

Abstract
Only a handful of NSCLC patients have been included in Dendritic Cell (DC) vaccine clinical trials. We had previously reported a series of 16 individuals with stage IA to IIIB NSCLC who received autologous DC vaccines matured with Dendritic Cell / T Cell-derived Maturation Factor (DCTCMF). Here we report the results of a continuation study with similar inclusion criteria, immunization protocol, and analysis, using an immature DC vaccine. Of the 14 participants, seven had undergone surgical resection (stage I/II), with or without adjuvant therapy, and seven with unresectable stage III had been treated with chemo-radiation alone. Autologous DCs were pulsed with apoptotic bodies derived from an allogeneic NSCLC cell line that over-expresses Her2/neu, CEA, WT1, Mage2, and survivin. DCs were not exposed to any maturation stimulus. Individuals received two intradermal vaccines (average 8.1x107 DC per immunization) one month apart. Immune responses were measured by IFN-γ ELISPOT, comparing relative number of antigen-reactive T-cells from pre-vaccine to timepoints post immunization. Immunologic responses were seen in 4/7 stage III unresectable, and 6/7 stage I/II surgically resected patients, including 3/3 resected patients who had also received adjuvant chemo-radiation. There were no related adverse events. One of seven surgically resected patients recurred and 4/7 stage III patients progressed. 3/5 patients with progressive disease showed no immunologic response. Data indicate that immature DC pulsed with apoptotic tumour cells have similar biologic activity to a DCTCMF-matured DC preparation delivered in a similar clinical protocol. Therapeutic efficacy is unknown and clinical outcomes are anecdotal.
Keywords: Dendritic Cell Vaccines, Lung Cancer, NSCLC, Clinical Trial
Introduction
Three year survival from unresectable stage IIIA/B NSCLC does not exceed 25% and mortal recurrences in surgically resectable patients approach 50% [1,2]. Tumour vaccines may have an adjuvant role in surgically resectable and unresectable NSCLC by consolidating responses to conventional therapy. Dendritic cells (DC) are potent antigen presenting cells that have been under intensive investigation as components of tumour vaccines [3-5]. There is not a standardized methodology for preparing vaccines and many questions remain about the optimal source or type of antigen and maturation state of DCs. Regardless, numerous DC vaccine trials have shown biologic activity suggesting additional investigation is warranted [3-6]. Literature also indicates that a percentage of individuals may derive therapeutic benefit, although, as expected from phase I/II trials, reports of clinical efficacy are anecdotal [3-6]. This study evaluates immunologic responses to immature, antigen-pulsed autologous DC vaccines in two distinct groups of non-small cell lung cancer patients. Data are analyzed in context of prior results using a DC preparation matured with Dendritic Cell / T Cell-derived Maturation Factor (DCTCMF) [7].
Methods
Human Subjects/patient characteristics
Individuals with histologically confirmed stage I-IIIB NSCLC who had completed definitive medical, surgical, or multimodality therapy, and had stable clinical disease at screening, were eligible for the study. Participants were consented under a protocol approved by the University of Kentucky's Medical IRB. Individuals were eligible to enter the study anytime from 6 weeks to 3 years following definitive therapy (average 9 months). The treatment group was heterogeneous with respect to stage, histology, treatment of primary disease, and risk of recurrence. Patient characteristics are summarized in Table 1.
Table 1. Patient characteristics.
Patients are stratified by therapy received for lung cancer - surgery alone (Surgical), surgery plus adjuvant medical therapy (Multi-modality) or medical therapy alone (Nonsurgical). Therapy roughly corresponds with stage – Surgical and Multimodality: stage IA-IIB vs. Nonsurgical: stage IIIA/B. TNM stage classification is shown for each subject. Histologic subtypes of NSCLC include Adenocarcinoma (Ad), Squamous cell carcinoma (Sq), Bronchoalveolar cell carcinoma (BAC), and Large cell carcinoma. Specific therapy(ies) received included: Surgery (Sg), Chemotherapy (Chemo) and Radiation Therapy (XRT). The latter two were delivered as initial definitive cancer therapy (in Nonsurgical patients), or in an adjuvant setting following surgical resection (Adj). Also shown is months from last treatment to date of first vaccine.
|
Patient number |
Age | Histology | Stage | Treatment |
Months from last treatment to vaccine |
|---|---|---|---|---|---|
| Surgical | |||||
| DC17 | 70 | Large cell | T2N0M0 | Sg | 24 |
| DC20 | 62 | Ad | T1N0M0 | Sg | 12 |
| DC26 | 66 | Ad | T1N0M0 | Sg | 17 |
| DC27 | 65 | BAC | T1N0M0 | Sg | 24 |
| Multi-modality | |||||
| DC23 | 56 | Ad/Sq | T2N0Mx | Sg, Adj-Chemo/XRT | 7 |
| DC28 | 60 | Large cell | T2N0M0 | Sg, Adj-Chemo/XRT | 12 |
| DC29 | 67 | Sq | T2N1M0 | Sg, Adj-Chemo | 5 |
| Nonsurgical | |||||
| DC18 | 59 | Ad | T2N3M0 | Chemo/XRT | 24 |
| DC19 | 67 | Sq | T4NxMx | Chemo/XRT | 5 |
| DC21 | 51 | BAC | T4N0M0 | Chemo (prior resection) | 4 |
| DC22 | 48 | Ad/Sq (BAC features) | T2N2M0 | Chemo/XRT | 7 |
| DC24 | 50 | Large cell | T1N2M0 | Chemo/XRT | 5 |
| DC25 | 63 | Sq | T2N2Mx | Chemo/XRT | 3 |
| DC30 | 58 | Ad | T2N3M0 | Chemo/XRT | 12 |
Trial Design
The trial was nonrandomized. Measurable immunologic response to vaccine was the major endpoint. Comparative immunologic data was central to the study. Individuals were stratified by stage and prior therapy (surgically resected I-II vs non-surgical stage III) to assess inhibitory effects of persistent tumour burden on immunologic responses. Secondary stratification looked at individuals who received prior chemotherapy and radiation versus surgical resection only. Small sample size and patient heterogeneity would preclude meaningful assessment of therapeutic effects. Clinical tolerability was determined by routine safety labs and clinical events described by the National Cancer Institute, Cancer Therapy Evaluation Program (CTEP), Common Terminology Criteria for Adverse Events (CTCAE).
Leukapheresis
A single mononuclear cell harvest was performed on each subject. The Cobe Spectra Apheresis System was used for all procedures. Three total blood volumes were processed at each procedure. A majority of individuals required placement of a femoral Udall catheter for access.
DC preparation
DC vaccines were prepared in compliance with FDA recommendations (BBIND-11543). CD14+ cells were isolated using a standard 4-hour plastic adherence step. CD14+ cells (90-95% pure) were cultured in XVIVO-15 medium with 2mM l-glutamine, 100μM nonessential amino acids, 1mM sodium pyruvate, and 20mM hepes buffer without addition of human serum or antibiotics. Initial culture was supplemented with 20ng/ml GMCSF (Berlex Inc., Seattle WA; GMP quality) and 20ng/ml IL-4 (R&D Systems Inc Minneapolis, MN; GMP quality), and cytokines were replenished on days 2 and 5. Tumour antigens (described below) were added at day 7. On day eight 1650-pulsed DCs were harvested and cryopreserved for safety testing in 90% human serum and 10% DMSO (v/v) using controlled rate freezer. A representative aliquot of the cryopreserved product was thawed at 24 hrs and sterility testing performed at the University of Kentucky Clinical Microbiology Laboratory. All products were free of bacterial or fungal contamination over two weeks, negative for mycoplasma and contained <5Eu endotoxin. The day of delivery, the product was thawed, washed 3 times in sterile PBS, and assessed for viability using trypan blue exclusion technique. In all cases viability was greater than 70%. Following these steps 108 antigen pulsed DCs were resuspended in 3.0 cc of sterile saline for immediate injection.
Antigen source and preparation
To generate a pluripotent vaccine, the adenocarcinoma cell line 1650 was used as a source of tumour-rejection antigens [7,8]. This tumour cell line, stable in long term culture, over-expresses Her-2/neu, CEA, Mage 2, WT-1 and survivin [7,8]. The line was confirmed sterile by Clinical Laboratory analysis and was confirmed mycoplasma negative at regular intervals using PCR (Gen-Probe Inc., San Diego, CA). Just prior to DC pulsing, 1650 were apoptosed, then lethally irradiated as previously described. In brief, 1650 (2×106 cells/ml) were placed 6.5cm from a 20 watt UV-b light source for 2 minutes. Cells were then irradiated with 10,000 rads using a Cesium-137 source. Irradiated 1650 were nonviable when monitored in extended culture. Apoptosis was confirmed by annexin-V staining. Apoptosed-irradiated 1650 were added to day 7 DC cultures at a DC:1650 ratio of 1:1 and incubated overnight (12hrs) at 37°c. Post incubation, the DCs were harvested, washed, counted, then cryopreserved as described above.
Immunization protocol
A prime vaccine and a single boost were given one month apart intradermally in the thigh. The target dose was 108 antigen-pulsed DCs in 3 cc volume. Average number of DCs injected in 14 prime injections was 8.2×107 (viability 84%) and 14 boost injections was 7.9×107 (viability 84%). Patients were monitored in the outpatient clinic for 2 hrs following immunization for immediate unanticipated adverse events.
Clinical evaluation
Follow-up by primary treating physicians included routine history and physical, CXR and/or Cat Scans at regular intervals post therapy or as directed by signs or symptoms of tumour recurrence.
Immunologic assessment
Serial blood samples were drawn for immunologic testing (prevaccine, wk1, wk4, wk5, wk8, wk12 and wk16 to complete the initial series; samples were also drawn at 6 and 12 months post vaccine). Peripheral blood mononuclear cells (PBMC) were isolated using standard Ficoll Hypaque separation and expanded using phytohemaggluttinin (PHA; 5μg/ml × 10d) as previously described [7]. PHA expansion was done to preserve the irreplaceable resource of samples from immunized patients that could be used for repeat immunologic analysis and/or related investigation. Although PHA expansion will increase the absolute magnitude of observed responses when directly compared to the unstimulated PBMC, data show excellent agreement of the relative measures and patterns of response [9]. The reliability of PHA expanded PBMC in immunological monitoring has been validated in our unpublished studies and in the literature [9]. Prior to analysis, lymphocytes were thawed, assessed for viability and rested in IL-2 free medium for two days. IFN-γ ELISPOT assays were performed using 1650-antigen pulsed autologous DCs as targets for immune reactivity and the controls of DCs alone and cells alone as previously described [7]. PHA stimulated normal donor PBMC were used as positive control in all assays.
Statistical methods
Significance was determined by ANOVA.
Results
DC Characteristics
The cell surface phenotype of the final vaccine product was as follows: CD83 5%±3%, CD80/CD86 33%±10%, CD40 77%±8%, (Figure 1A). The vaccines produced variable but low levels of IL10 (35±32 pg/ml) and IL12p40 (15±22 pg/ml), near baseline levels of IL-12p70 and TNF alpha, and moderate levels of IL6 (456±180 pg/ml) (Figure 1B). Additional comparisons of DC phenotype revealed similar levels of the maturation markers CD80 and CD83 pre and post antigen pulsing (pre: CD80/86= 28 +/− 17 and CD83= 5 +/− 2), but interestingly showed an increase in CD40 from 16%±17% to 77%±8%, suggesting a limited physiologic effect from culture with apoptosed-irradiated 1650 tumour cells. Experiments also showed these cells did upregulate CD80 and CD83 and were capable of producing significant amounts of IL-12p70 when matured with LPS/IFN-γ for 24 hrs in vitro (data not shown).
Figure 1. Phenotypic characteristics of DC vaccines.
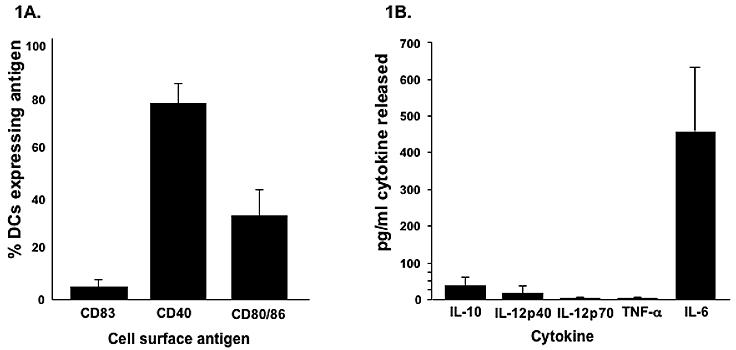
Data represent mean of 14 DC preparations ± SD (A) percent of cells expressing the maturation marker CD83, CD40, and the ratio of CD80/CD86 (B) cytokine secretion (106 DCs/ml/24 hr) of IL-10, IL-12p40, IL-12p70, TNF alpha, and IL6
Adverse effects
Most subjects noted mild erythema and indurations at the injection site for 24hr post immunization. In contrast to the previous vaccine, the delayed skin reaction, which appeared at the injection site 24-48 hr after immunization in ½ the subjects with the prior vaccine, was not seen in this series of subjects. That reaction may have been related to the more advanced maturation state of the DCs or from allo-antigens found in allogeneic human serum used in the preparation of that vaccine [7]. Safety labs checked following immunization revealed no abnormalities in haematological parameters or serum chemistries.
Immunologic responses to vaccines
IFN-γ ELISPOT was performed at all available timepoints in all individuals (Figure 2). A complete series of datapoints over 24 weeks were not available for all subjects. Measurements were made of the number of IFN-γ producing lymphocytes (spots) in 24hr culture with 1650-antigen pulsed DC targets (similar to vaccine product). Controls included lymphocytes plus DC without antigen and lymphocytes alone. Comparisons were made between each post-vaccine measurement and the pre-vaccine response for each condition. Comparisons were also made between conditions at individual timepoints (1650-pulsed DC targets vs. controls).
Figure 2. IFN-γ ELISPOT.
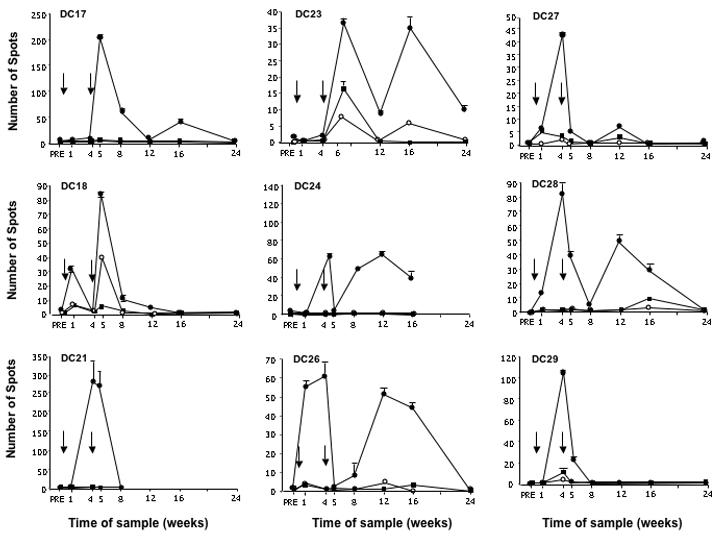
Measurements of lymphocytes (“spots”) from available timepoints responding to: (●)1650-pulsed autologous DCs, (■) DCs alone, (○) lymphocytes alone. Arrows indicate time of vaccine delivery.
Five individuals showed no significant increases in number of spots above baseline to 1650-pulsed DC targets nor increases above controls (not shown). Nine individuals showed clearly elevated reactions above baseline and timepoint controls (Figure 2). Pre-vaccine baselines ranged from 0 to 4 spots. Peak 1650-specific reactions ranged from 34±5 to 273±63 spots (p≤0.01 compared to controls). Patterns of reactivity varied between individuals. Seven of nine observed responses were biphasic. One of nine showed only one dominant peak and no data were available beyond week 8 on another. Peak reactions were seen at one or four weeks post prime and/or boost immunization, were measurable above baseline for four to eight weeks, and returned to baseline by 24 weeks.
Correlation of immune response with clinical criteria
Immunologic responses were seen in 4/7 stage III unresectable, and 6/7 stage I/II surgically resected patients, including 3/3 resected patients who had also received adjuvant chemotherapy and radiation. Immunologic responses were seen among all histologic sub-classifications of NSCLC.
Clinical Outcomes
Clinical follow-up is available on all individuals for a minimum of twelve months from primary immunization. Data are shown in Table 2. Five of fourteen individuals to date have documented disease recurrence or progression. One of seven surgically resected patients recurred and 4/7 stage III patients progressed, three of whom died two to seven months following detection and palliation of metastatic disease (DC19, DC21, DC25). One individual (DC30) with unresectable stage IIIB NSCLC was treated with whole brain radiation for two brain metastasis one month from the second vaccine and survives 9 months from that diagnosis without evidence of systemic or CNS disease. Another individual (DC23) with resected stage IB NSCLC was diagnosed with skeletal metastases two months from the second vaccine, was treated with chemotherapy radiation and Tarceva, and shows no evidence of disease 14 months after detection. Three of five patients with progressive disease showed no immunologic response. Two patients with unresectable stage III disease who had an immunologic response to immunization have radiographically stable disease at 23 and 51 months following initial chemo/radiation, although a third patient with stable unresectable stage III disease at 27 months from therapy showed no immunologic response.
Table 2. Patient outcomes and immune response to vaccines.
Subjects are stratified by clinical status at the time of the report. Upper table: no evidence of disease (NED; n=9). Lower table: recurrence / progression (n=5). Site of recurrent / progressive disease and status of the subject at the time of the repost is noted. Immunologic response measured by IFN-γ ELISPOT is indicated by positive ( + ; n=9) or negative ( − ; n=5). Stage grouping of TNM subsets is shown for each subject. Also shown is months from completion of definitive therapy and months from second vaccine at the time of the report.
|
Patient number |
Stage |
Months from conventional treatment |
Months from 2nd vaccine |
Recurrence from 2nd vaccine (months) |
Status at time of report |
Immunologic Response |
|---|---|---|---|---|---|---|
| DC17 | IB | 51 | 23 | - | NED | + |
| DC18 | IIIB | 51 | 24 | - | NED | + |
| DC20 | IA | 33 | 22 | - | NED | − |
| DC22 | IIIA | 27 | 18 | - | NED | − |
| DC24 | IIIA | 23 | 18 | - | NED | + |
| DC26 | IA | 32 | 14 | - | NED | + |
| DC27 | IA | 38 | 13 | - | NED | + |
| DC28 | IB | 28 | 13 | - | NED | + |
| DC29 | IIB | 17 | 12 | - | NED | + |
| DC19 | IIIB | Expired 12 |
Expired 6 |
3 | Progression Loco-regional Expired |
− |
| DC21 | IIIB/IV | Expired 6 |
Expired 2 |
2 | Progression Loco-regional Expired |
+ |
| DC23 | IB | 25 | 17 | 3 | Metastasis Skeletal Stable |
+ |
| DC25 | IIIA | Expired 7 |
Expired 2 |
<1 | Progression Loco-regional Expired |
− |
| DC30 | IIIB | 21 | 10 | 1 | Metastasis Brain Stable |
− |
Discussion
Based on antigen specificity of the immune system and safety profile of cancer vaccines, effective immunotherapy would be an ideal adjuvant following initial clinical responses to definitive therapy [10]. We have previously shown that biologically active autologous DC vaccines can be produced for a variety of NSCLC patients [7]. In the current series of patients we evaluate responses to immature DC vaccines. In this protocol we again used allogeneic tumour to produce a multivalent vaccine. This allowed us to immunize individuals who did not have autologous tumour available for vaccine construction. This also facilitates comparison of immunologic reactions across a heterogeneous patient group of subjects and between DC vaccine preparations [7].
Vaccines were well tolerated. Immunologic responses were noted in 6/7 surgically resected patients, three of who had received adjuvant medical therapy, and 3/7 stage III unresectable patients. Sample size and patient heterogeneity precluded meaningful statistical assessment of clinical outcomes. We stratified patients by surgical vs. non-surgical therapy for anecdotal review of clinical events. For historical clinical comparison, 5-year survival for surgically resected stage I/II disease is 60-70%, where by contrast, 75-80% of stage III unresectable patients are expected to die from their disease within 3 years of definitive therapy [1,2]. In the current series, six of seven surgically resected patients are without evidence of disease 17 to 51 months from definitive therapy (surgical resection or adjuvant therapy). Five of these six showed immunologic response following immunization. One of seven surgically resected subjects who showed a positive immunologic response to vaccine was diagnosed with skeletal metastases 3 months from the second vaccine and is alive 14 months from that diagnosis without current radiographic evidence of disease. Among the seven non-surgical patients (stage IIIA/B) in this recent series, four were diagnosed with progressive disease 2 weeks to 3 months from the second vaccine, three of whom have died, and one of whom has no evidence of disease 9 months after whole brain radiation for two brain metastasis. The three other non-surgical stage III patients survive with no evidence of disease at 23, 27 and 51 months from completion of chemotherapy/radiation for primary disease. No immunologic response was noted in three of five individuals with recurrence/progression, although among the three unresectable patients without evidence of progressive disease, only two showed immunologic response following immunization. It is important to iterate that our IFN-γ ELISPOT readout assay, that targets antigens used in the vaccine, is not necessarily indicative of clinical benefit. Conversely, neither are we sure that lack of measurable reactions by IFN-γ ELISPOT indicates lack of therapeutic benefit [6].
A number of variables related to both the host environment and the vaccine itself may be relevant to potential therapeutic efficacy [6]. Immunologic resistance of tumour to immune effector cells at the local level remains a potential limitation to vaccine efficacy. We continue to question whether metastasis in typically immune privileged sites, specifically the brain, are accessible to immune effector cells [7]. Additionally, our choice of antigens, that includes CEA, HER2/neu, WT-1, survivin, and Mage-2 may not have been optimal for all individuals. Notably, characterization of tumours for relevant antigens was not possible for a majority of subjects. Retrospective analysis of specific antigens expressed by some patient tumours is being considered. Regardless of the antigen relevance, and independent of the antigen specificity of the responses, both protocols used for DC vaccine production indicate autologous DC vaccines are capable of inducing immune responses in patients with all stages of lung cancer.
In context, our primary interest in this study was evaluation of immunologic response to DC vaccines. The distinguishing feature of this vaccine, compared to the previous, is that no maturation factor was added to the culture, which historically defines the vaccine as an immature DC preparation [3]. Importantly, the 64% immunologic response rate in the current series of 14 patients indicates biologic activity of this classic immature DC vaccine is comparable to the DCTCMF-matured DC vaccines used in the previous series of 16 patients [7]. The comparison is useful, and the results are instructive, in light of concerns that immature DC vaccines could be easily skewed to produce greater amounts of IL-10, thereby inducing tolerance rather than immune stimulation [11-16]. The data also appear to contrast with other small comparative human DC vaccine trials in the literature that suggest immature DC vaccines are less effective at immune induction than matured DC vaccines, possibly related to ineffective migration of antigen-pulsed vaccines in vivo [3,17-19]. Although the final vaccine preparation used here closely resembles the immature DC preparations described in those studies, a special note should be made of literature that indicates apoptotic tumour cells, but not tumour peptides, can provide a weak maturation stimulus in DC culture [20-23]. Importantly, neither CD80/86 nor CD83, both key determinants of DC maturation, were altered by ingestion of apoptotic bodies. The discrepancy between our observations and the current literature, however, might be explained if immature DC vaccines pulsed with apoptotic tumour cells behave differently in vivo than immature peptide-pulsed DC vaccines because they are “partially” matured. Thus the upregulation of CD40 following antigen pulsing is an intriguing finding that might suggest CD40 is a pivotal determinant of DC vaccine potency in vivo. Regardless, based on the immunologic responses seen in this study, we must hypothesize that immature DCs pulsed with apoptotic tumour cells do effectively complete their maturation and present antigen in vivo, and we should certainly question conjecture that a classically matured DC vaccine is required for maximal immune induction [15-24].
Assuming immunologic response can be validated as a measure of clinical benefit, the kinetics of observed responses could be relevant to putative therapeutic effects. Assigning significance to response patterns in this and other studies, however, is problematic in context of small study numbers, inherent biological variability, the partial view of the immune system provided by peripheral blood monitoring, and the semiquantitative nature of monitoring assays. Comparison of the two vaccine preparations used at our institution is facilitated by the consistency of antigen, dose, immunization schedule, and monitoring techniques [7]. By contrast cross-trial comparison is obfuscated by protocol differences, even when similar monitoring techniques were used [6]. Findings in this study that are consistent with other vaccine studies include the decline to baseline after peak response and evidence of recall on subsequent immunization [6]. The return to baseline after an increase of reactive T cells seen with each of the responders might be viewed as a transient response, or alternatively, that peripheral blood simply offers a measure of antigen reactive T cells in transit. Since we are not measuring T cell reactivity in tissue or nodal sites, interpretation is speculative. We also observed a biphasic, or recall response in 7/8 assessable responders (a ninth responder, DC21, expired before a second peak might have been observed). The reason that only a single peak was observed in DC29 is unclear. Additionally, despite the fact that the ELISPOT is only a semiquantitative assay, the diminished magnitude of measured response to a second immunization in two of those eight responders (DC17 and 27) may be a related phenomenon. An intriguing speculation is that T-regulatory cells induced by the prime immunization dampen the response to second immunization. Since pre-existing elevations in T regulatory cells in some cancer patients may also explain why some subjects show no response to immunization, comprehensive investigation is warranted; additional analysis of serial samples and correlation with immunologic reactivity is underway. Interestingly, ELISPOT measurements seen with the immature DC preparation are more consistently elevated over multiple time-points (over four to eight weeks) when compared to the DCTCMF-matured vaccines from the initial series. Although this may suggest that responses to the immature vaccine are protracted, the subject numbers are too small, and response variability too great, to statistically compare the kinetics of the two response patterns. Another notable difference in results between the current and the previous series is the relative absence of the 1650-antigen independent response seen in the 5/16 individuals from the initial series. We now suspect that particular response pattern was an allo-response to MHC antigens from allogeneic human serum that had been added to optimize DC culture conditions. The preparation used in the current series of subjects was grown in serum-free conditions.
In summary, the current study shows an immature DC vaccine preparation, pulsed with apoptotic tumour cells, has similar biologic efficacy to a DCTCMF-matured preparation in NSCLC patients. The reliability of these vaccines to induce immune responses presents an avenue to study anti-tumour immunity in lung cancer patients. Although vaccines may ultimately find a permanent role as adjuvants that consolidate responses to definitive medical, surgical or multimodality therapy for all stages of NSCLC, cost and required effort will likely preclude design of a large randomized therapeutic trial with DC vaccines. Nonetheless, this vaccine provides an acceptable immunologic standard with which to compare other vaccines that might be applied in a less resource intensive therapeutic protocol.
Acknowledgements
Research Support: Studies were supported by the Cancer Treatment Research Foundation Grant #G-01-009, National Institutes of Health R21-CA91624-02, Kentucky Lung Cancer Research Association, and the Veteran's Administration.
We would like to thank: Jennie Batsel, Scarlet Hawthorne, Jennie Bowden and Larry Dickson of the UK Leukapheresis Center for their outstanding care of the research patients.
Acknowledgements of research support: These studies were supported by the Cancer Treatment Research Foundation Grant #G-01-009, National Institutes of Health R21-CA91624-02, Kentucky Lung Cancer Research Association, and the Veteran's Administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
None declared - the authors of this manuscript, EA Hirschowitz, T Foody, GE Hidalgo and JR Yannelli, have no conflicts of interest that that could inappropriately influence this work.
References
- 1.Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004;350:379–392. doi: 10.1056/NEJMra035536. [DOI] [PubMed] [Google Scholar]
- 2.Arriagada R, Bergman B, Dunant A, et al. International Adjuvant Lung Cancer Trial Collaborative Group. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. N Engl J Med. 2004;350:351–360. doi: 10.1056/NEJMoa031644. [DOI] [PubMed] [Google Scholar]
- 3.Cranmer LD, Trevor KT, Hersh EM. Clinical applications of dendritic cell vaccination in the treatment of cancer. Cancer Immunol Immunother. 2004;53:275–306. doi: 10.1007/s00262-003-0432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mocellin S, Mandruzzato S, Bronte V, et al. Part I: Vaccines for solid tumours. Lancet Oncol. 2004;5:681–689. doi: 10.1016/S1470-2045(04)01610-9. [DOI] [PubMed] [Google Scholar]
- 5.Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opin Immunol. 2005;17:163–169. doi: 10.1016/j.coi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Keilholz U, Weber J, Finke JH, et al. Immunologic monitoring of cancer vaccine therapy: results of a workshop sponsored by the Society for Biological Therapy. J Immunother. 2002;25:97–138. doi: 10.1097/00002371-200203000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Hirschowitz EA, Foody T, Kryscio R, et al. Autologous dendritic cell vaccines for non-small-cell lung cancer. J Clin Oncol. 2004;22:2808–2815. doi: 10.1200/JCO.2004.01.074. [DOI] [PubMed] [Google Scholar]
- 8.Wroblewski JM, Bixby DL, Borowski C, et al. Characterization of human non-small cell lung cancer (NSCLC) cell lines for expression of MHC, co-stimulatory molecules and tumour-associated antigens. Lung Cancer. 2001;33:181–194. doi: 10.1016/s0169-5002(01)00210-0. [DOI] [PubMed] [Google Scholar]
- 9.Godoy-Ramirez K, Franck K, Mahdavifar S, et al. Optimum culture conditions for specific and nonspecific activation of whole blood and PBMC for intracellular cytokine assessment by flow cytometry. J Immunol Methods. 2004;292:1–15. doi: 10.1016/j.jim.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 10.Hirschowitz EA, Hiestand DM, Yannelli JR. Active Immunotherapy For Lung Cancer. Journal of Thoracic Oncology. 2006;1:93–104. [PubMed] [Google Scholar]
- 11.Kalinski P, Hilkens CM, Wierenga EA, et al. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20:561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 12.Kapsenberg ML, Hilkens CM, Wierenga EA, et al. The paradigm of type 1 and type 2 antigen-presenting cells. Implications for atopic allergy. Clin Exp Allergy. 1999;29:S33–S36. [PubMed] [Google Scholar]
- 13.Rissoan MC, Soumelis V, Kadowaki N, et al. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–1186. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 14.Groux H, Bigler M, de Vries JE, et al. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 16.Itano AA, Jenkins MK. Antigen presentation to naive CD4 T cells in the lymph node. Nat Immunol. 2003;4:733–739. doi: 10.1038/ni957. [DOI] [PubMed] [Google Scholar]
- 17.Jonuleit H, Giesecke-Tuettenberg A, Tuting T, et al. A comparison of two types of dendritic cell as adjuvants for the induction of melanoma-specific T-cell responses in humans following intranodal injection. Int J Cancer. 2001;93:243–251. doi: 10.1002/ijc.1323. [DOI] [PubMed] [Google Scholar]
- 18.de Vries IJ, Lesterhuis WJ, Scharenborg NM, et al. Maturation of dendritic cells is a prerequisite for inducing immune responses in advanced melanoma patients. Clin Cancer Res. 2003;9:5091–5100. [PubMed] [Google Scholar]
- 19.De Vries IJ, Krooshoop DJ, Scharenborg NM, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63:12–17. [PubMed] [Google Scholar]
- 20.Chen Z, Moyana T, Saxena A, et al. Efficient antitumour immunity derived from maturation of dendritic cells that had phagocytosed apoptotic/necrotic tumour cells. Int J Cancer. 2001;93:539–548. doi: 10.1002/ijc.1365. [DOI] [PubMed] [Google Scholar]
- 21.Ishii S, Hiroishi K, Eguchi J, et al. Dendritic cell maturation induced by delivery of ultraviolet-mediated apoptotic colorectal cancer cell lines. Anticancer Res. 2003;23:2457–2463. [PubMed] [Google Scholar]
- 22.Ip WK, Lau YL. Distinct maturation of, but not migration between, human monocyte-derived dendritic cells upon ingestion of apoptotic cells of early or late phases. J Immunol. 2004;173:189–196. doi: 10.4049/jimmunol.173.1.189. [DOI] [PubMed] [Google Scholar]
- 23.Rovere P, Vallinoto C, Bondanza A, et al. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- 24.Adema GJ, de Vries IJ, Punt CJ, et al. Migration of dendritic cell based cancer vaccines: in vivo veritas? Curr Opin Immunol. 2005;17:170–174. doi: 10.1016/j.coi.2005.01.004. [DOI] [PubMed] [Google Scholar]