health and the ethical guidelines of the International Association for the Study of Pain.
Drugs and method of administration
The drugs, 2′,3′-dideoxycytidine (ddC) and the bicyclam, AMD3100, were employed in this study, and were all purchased from Sigma (St Louis, MO, USA). All drugs were freshly prepared in saline on the day of the experiment. ddC- and vehicle-treated groups were given a one-time intraperitoneal (i.p.) injection of ddC (25mg/kg) or saline (vehicle), respectively. After hyperalgesia and allodynia were established, the animals were given an i.p. injection of AMD3100 (5mg/kg).
Von Frey test for mechanical allodynia
The von Frey test was performed on the area of the hind paws as previously described (LaMotte et al., 1998; Song et al., 1999; Zhang et al., 1999; Ma et al., 2003). Briefly, the rat was placed on a metal mesh floor and covered with a transparent plastic dome where the animal rested quietly after an initial few minutes of exploration. Animals were habituated to this testing apparatus for 15 minutes a day, two days prior to pre-injection behavioral testing. Following acclimation, each filament was applied to six spots spaced across the glabrous side of the hind paw; two distinct spots for the distribution of each nerve branch (saphenous, tibial and sural). Mechanical stimuli were applied with seven filaments, each differing in the bending force delivered (10, 20, 40, 60, 80, 100, and 120 mN), but each fitted a flat tip and a fixed diameter of 0.2 mm. The force equivalence of mN to grams is: 100 mN=10.197 grams. The filaments were tested in order of ascending force, with each filament delivered for 1 second in sequence from the 1st to the 6th spot alternately from one paw to the other. The interstimulus interval was 10-15 seconds. A cutoff value of 120 mN was used; animals that did not respond at 120 mN were assigned that value (Ma et al., 2003).
Measurements were taken on 3 successive days before rats were subjected to either a ddC injection or a vehicle injection. Post-injection tests were made 3, 7, 10, and 14 days after surgery and weekly thereafter for the duration of the experiment.
Stimuli were applied randomly to left and right hind paws to determine the stimulus intensity threshold stiffness required to elicit a paw withdrawal response. The incidence of foot withdrawal was expressed as a percentage of six applications of each filament as a function of force. A Hill equation was fitted to the function (Origin version 6.0, Microcal Software) relating the percentage of indentations eliciting a withdrawal to the force of indentation. From this equation, the threshold force was obtained and defined as the force corresponding to a 50% withdrawal rate. A threshold that exhibits at least a -20 mN difference from the baseline threshold of testing in a given animal is representative of neuropathic pain (Ma et al., 2003).
Threshold values were statistically analyzed for each foot separately and the significance of differences between the average of at least two pre-injection tests and the mean obtained for each post-injection test. In all tests, baseline data were obtained for the ddC-treated and shamtreated groups before drug or vehicle administration. Within each treatment group, post-administration means were compared with the baseline values by repeated measures analyses of variance (RMANOVA) followed by post hoc pairwise comparisons (Student-Newman-Keuls Method). A probability level of .05 indicates significance.
Tissue processing
Adult female Sprague-Dawley rats were deeply anesthetized with isoflurane and transcardially perfused with saline followed by 4% paraformaldehyde. Following transcardial perfusion, both left and right sciatic nerves were removed and immersion postfixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). After 24 h at 4°C, the tissue was rinsed in phosphate buffer, and osmicated with 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 hour. The tissue was then dehydrated through graded alcohols and embedded in plastic embedding media (EPON epoxy resins, Resolution Performance Products, Houston TX). The sections were cut 1 μm thick and stained with toluidine blue. These semithin plastic sections were coverslipped with DPX plastic polymer and photographed at high magnification with brightfield illumination. Some axonal morphological changes were examined using transmission electron microscopy (TEM).
Axon Pathology
Vehicle- and ddC-treated rats (n=3 animals per condition) were sacrificed and processed as above. At least 1000 sciatic nerve myelinated axons were assayed per animal. Myelinated axons examined in ddC-treated nerves were limited to areas of obvious pathology (see Figure 2). Quantification of axon diameter, myelin sheath and axoplasm area was determined using ImagePro Plus (Media Cybernetics, Silve r Spring, MD). Individuals conducting axon quantification were blinded to the treatment conditions. Data are represented as means ± SEM%. Once initialized, the images were altered to maximize dark/light contrast, and the program’s contrast sensitivity was adjusted to maximize recognition of both thinly and thickly myelinated fibers. The area of the myelin, axonal cytoplasm, and the mean diameter of each axon was measured. Extraneous dark objects, incomplete axons/myelin sheaths, hypertrophied myelin with no visible cytoplasmic lumen, and split axons were excluded from the data. All nonmyelinated axons below 2 μms in diameter were excluded from the data analysis.
Figure 2. ddC induced neuropathic pain is accompanied by nerve pathology.

The sciatic nerve was removed and analyzed for resulting pathology three days after a single i.p injection of ddC. A & B) Low and high power photomicrographs of the sciatic nerve from a vehicle-treated rat. C & D) Low and high power photomicrographs of a PID3 sciatic nerve after a single i.p. injection of ddC. Note the increased myelin sheath thickness of the largest diameter nerve fibers in the sciatic nerve. The mean diameter of normal and pathological axons did not differ, suggesting that the increase in myelin area diminishes the area of the axon cytoplasm. Also observed is a degeneration of the Remak bundles associated with unmyelinated axons as well (see arrows). Scale bar for A and C is 20μm; B and D is 5μm.
Immunocytochemical staining
Adult female Sprague-Dawley rats were deeply anesthetized with isoflurane and transcardially perfused with saline followed by 4% paraformaldehyde. Sagittal sections of the DRG were serially cut at 14 μm onto Super-Frost microscope slides (Fisher Scientific, Pittsburgh PA). At least 6 sections per lumbar DRG were obtained for immunocytological analysis. Tissue was processed such that DRG sections on each slide were at intervals of 80 μm. Slides were incubated with blocking buffer (3% BSA/3% horse serum/0.4% Triton) for 1 hour, followed by overnight incubation with the mouse monoclonal antisera generated against the rat macrophage marker, ED-1 (1:500; Serotec, Raleigh, NC), at room temperature. After primary incubation, secondary antibodies (anti-mouse conjugated to CY3, made in donkey at 1:800; Jackson ImmunoResearch, West Grove, PA) were used to visualize cells. Slides were washed in PBS for 5 min each (x3) and coverslipped with a PBS/glycerol solution. All tissue sections were also stained with Hoechst 33258 nuclear marker nuclear label (Invitrogen Corporation, Carlsbad CA).
In situ hybridization
In situ hybridization histochemistry for chemokine receptors was performed using digoxigeninlabeled riboprobes. Adult female Sprague-Dawley rats were sacrificed using carbon monoxide. DRGs from the injected animals were rapidly removed, embedded in OCT compound (Tissue Tek, Ted Pella, Inc., Redding, CA) and frozen. Sections were cut serially at 12 μm. The CXCR4 and SDF-1 probes were generated as described previously (Lu et al., 2002). The CCR2 probe was prepared as described (White et al., 2005c). Briefly, an 848-bp CCR2 cDNA fragment (nucleotides 489-1336 of GenBank no. U77349) was cloned by PCR using rat spleen cDNA. The resulting PCR product was subcloned into pGEM-T Easy and sequenced to ensure identity for riboprobe use. The CCR2 template was linearized with SacII to generate a probe of 950 bases by using SP6 polymerase. Signals were visualized by using NBT/BCIP reagents (Roche Applied Science, Indianapolis, IN) in the dark for 2-20 h depending upon the abundance of the RNA. The in situ image was captured using a Retiga EX charge-coupled device camera (Qimaging, Burnaby, BC). CCR2 mRNA expression studies were used for receptor localization because of the failure of immunocytochemistry to detect neuronal CCR2 protein.
For the CCR5 probe, we used CD1 mouse brain cDNA. The CCR5 fragment used the forward primer 5′-tgg att atg gta tgt cag cac cc-3′and the reverse primer 5′-tcg att atg gta tgt cag cac cc-3′. All PCR fragments were subcloned into a pCR II-TOPO vector, and were verified by restriction analysis and automated DNA sequencing (Perkin Elmer, Wellesley, MA). The plasmid templates were linearized by restriction enzyme digestion. Then transcription was labeled by digoxygenin (Roche Applied Science, Indianapolis, IN).
Preparation of dorsal root ganglion neurons
The L4/L5 DRGs was acutely dissociated with methods described by Ma and LaMotte (Ma and LaMotte, 2005). Briefly, L4 and L5 DRG were removed from control or drug-treated animals. The DRGs were treated with collagenase A and collagenase D in DMEM for 20 minutes (1mg/ml; Roche Applied Science, Indianapolis, IN), followed by treatment with papain (30units/ml, Worthington Biochemical, Lakewood, NJ) in HBSS containing .5mM EDTA and cysteine at 35°C. The DRGs, containing both neurons and non-neuronal cells, were then dissociated via mechanical trituration in DMEM containing 1mg/ml bovine serum albumin and trypsin inhibitor (1mg/ml, Sigma-Aldrich, St. Louis, MO). The culture media was supplemented with 10% fetal bovine serum, penicillin and streptomycin (100ug/ml and 100 U/ml). The cells were then plated on coverslips coated with poly-L-lysine and laminin (1mg/ml) and incubated for 2 hours before more culture media was added to the wells. The cells were then allowed to sit undisturbed for 12-15 hours to adhere at 37°C (with 5% CO2).
Intracellular Ca2+ imaging
The dissociated DRG cells were loaded with fura-2 AM (3uM, Invitrogen, Carlsbad, CA) for 25 minutes at room temperature in a balanced salt solution (BSS) [NaCl (140mM), Hepes (10mM), CaCl2 (2mM), MgCl2 (1mM), Glucose (10mM), KCl (5mM)]. The cells were rinsed with the BSS and mounted onto a chamber that was placed onto the inverted microscope and continuously perfused with BSS at a rate of 2ml/min. Intracellular calcium was measured by digital video microfluorometry with an intensified CCD camera coupled to a microscope and MetaFluor software. Cells were illuminated with a 150W xenon arc lamp, and the excitation wavelengths of the fura-2 (340/380nm) were selected by a filter changer. Chemokines were applied for two minutes directly into the coverslip bathing solution after the perfusion was stopped. If no response was seen within 1 minute, the chemokine was washed out. For all experiments, MCP-1 (100nM), SDF1α (100nM), capsaicin (100nM), high K+ (50mM) and ATP (100μM) were added to the cells. The chemokines used were purchased from R & D Systems, and all were used at a concentration of at least 100nM to simulate conditions during a pathologic state (Charo and Ransohoff, 2006). They were reconstituted in 0.1%BSA/PBS, and aliquots were stored at -20°C.
The significance of differences between the control group, and the various treatment groups were statistically analyzed. Within each treatment group, post-drug administration means were compared with the baseline/control values by analysis of variance (ANOVA), followed by a Dunnett’s Multiple Comparison test. A probability level of .05 indicates significance.
RESULTS
ddC is an antiretroviral drug representative of the class of compounds that have been widely used to treat HIV-1 infection. Many of these drugs, including ddC, produce long lasting pain hypersensitivity in humans (Berger and Levy, 1993; Dalakas et al., 2001) and rodents, but no changes in motor coordination (Joseph et al., 2004). To study changes in pain sensitivity following an injection of ddC, we investigated alterations in the threshold force of indentation (produced by von Frey filaments) necessary for eliciting a flexion hindpaw withdrawal reflex.
The time-course of peripheral neuropathy development in rats following a single administration of ddC was determined over a 7-week period (n=12). A single administration of ddC (25mg/kg) produced a significant bilateral decrease in paw withdrawal threshold to von Frey hair stimulation from post-injection day (PID) 3 through the last day of testing at PID42 (p<0.001) (Fig. 1). Though there were significant differences in the paw withdrawal response to physical stimulus following a single i.p. injection of ddC, there were no changes in grooming behavior or physical appearance. This pattern of behavior is consistent with previous observations in the literature (Joseph et al., 2004; Joseph and Levine, 2006). Saline vehicle injection did not produce changes in the paw withdrawal threshold to von Frey stimulation (74.81±1.45 mN; n=6).
Figure 1. The effects of ddC on paw withdrawal thresholds in response to mechanical stimulation.

Baseline responses to tactile stimulation with von Frey filaments were determined before NRTI injection and for 3-42 days following NRTI treatment. Each bar represents the combined bilateral changes in mean threshold force (±SE) eliciting a withdrawal to mechanical indentation of the hindpaw. The threshold force was significantly reduced starting 3 days after the intraperitoneal injection of ddC, and lasted through 42 days (*, p<0.001, n=12).
ddC-induced neuropathic pain is accompanied by nerve pathology
Compared with vehicle treatment, a single injection of ddC resulted in neuropathological changes in peripheral nerves as early as PID3 (Fig. 2A-D), which were present for at least 48 days (n=8, data not shown). We observed that the largest diameter nerve fibers in the sciatic nerve exhibited redundant myelin loop formation, a type of hypermyelination more precisely known as tomaculae formation (Fig. 2B,D) (Finsterer 2004;Vital 2004). These myelin structures were present over the entire length of a paranodal segment. Interestingly, this myelin-associated pathology was present only in peripheral nerve fibers distal to the DRG and was not observed in the dorsal roots. The tomaculae present in ddC-treated rats is similar to myelin pathology observed in TGFβ1 mutant mice (Day et al., 2003). In addition to the tomaculae formation, axonal cytoplasm was diminished by 40% in the hypermyelinated axons of PID3 animals (n=3) when compared with vehicle-treated animals (n=3) [Supplementary Fig. 1]. This myelin effect and reduced axoplasm has also been observed in PMP220/0 (Adlkofer et al., 1995), neuregulin 1 type III transgenic mice (Michailov et al., 2004) and myelin-associated glycoprotein (MAG) deficient mice (Cai et al., 2002). Despite the observed pathology, individual nerve fiber diameters were within the normal range with no evidence of demyelination/remyelination or axonal degeneration in the peripheral nerve of ddC-treated animals. Also, as previously reported (Keswani et al., 2006), Remak bundles associated with unmyelinated axons were abnormal with the administration of the NRTI. Under normal conditions, multiple unmyelinated axons are enclosed by the basal lamina of a single Schwann cell (Fig. 2B, arrow). However, after administration of ddC, the integrity of the Schwann cells was lost (Fig. 2D, arrow). ED-1 immunopositive macrophages were largely absent from the DRG and associated nerve with the few exceptions of resident endoneurial macrophages (Supplementary Fig. 2). The sensory abnormalities and histopathological myelin changes observed in ddC-treated rats are similar to those found in individuals with hereditary neuropathy with liability to pressure palsy (also called tomaculous neuropathy), a condition known to be due to a 17p11.2 chromosomal deletion (Lonnqvist and Pihko, 2003; Chance, 2006).
CXCR4 and SDF-1 upregulation in the DRG following ddC administration
We examined the state of CXCR4 mRNA expression in the DRG following a single injection of ddC. Under normal conditions, significant basal expression of CXCR4 receptor mRNA was observed mainly in non-neuronal cells of the DRG, as previously reported (Kury et al., 2002) (Fig. 3A), although expression in a few small neurons was also noted (n=4). At 1, 7 and 14 days post-injection, there was a considerable upregulation of the CXCR4 mRNA in non-neuronal cells of the DRG (Fig. 3B-D; n=4 for each time point). In addition, increased expression of CXCR4 receptor mRNA was noted in neurons of several sizes and in the peripheral nerve following ddC treatment (Supplementary Fig. 3). As noted previously, a lack of ED1-positive cells (macrophage marker) in either the ganglia or associated nerve (Supplementary Fig. 2) suggests that these CXCR4 positive cells were not infiltrating leukocytes.
Figure 3. CXCR4 receptor mRNA expression in the DRG following ddC administration.

We used in situ hybridization to visualize changes in CXCR4 receptor mRNA levels. A) Lumbar DRG removed from saline-treated rat exhibited a basal level of CXCR4 mRNA receptor expression. This was mainly observed in non-neuronal cells (densely black small cells), however, a few neurons were also positive. B) At one day post-injection, there was an upregulation of the CXCR4 receptor mRNA in the DRG. C and D) At 7 and 14 days post-injection, there was an even greater level of CXCR4 mRNA expression in the DRG. This appeared to be mainly in non-neuronal cells; however, there is some neuronal CXCR4 expression as well. (Arrows: neurons negative for CXCR4 mRNA transcripts, arrowheads: CXCR4 mRNA transcript positive neurons). Scale bar is 250 μm for panels A-C. Panel D is 75 μm (n=4 for each time point).
The sole ligand for the CXCR4 receptor is the chemokine, SDF-1/CXCL12, which is also expressed in non-neuronal cells of the DRG under normal conditions (Fig. 4A,B) (Gleichmann et al., 2000). At 7 (Fig. 4C,D) and 14 days post-injection (Fig. 4E,F), an upregulation of SDF1 mRNA was also observed, mostly in non-neuronal cells, although occasionally neurons were also positive for SDF1.
Figure 4. SDF1 mRNA expression in the DRG following ddC injection.
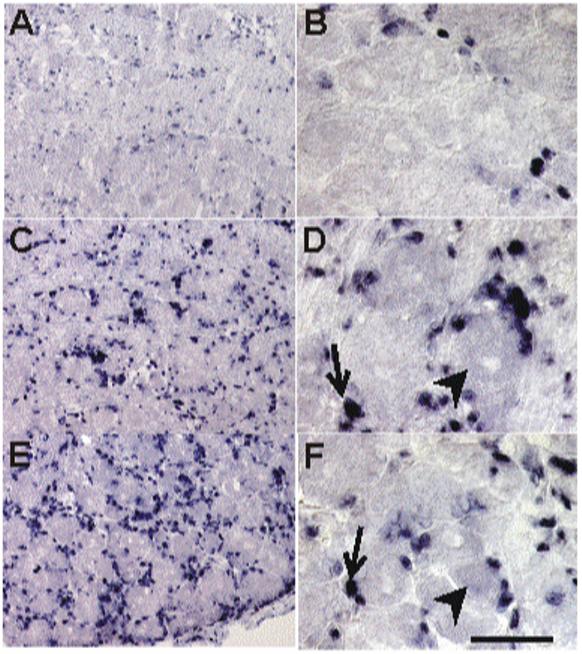
In situ hybridization was used to assess the expression pattern of SDF1 mRNA. A) Low power and (B) high power photomicrograph of basal expression of SDF1 mRNA was observed in the lumbar DRG from saline injected rats, mainly in non-neuronal cells (small, densely stained cells). After a single injection of ddC, the level of SDF1 mRNA expression increased at post-injection day (PID) -7 (C, low power magnification; D, high power magnification) and PID14 days (E, low power magnification; F, high power magnification). Most of the expression appeared to be in non-neuronal cells, however some neurons were also positively labeled. (Arrows, glia positive for SDF1 mRNA; arrowheads, neurons positive for SDF1 mRNA). Scale bar A, C and E is 100 μm; B, D and F is 50 μm (n = 4 for each time point).
We also performed in situ hybridization on the same DRG tissue using probes for the CCR2 and CCR5 chemokine receptors. Despite increased expression of these chemokine receptors in other chronic pain models (White et al., 2005c; Bhangoo et al., 2006), neither CCR2 nor CCR5 receptors were present in naïve nor ddC-treated animals (data not shown).
SDF-1 application increased [Ca2+]i in DRG cells following ddC exposure
Activation of chemokine receptors results in the excitation of DRG neurons and in an increase in [Ca2+]i in neurons and other chemokine sensitive cells (Oh et al., 2001). Using Ca2+ imaging, it is possible to monitor this [Ca2+]i increase, and so test for the presence of functional receptors. This technique can be used to not only confirm the presence of functional receptors, but also to quantify the number of cells expressing receptors, and to characterize these cell types. To further confirm the functional nature of the observed CXCR4 mRNA upregulation, we used fura-2 imaging of SDF1-induced increases in [Ca2+]i in acutely dissociated DRG cells as an indicator of functional CXCR4 receptor expression. In all experiments the chemokines, MCP-1 (CCR2 agonist, 100nM) and SDF1 (CXCR4 agonist, 100nM) were applied to the acutely isolated cells. The concentrations of chemokines used were at the maximum of their dose response curves to ensure the activation of any receptors present (Oh et al., 2001; White et al., 2005c; Fields and Burnstock, 2006). Following chemokine application, capsaicin (Trp V1 agonist), high K+ (activates voltage dependent Ca2+ channels) and ATP (activates purinergic receptors) were added to further characterize the phenotype of the imaged cells. A response to capsaicin and/or high K+ is indicative of a neuron, while a response to only ATP indicates a non-neuronal cell, such as a glial cell. We observed that after a single injection of ddC, there was a considerable increase in the number of acutely isolated DRG cells responding to SDF1 application at 7 and 14 days post-injection (Fig. 5A-D, table 1A,B), indicating an increase in CXCR4 receptor expression. In support of the in situ hybridization data, a majority of the responding cells were non-neuronal, based on their lack of response to capsaicin and/or high K+ (Fig. 5C,D). However, an overall increase in the SDF-1 sensitivity of cells classified as neurons was also noted (Fig. 5B, table 1A). Thus, the functional data supported the observations made using in situ hybridization. In contrast to the increased effects of SDF-1, no significant increases were observed in the number of cells responding to MCP-1 over the same time period (Fig. 5, table 1A).
Figure 5. SDF1 increases [Ca2+]i levels in DRG cells following ddC administration.

Examples of [Ca2+]i responses of cells acutely isolated from rat DRG at 7 and 14 days post-ddC injection. A) Under normal conditions, neuronal cells (capsaicin and/or K+ sensitive) were generally unresponsive to SDF1 (100nM) application. At post-ddC injection day 7 (PID7), the number of cells responding to SDF1 application increased (see table 1). Some of those responding cells were classified as neurons (B), while most responding to SDF1 application were classified as glial cells (capsaicin,K+ insensitive,ATP sensitive) (C and D) The percentage of cells responding to SDF1 at PID 14 also increased (see table 1 and D). For all experiments, MCP1 (100nM) was also applied. The percentage of cells responding to MCP-1 did not change (see table 1) [MCP-1 (M), SDF-1(S), Capsaicin (C), high K (K) and ATP (A)].
Table 1. Percentage of cells responding to SDF1 application.
We used calcium imaging to monitor the change in [Ca2+]i after chemokines are applied to acutely dissociated DRG from rats treated with vehicle or ddC. In all experiments, SDF1 and MCP1 were applied to the cells, followed by capsaicin, high K+ and ATP to characterize the cells. The number of cells responding to SDF1 application increased significantly by 14 days after ddC treatment. The number of cells responding to MCP1 application did not change in all experiments, suggesting CCR2 signaling is not part of the pain mechanism. Analysis of the cells responding to SDF1 application at PID7 and 14 revealed that most were non-neuronal based on their lack of response to capsaicin and high K+.
Table 1A. Percentage of neurons responding after chemokine application
Table 1B. Identity of cells responding to chemokine application
| Condition | SDF1 responsive neurons | MCP-1 responsive neurons |
|---|---|---|
| Control - Saline injected (n=61) | 4.9% (n=3) | 4.9% (n=3) |
| ddC injection - PID 7 days (n=74) | 16.2% * (n=12) | 4.1% (n=3) |
| ddC injection - PID 14 days (n=58) | 36.2% * (n=21) | 3.4% (n=2) |
| Cell type | PID7 | PID14 |
|---|---|---|
| SDF1 responsive neurons | 16.2% (n=12) | 36.2% (n=21) |
| Non-neuronal cells | 83.3% (n=62) | 63.8% (n=37) |
p<0.01
Effect of AMD3100 on ddC-induced allodynia
In order to establish whether the observed increase in CXCR4 signaling was a causal factor in the observed ddC associated pain hypersensitivity, we studied the effects of the selective CXCR4 antagonist, AMD3100 on rats with established ddC-induced allodynia. Seven and 14 days after a ddC injection, rats were given an intraperitoneal injection of AMD3100 (5mg/kg). Behavior was tested at 1, 4 and 24 hours following drug administration. We observed that AMD3100 effectively attenuated nociceptive pain behavior in the rats on both PID7 and PID14 (Fig. 6). At 24 hours, animal behavior was not significantly different from ddC-induced allodynic levels (data not shown). Although acute administration of AMD3100 was effective in diminishing tactile allodynia, daily administration of the same dosage did not prevent ddC-induced nociceptive pain behavior (data not shown).
Figure 6. The effect of the CXCR4 antagonist, AMD3100, on ddC-induced neuropathic pain.

After the induction of neuropathic pain using ddC, AMD3100 was administered and bilateral pain behavior was assessed using the Von-Frey filament test. Behavior was tested at 1, 4 and 24 hours after the administration of AMD3100 (5mg/kg) on post-injection day (PID) -7 and PID14. Following a single injection of ddC, the bilateral paw withdrawal threshold required to elicit a response was significantly reduced at post-injection PID 6 and PID13 compared to vehicle-treated rats (n=6;*p<0.001). Following administration of AMD3100 on PID7 and again at PID14, bilateral paw withdrawal thresholds increased to pre-ddC levels and attenuation of nociceptive behavior lasted for at least 4 hours. Twenty-four hours after AMD3100 treatment, paw withdrawal threshold levels did not differ from PID6 or PID13.
DISCUSSION
The results of the experiments reported here clearly implicate SDF1/CXCR4 signaling in the production of NRTI-induced neuropathic pain. Moreover, these data confirm previous observations that chemokine signaling can directly affect the cellular elements of peripheral sensory nerves, both the DRG neurons themselves as well as associated glia and microglia. Oh et al (Oh et al., 2001) demonstrated that cultured DRG neurons could express chemokine receptors and that both chemokines and gp120 could directly excite these neurons. The excitation may be primarily produced through transactivation of TRP channels, resulting from receptor-induced activation of phospholipase C and relief of the tonic TRP channel block by PIP2 (Zhang et al., 2005). This DRG excitation is associated with the release of transmitters such as substance P and CGRP (Oh et al., 2001; Qin et al., 2005).
In studies carried out in vivo using rodent models of neuropathic pain, it has generally been observed that most chemokines and their receptors are not expressed in the DRG at very high levels, but are upregulated in association with induction of states of pain hypersensitivity (Abbadie et al., 2003; Tanaka et al., 2004; White et al., 2005b; White et al., 2005c; Bhangoo et al., 2006; Xie et al., 2006; Zhang and De Koninck, 2006). Upregulation of the chemokine MCP-1 and its receptor CCR2 by DRG neurons in models of spinal stenosis and lysophatidyl choline-induced demyelination are examples of this phenomenon (White et al., 2005b; White et al., 2005c; Bhangoo et al., 2006; Sun et al., 2006). On the other hand, the pattern of CXCR4 receptor expression is somewhat different. These chemokine receptors are expressed by numerous DRG satellite glial cells and Schwann cells as well as a limited number of neurons under normal conditions (Kury et al., 2002; Kury et al., 2003; White et al., 2005b; Bhangoo et al., 2006). Glial cells primarily express the ligand for this receptor, the chemokine SDF-1/CXCL12, although it is also observed in a few DRG neurons (Gleichmann et al., 2000; White et al., 2005b; Bhangoo et al., 2006). We observed that rats treated with ddC produced a substantial upregulation of CXCR4 mRNA expression in both neurons and glia. In addition, there was also an increase in levels of SDF-1 mRNA in glial cells. The functional nature of this increased CXCR4 receptor expression was clear from the Ca2+ imaging studies using acutely isolated cells. These experiments demonstrated an upregulation of functional CXCR4 receptors primarily in glial cells, but also in some neurons. Finally, the role of upregulated CXCR4 signaling in the genesis of ddC-induced pain hypersensitivity is clear from experiments demonstrating the effectiveness of the CXCR4 antagonist, AMD 3100, in temporarily reversing pain under these conditions, although daily administration at the same dose was ineffective at diminishing chronic pain behavior. The study of AMD3100 dose response curves and effects of continuous infusion provide a framework for future trials in this toxic neuropathy.
One difference between the patterns of chemokine signaling in this case compared to previous studies of neuropathic pain, is that ddC administration only results in the upregulation of CXCR4 receptors. No increase in CCR2 or CCR5 receptor expression was observed. Upregulation of all of these receptors by DRG neurons is clearly observed in association with pain hypersensitivity as a result of lysophophatidylcholine-induced focal demyelination of the sciatic nerve, for example (White et al., 2005b; Bhangoo et al., 2006). Indeed, we have observed that AMD3100 is ineffective at attenuating pain hypersensitivity in association with lysophophatidylcholine treatment (unpublished observations). Thus, ddC-induced pain hypersensitivity appears to be uniquely CXCR4 dependent.
Although these data clearly implicate CXCR4 signaling in the genesis of ddC-associated neuropathy, the precise sequence of cellular events through which the drug produces increased SDF-1/CXCR4 expression is not entirely clear. Furthermore, why does upregulation of this chemokine and its receptor result in pain hypersensitivity? One possibility is that pain hypersensitivity is mediated by the CXCR4 receptors expressed by DRG neurons. Previous studies have demonstrated that SDF-1 and other chemokines can directly excite DRG neurons (Oh et al., 2001; White et al., 2005c; Sun et al., 2006). Thus, increased numbers of CXCR4 receptors together with increased SDF-1 expression by, and released from surrounding glial cells, may produce enhanced levels of sensory neuron excitation. A role for CXCR4 receptors expressed by glial cells may also be possible however, if SDF-1 controls its own release from these cells in an autologous manner. Alternatively, activation of Schwann cell CXCR4 receptors may also increase the release of other excitatory substances, as suggested elsewhere (Keswani et al., 2003). Kury et al (2003) previously demonstrated increased expression of CXCR4 by Schwann cells in the sciatic nerve using a model of Wallerian degeneration. Upregulation of CXCR4 was associated with increased levels of the transcription factor, Mash2 (Kury et al., 2002). Interestingly, in studies on non-neuronal cells, Mash 2, CXCR4 and SDF-1 have been shown to be downstream targets of the hypoxic induction transcription factor-1 (HIF-1) (Staller et al., 2003; Zagzag et al., 2005). As the initial site of action of NRTIs is thought to be the mitochondria and mitochondrial function is closely linked to the activity of HIF-1, it is certainly possible that these cellular elements are linked in the regulation of CXCR4 receptors in the present case.
Upregulated CXCR4 signaling is unique in this model of neuropathic pain because not only is it observed with administration of a drug used in HIV treatment, but the CXCR4 receptor is a co-receptor for the viral coat protein of the HIV1 virus, gp120. Therefore, it is possible that increased numbers of CXCR4 receptors could enhance the effects of either free or virion associated gp120. We have previously demonstrated that like SDF-1, T-tropic gp120 can directly excite cultured DRG neurons and can induce pain when injected into the rat paw (Oh et al., 2001). Furthermore, gp120 binding to CXCR4 receptors expressed by glial cells could also participate by stimulating the release of SDF-1 or other potential excitatory mediators (Bezzi et al., 2001). In support of this idea it has previously been demonstrated that gp120 can enhance transmitter release from cultured astrocytes (Bezzi et al., 2001). This latter scenario is also supported by recent data using cultured DRG neurons and Schwann cells (Keswani et al., 2003; Melli et al., 2006). According to these studies CXCR4 receptors expressed by both Schwann cells and DRG neurons are important in mediating the effects of gp120. Taken together, it is possible that the upregulated CXCR4 signaling observed following NRTI treatment, with the added proalgesic effects of gp120 and HIV1 infection could have a synergistic effect with respect to neuropathic pain. In a recent publication by Keswani et al. (2006), it was shown that when the NRTI, ddI, is administered to transgenic mice overexpressing gp120, a sensory neuropathy characterized by distal degeneration of sensory axons, develops. This pathology is similar to that seen in HIV1 infected patients undergoing NRTI treatment. In addition, they noted that in the older untreated gp120 transgenic mice, there was evidence of distal axonal degeneration. This study further confirms that NRTI treatment in conjunction with the proalgesic gp120 protein can have a synergist effect in the pain syndrome. The mechanism behind this pain syndrome is still unclear, however, our data would suggest that the block of CXCR4 receptors would represent an important therapeutic intervention in NRTI-associated neuropathy. Indeed, our results demonstrate that, once established, block of CXCR4 receptors with AMD3100 does attenuate ddC-induced pain hypersensitivity.
Supplementary Material
A single injection of ddC resulted in pathological changes in the sciatic nerve of rats sacrificed at PID3. Hypermyelination in the sciatic nerve was observed in drug-treated animals resulting in a significant increase in the total myelin area of measured axons. The overall diameter of normal and pathological axons did not differ. In addition, there was a 40% reduction in the total axonal cytoplasm area in the sciatic nerve of ddC-treated rats, suggesting that the increase in myelin area diminishes the area of the axon cytoplasm (*, p<0.05). Quantification of axon diameter, myelin sheath and axoplasm area was determined using ImagePro Plus (Media Cybernetics, Silver Spring, MD) (n=3 for each treatment conditions).
Using immunohistochemistry we observed that lumbar DRG from rats treated with ddC (A) or saline vehicle (C) exhibit few ED-1 immunopositive macrophages at PID7 (red arrows). These results did not differ from lumbar DRG derived from naïve rats. Panels B and D are tissue sections labeled with Hoescht nuclear stain that correspond with ddC (A) and saline-treated (C) animals Scale bar is 75 μm.
We used in situ hybridization to visualize changes in CXCR4 receptor mRNA levels in the peripheral nerve after ddC administration. A) Lumbar DRG removed from saline-treated rats exhibited a basal level of CXCR4 mRNA expression in Schwann cells. B and C) At 7 and 14 days post-ddC injection, respectively, there was an increase in CXCR4 mRNA expression in Schwann cells of the peripheral nerve (arrows: Schwann cells positive for CXCR4 mRNA) (n=4 for each time point).
Footnotes
Corresponding Author Fletcher A. White, Cell Biology, Neurobiology & Anatomy, Loyola University - Chicago, Maywood, IL, USA. Phone: 708.216.6728, Fax: 708.216.6731, Email:fwhite@lumc.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- Belmadani A, Tran PB, Ren D, Miller RJ. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26:3182–3191. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger JR, Levy RM. The neurologic complications of human immunodeficiency virus infection. Med Clin North Am. 1993;77:1–23. doi: 10.1016/s0025-7125(16)30269-3. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bhangoo S, Jung H, Chan DM, Ripsch M, Ren D, Miller RJ, White FA. Peripheral demyelination injury induces upregulation of chemokine/receptor expression and neuronal signaling in a model of neuropathic pain. 2006. Society for Neuroscience Abstract Viewer/Itinerary Planner 250.3.
- Bodner A, Toth PT, Miller RJ. Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol. 2004;188:246–253. doi: 10.1016/j.expneurol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Cai Z, Sutton-Smith P, Swift J, Cash K, Finnie J, Turnley A, Thompson PD, Blumbergs PC. Tomacula in MAG-deficient mice. J Peripher Nerv Syst. 2002;7:181–189. doi: 10.1046/j.1529-8027.2002.02023.x. [DOI] [PubMed] [Google Scholar]
- Chance PF. Inherited focal, episodic neuropathies: hereditary neuropathy with liability to pressure palsies and hereditary neuralgic amyotrophy. Neuromolecular Med. 2006;8:159–174. doi: 10.1385/NMM:8:1:159. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Peripheral neuropathy and antiretroviral drugs. J Peripher Nerv Syst. 2001;6:14–20. doi: 10.1046/j.1529-8027.2001.006001014.x. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Semino-Mora C, Leon-Monzon M. Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3′-dideoxycytidine (ddC) Lab Invest. 2001;81:1537–1544. doi: 10.1038/labinvest.3780367. [DOI] [PubMed] [Google Scholar]
- Day WA, Koishi K, McLennan IS. Transforming growth factor beta 1 may regulate the stability of mature myelin sheaths. Experimental Neurology. 2003;184:857–864. doi: 10.1016/S0014-4886(03)00308-X. [DOI] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichmann M, Gillen C, Czardybon M, Bosse F, Greiner-Petter R, Auer J, Muller HW. Cloning and characterization of SDF-1gamma, a novel SDF-1 chemokine transcript with developmentally regulated expression in the nervous system. Eur J Neurosci. 2000;12:1857–1866. doi: 10.1046/j.1460-9568.2000.00048.x. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–114. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Khasar SG, Levine JD. Novel mechanism of enhanced nociception in a model of AIDS therapy-induced painful peripheral neuropathy in the rat. Pain. 2004;107:147–158. doi: 10.1016/j.pain.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12(Suppl 1):878–892. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Keswani SC, Polley M, Pardo CA, Griffin JW, McArthur JC, Hoke A. Schwann cell chemokine receptors mediate HIV-1 gp120 toxicity to sensory neurons. Ann Neurol. 2003;54:287–296. doi: 10.1002/ana.10645. [DOI] [PubMed] [Google Scholar]
- Kury P, Greiner-Petter R, Cornely C, Jurgens T, Muller HW. Mammalian achaete scute homolog 2 is expressed in the adult sciatic nerve and regulates the expression of Krox24, Mob-1, CXCR4, and p57kip2 in Schwann cells. J Neurosci. 2002;22:7586–7595. doi: 10.1523/JNEUROSCI.22-17-07586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kury P, Koller H, Hamacher M, Cornely C, Hasse B, Muller HW. Cyclic AMP and tumor necrosis factor-[alpha] regulate CXCR4 gene expression in Schwann cells. Mol Cell Neurosci. 2003;24:1–9. doi: 10.1016/s1044-7431(03)00132-5. [DOI] [PubMed] [Google Scholar]
- LaMotte RH, Song XJ, Greenquist K, Zhang JM. Withdrawal thresholds to mechanical indentation and to controlled-temperature heating of the rat hind paw. 1998. Society for Neuroscience Abstract Viewer/Itinerary Planner:629.625.
- Lonnqvist T, Pihko H. Hereditary neuropathy with liability to pressure palsies (HNPP) in a toddler presenting with toe-walking, pain and stiffness. Neuromuscul Disord. 2003;13:827–829. doi: 10.1016/s0960-8966(03)00134-2. [DOI] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci U S A. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, LaMotte RH. Enhanced excitability of dissociated primary sensory neurons after chronic compression of the dorsal root ganglion in the rat. Pain. 2005;113:106–112. doi: 10.1016/j.pain.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Ma C, Shu Y, Zheng Z, Chen Y, Yao H, Greenquist KW, White FA, LaMotte RH. Similar Electrophysiological Changes in Axotomized and Neighboring Intact Dorsal Root Ganglion Neurons. J Neurophysiol. 2003;89:1588–1602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- Melli G, Keswani SC, Fischer A, Chen W, Hoke A. Spatially distinct and functionally independent mechanisms of axonal degeneration in a model of HIV-associated sensory neuropathy. Brain. 2006;129:1330–1338. doi: 10.1093/brain/awl058. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. PNAS. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave K-A. Axonal Neuregulin-1 Regulates Myelin Sheath Thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and Glycoprotein120 Produce Pain Hypersensitivity by Directly Exciting Primary Nociceptive Neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Wan Y, Wang X. CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. Journal of Neuroscience Research. 2005 doi: 10.1002/jnr.20612. dx.doi.org/10.1002/jnr.20612:NA. [DOI] [PubMed] [Google Scholar]
- Song XJ, Hu SJ, Greenquist KW, Zhang JM, LaMotte RH. Mechanical and thermal hyperalgesia and ectopic neuronal discharge after chronic compression of dorsal root ganglia. J Neurophysiol. 1999;82:3347–3358. doi: 10.1152/jn.1999.82.6.3347. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006 doi: 10.1152/jn.00222.2006. 00222.02006. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors in the brain: a developing story. J Comp Neurol. 2003a;457:1–6. doi: 10.1002/cne.10546. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ. Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci. 2003b;4:444–455. doi: 10.1038/nrn1116. [DOI] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov. 2005a;4:834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Ripsch M, Bhangoo S, Ren D, Weiss C, Miller RJ. Regulation of chemokines/receptors in the dorsal root ganglion following focal demyelination of the sciatic nerve. 2005b. Society for Neuroscience Abstract Viewer/Itinerary Planner 748.9.
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is upregulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005c;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie WR, Deng H, Li H, Bowen TL, Strong JA, Zhang JM. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflammatory irritation of the spinal ganglia. Neuroscience. 2006;142(3):809–822. doi: 10.1016/j.neuroscience.2006.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagzag D, Krishnamachary B, Yee H, Okuyama H, Chiriboga L, Ali MA, Melamed J, Semenza GL. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-offunction induces expression of a ligand and its receptor. Cancer Res. 2005;65:6178–6188. doi: 10.1158/0008-5472.CAN-04-4406. [DOI] [PubMed] [Google Scholar]
- Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97:772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Song XJ, LaMotte RH. Enhanced excitability of sensory neurons in rats with cutaneous hyperalgesia produced by chronic compression of the dorsal root ganglion. J Neurophysiol. 1999;82:3359–3366. doi: 10.1152/jn.1999.82.6.3359. [DOI] [PubMed] [Google Scholar]
- Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci U S A. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A single injection of ddC resulted in pathological changes in the sciatic nerve of rats sacrificed at PID3. Hypermyelination in the sciatic nerve was observed in drug-treated animals resulting in a significant increase in the total myelin area of measured axons. The overall diameter of normal and pathological axons did not differ. In addition, there was a 40% reduction in the total axonal cytoplasm area in the sciatic nerve of ddC-treated rats, suggesting that the increase in myelin area diminishes the area of the axon cytoplasm (*, p<0.05). Quantification of axon diameter, myelin sheath and axoplasm area was determined using ImagePro Plus (Media Cybernetics, Silver Spring, MD) (n=3 for each treatment conditions).
Using immunohistochemistry we observed that lumbar DRG from rats treated with ddC (A) or saline vehicle (C) exhibit few ED-1 immunopositive macrophages at PID7 (red arrows). These results did not differ from lumbar DRG derived from naïve rats. Panels B and D are tissue sections labeled with Hoescht nuclear stain that correspond with ddC (A) and saline-treated (C) animals Scale bar is 75 μm.
We used in situ hybridization to visualize changes in CXCR4 receptor mRNA levels in the peripheral nerve after ddC administration. A) Lumbar DRG removed from saline-treated rats exhibited a basal level of CXCR4 mRNA expression in Schwann cells. B and C) At 7 and 14 days post-ddC injection, respectively, there was an increase in CXCR4 mRNA expression in Schwann cells of the peripheral nerve (arrows: Schwann cells positive for CXCR4 mRNA) (n=4 for each time point).