

Abstract
In addition to their conventional G-C/T target sequences, Sp1 family transcription factors (Sp-factors) can interact with a subset of the target sequences for NFκB. Due to the low level of bona fide NFκB activity in most resting cells, this interaction between Sp-factors and κB-sites could play important roles in cell function. Here we used mutagenesis of a canonical κB element from the immunoglobulin and HIV promoters to identify the GC-rich sequences at each end required for Sp-factor targeting. Through screening of multiple κB-elements, a sequence element located in the second intron of superoxide dismutase-2 (SOD2) was identified as a good candidate for both NFκB and Sp-factor binding. In neurons, the prominent proteins interacting with this site were Sp3 and Sp4, whereas Sp1, Sp3, and NFκB were associated with this site in astroglia. The neuronal Sp-factors repressed transcriptional activity through this κB-site. In contrast, astroglial Sp-factors activated promoter activity through the same element. NFκB contributed to control of the SOD2 κB element only in astrocytes. These findings imply that cell-type specificity of transcription in the CNS—particularly with regard to κB elements—may include two unique aspects of neurons: 1) a recalcitrant NFκB and 2) the substitution of Sp4 for Sp1.
Transcription factors belonging to the family typified by Sp1 are ubiquitously expressed in mammalian cell types. Sp-factors are involved in the expression of a large number of genes, including most of those known as housekeeping genes; therefore, these factors might participate in every aspect of cellular activity. Indeed, null mutation of Sp1 in mouse is embryonic lethal (1); similarly, mice genetically ablated for Sp3 die soon after birth (2). The abnormalities arising from Sp4 ablation are most apparent in the nervous system (3,4), consistent with the finding that Sp4 expression is highly enriched in neurons (4–6). Other zinc-finger transcription factors with homology to Sp1, including Sp2, have DNA-binding preferences quite distinct from those for Sp1, -3, and -4 (7). Collectively, these findings indicate that Sp-factors are not redundant in their roles and that none is dispensible. Abundant evidence shows that each Sp-factor possesses discrete functional properties (8). Sp1 is generally considered a transcriptional activator, although an inhibitory region has recently been mapped in its extreme aminoterminus (7,9). Sp3 can be an activator or inhibitor dependent on its translation initiation sites, posttranslational modifications, and the sequence context of a given cis element (10). Sp4 also exhibits some flexibility with regard to transactivation, but the determinants of its activity remain obscure. The importance of Sp-factors has also become manifest in two forms of human disease. Leaching Sp1 from chromatin by a CUG expansion appears to be a key step for the development of myotonic dystrophy type I (11). Similarly, sequestering Sp-factor activity by mutant huntington protein might be the one of the primary cellular events in the pathogenesis of Huntington’s disease (12,13).
Apart from binding to their consensus GC or GT rich sequences, Sp-factors are able to bind to non-canonical sequences, in particular to some κB-elements (14–18). In neurons, the interaction between Sp-factors and κB-elements could be especially important for cell function because of the paucity and/or recalcitrance of NFκB activity in neurons (5,17,19–22). The prominent proteins binding to κB-elements in neurons are Sp-factors, and the activity of Sp-factors can be diminished by toxic levels of glutamate (17,21). In mixed neuron-glia cultures, glutamate induces NFκB activity in the glia but not in the neurons; NFκB is unresponsive to glutamate in pure cultures of glia or neurons (19). For these reasons, and to further elucidate the disparate gene-regulatory mechanisms utilized by neurons and glia, it is important to characterize in neurons the influence of Sp-factors on the transcription of genes that are responsive to NFκB in other cell types.
One gene typically controlled by NFκB factors and playing an important role for cell survival is superoxide dismutase-2 (SOD21; also known as manganese SOD) (23). SODs are a group of enzymes engaged to fend off cellular stress initiated by reactive oxygen species (ROS). SOD2 is located in the mitochondrial matrix and plays an indispensable role in protecting cells from a myriad of insults (24–27). In many types of cells, ROS can activate NFκB, which in turn upregulates the compensatory expression of prosurvival genes. These may include SOD2, as its promoter contains a functional κB cis element. Interestingly, the functional κB-site has been mapped into the second intron of both human and mouse SOD2 genes (23,28,29). This intron also harbors other essential enhancer elements (C/EBP-1, C/EBP-2 and C/EBP-x) besides the κB site. Similar gene organization is found in the rat SOD2 gene, where the κB site is conserved with a slight variation from human and mouse counterparts (Table 1).
Table 1.
κB-oligonucleotides used in EMSA studies
| Name | Sequence |
|---|---|
| Ig/HIV-κB | AGTTGAGGGGACTTTCCCAGGC |
| mut1 | AGTTGAcGGGACTTTCCCAGGC |
| mut2 | AGTTGAGcGGACTTTCCCAGGC |
| mut3 | AGTTGAGGcGACTTTCCCAGGC |
| mut4 | AGTTGAGGGcACTTTCCCAGGC |
| κBΔC | ATTGGGGACTTTCCAGGC |
| IL6 | AATGTGGGATTTTCCCATGA |
| APP1 | GAGACGGGGTTTCACCGTGTT |
| APP2 | GCATGGGGCTCCTCCCACCG |
| Bcl-x | GGCGGGGGGGACTGCCCAGGGAG |
| COX-2 | CGGGAGAGGGGATTCCCTGCGCC |
| MHC class I | CCAGCTTGGGGATTCCCCATCTCC |
| SOD2(rat) | GAGTAGGGGAAAAGCCCAGTTGG |
| SOD2(human) | AGACTGGGGAATACCCCAGTTGT |
| SOD2(mouse) | AAGCAGGGGAATAGCCCAGTTGG |
The core target for NFκB is underlined.
Based on our analysis of κB-sequences required for Sp-factors binding, we speculated that the SOD2-κB sequence could be an efficient site for both NFκB and Sp-factors binding. We found that the SOD2-κB site could be bound by both Sp-factors and NFκB in astrocytes, while in neurons the prominent binding factors were Sp3 and Sp4. Reporter assays showed that the intron was inhibitory in neurons and the κB site was solely responsible for this effect. In astrocytes, the intron acted as an activator driven by NFκB and Sp-factors (Sp1 and Sp3).
MATERIALS AND METHODS
Materials
The Sp1 consensus oligonucleotide and the oligonucleotide (Ig/HIV-κB) containing the κB enhancer shared by the immunoglobulin light chain and the HIV-LTR was obtained from Promega (Madison WI). Invitrogen (Carlsbad CA) supplied all other oligonucleotides. Antibodies against Rel family and Sp-family proteins were obtained from Santa Cruz: p50 (sc-114x), p65 (sc-372x), p52 (sc-297x), RelB (sc-226x), c-Rel (sc-71x), Sp1 (sc-59x), Sp3 (sc-644x), and Sp4 (sc-645x). Tumor necrosis factor (TNF, Cat# 400-14) was purchased from PeproTech Inc. (Rocky Hill NJ). Lipopolysaccharide (LPS) and glutamate were from Sigma (St Louis MO). Most of the oligonucleotides used in EMSA are delineated in Table 1; not listed: 50-14 (GTG ACG GGG AGG CCC CCA TAT), NFAT (GGA GGA AAA ACT GTT TCA TAC AGA AGG CGT), AP1 (CGC TTG ATG AGT CAG CCG GAA), and Oct1 (TGT CGA ATG CAA ATC CAT AGA A). Phosphothiate-modified oligonucleotides were used for Sp1 decoy (5′-CCA TAA GGG CGG GCA TTA GTC-3′) and its scrambled control (5′-GAC TGC AGT GAT CGA CTG ACG-3′).
Cell cultures
Primary neuronal cultures were established from the neocortices of 18-day Sprague-Dawley rat embryos as described previously (21). Neurons were maintained in Neurobasal medium containing B27 supplement (both from Invitrogen), 0.5 mM L-glutamine, and 10 μg/ml gentamycin sulfate. This serum-free medium combined with a transient (first five days) exposure to the mitotic inhibitor cytosine arabinoside (AraC, 3–10 μM) achieved cultures that were at least 99% neurons (19). All neuronal cultures used in this study were 8–10 days in vitro. Astrocyte cultures were established from Sprague-Dawley rat postnatal day-2 and maintained in minimal essential medium with Earle’s salts (MEM, Invitrogen) supplemented to 10% with fetal bovine serum (FBS, Invitrogen), as described previously (19). After reaching confluency, the astrocytes were denuded of microglia by shaking and vigorous lavage; the remaining cells had a morphology consistent with type I astrocytes and were subcultured for experiments by trypsinization. The NT2 (NTera2/D1) human neural-lineage teratocarcinoma cell line was obtained from Stratagene (La Jolla CA). The T98G human astroglioma cell line was obtained from American Type Culture Collection (Manassas VA); the N9 mouse microglial cell line was described previously (30). Mammalian cell lines were maintained in MEM + 10% FBS. NT2 cells were differentiated according to the rapid, neurosphere protocol developed by Cheung et al. (31). Specifically, the NT2 cells were grown in suspension and treated with retinoic acid (10 μM) for 14 days, followed by another 5 days of enrichment with AraC (1 μM) and uridine (10 μm) in Neurobasal/B27 medium; most cells were morphologically consistent with mature neurons after enrichment. Drosophila melanogaster SL2 cells were maintained at room temperature in Schneider’s insect medium (Sigma) with 10% FBS and 10 μg/ml gentamycin sulfate.
Plasmids
pGL3-basic was purchased from Promega. pGL3-RSI was constructed by PCR-cloning rat SOD2 intron 2 (RSI) into Mlu I and Bgl II sites of pGL3-basic. Relative to the κB site, the cloned sequence included 400 bp in the 5′ direction (primer: GCC GAC GCG TGC CAA CCA CAA CTT CTG GG; Mlu I site underlined) and 100 bp in the 3′ direction (primer: CCG GAG ATC TGT CTC CAC GGA AGG G; Bgl II site underlined) to the κB site and the clone was verified by sequencing. This sequence contains most of the elements responsive to TNF stimulus (23). GeneEditor™ in vitro Site-Directed Mutagenesis System (Promega) was used to mutate the κB site (GGGGAAAAGCCC → GGGGAAAAGatC) and the mutation was verified by sequencing. A truncated form of the SV40-promoter [SV40-promΔ; (17)], in which six tandem-repeat Sp1 sites were deleted, was derived from pGL3-prom (Promega). The TK promoter was obtained from pRL-TK (Promega). pGL3-RSI-promΔ and pGL3-RSImut-promΔ were constructed by inserting SV40-promΔ into the multiple cloning sites (Bgl II and Hind III sites) of pGL3-RSI and pGL3-RSImut, respectively. pGL3-RSI-TK and pGL3-RSImut-TK were constructed by the same strategy. The p65 and IκBα coding regions were amplified from NT2 cells with a high-fidelity PCR system (cat# 1732 641; Roche, Indianapolis IN) and cloned in-frame into pEFGP-N1 (p65) or pDsRed2-N1 (IκBα) (BD Biosciences-Clontech, Mountain View CA), creating p65-EGFP and IκB-DsRed2 fusion proteins. Clones were verified by sequencing and p65-EGFP proteins were located in nuclei while IκBα cotransfection retained all p65-EGFP in cytosol (32). The pRL-CMV (Promega) plasmid expressing Renilla luciferase constitutively was used as a control for transfection efficiency and viability. All plasmids were prepared with Qiagen Midiprep kits, and DNA quality and quantity were determined by both spectrophotometry and visual inspection in agarose gels.
Transient transfection and luciferase activity assay
Mammalian cells were cultured in 24-well plates and transfected using Lipofectamine 2000 (Invitrogen). Astrocytes were transfected when the cultures reached 60–70% confluency, whereas neurons were plated and transfected at a density of 1 × 105/cm2. One μg total DNA per well was used; neuron transfections: 0.5μg pGL3-basic or pGL3-promΔ series, 0.1 μg pGL3-TK series, and 0.1 μg pRL-CMV; astrocyte transfections: 0.6 μg pGL3-TK series and 0.1 μg pRL-CMV; pBluescript II (Stratagene) was used to adjust the total to 1 μg. When decoy oligonucleotides (50 nM final concentration) were used, pBluescript II was omitted. For p65 and IκBα cotransfection, 0.2 μg (per well) plasmid encoding these proteins was used. Transfections took place in the existing media (for neurons, Neurobasal with B27 supplement; for astrocytes, MEM + 10% FBS). After a 60–90 min of incubation, DNA and lipofectamine were washed off, and fresh maintenance medium was applied. Cells were harvested ~24 h after transfection for luciferase assay. Dual-luciferase reporter assay system (Promega) was used to detect both firefly and Renilla luciferase activities through a Veritas luminometer (Promega). All experiments were repeated at least five times. SL2 cells were transfected by pPACSp1 and pPACSp3 with a calcium phosphate method, which has been described previously (17).
Nuclear protein extraction and electrophoretic mobility shift assay (EMSA)
The detailed protocols have been described previously (17,19). N9 cells were activated with 10 nM LPS for 60 min and primary rat astrocytes were treated with TNF (100 ng/ml) for 60 min prior to nuclear protein extraction.
Chromatin immunoprecipitation (ChIP) assay
Cells were fixed with 1% formaldehyde in MEM for 10 min at room temperature, followed by two washes with PBS. Cells were scraped and harvested in PBS; this PBS and all subsequent buffers contained the following protease inhibitors: phenylmethylsulfonyl fluoride (PMSF, 0.5 mM), leupeptin (10 μg/ml), aprotinin (10 μg/ml), and calpain inhibitor ALLN (1 μM). After centrifugation, cell pellets were dissolved in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) and sonicated. The sonication conditions were optimized for each of the two cell types used. Chromatin was reproducibly reduced to fragments of 200–1000 bp by 8 and 12 sonication pulses in NT2 and T98G cells, respectively. Each pulse lasted 10–15 seconds and the energy output was set at 25% of sonicator maximum (F60 Sonic Dismembrator, Fisher Scientific). Cell debris was removed by centrifugation at 16,000 × g for 10 min at 4°C. Subsequent steps utilized buffers supplied in a kit by Upstate Biotech (Lake Placid NY), essentially following the manufacturer’s instructions. The cleared supernatants were diluted 10-fold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 167 mM NaCl, 16.7 mM Tris-HCl, pH 8.1). The solution was precleared with a slurry of protein A/G-agarose (Invitrogen) in TE buffer containing 200 μg/ml sheared salmon-sperm DNA (Boehringer Mannheim GmbH, Germany) and 1 mg/ml bovine serum albumin (BSA), incubating for 60 min at 4°C with rotation, followed by removal of the A/G-agarose by centrifugation. The precleared solution was incubated with 2 μg of primary antibody at 4°C overnight. Antibody complexes were recovered with protein A/G-agarose in salmon sperm DNA and BSA; the agarose pellets were then washed with 1 ml of the following buffers in order: once with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 150 mM NaCl, pH 8.1); once with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl, pH 8.1); once with LiCl wash buffer (250 mM LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1); three times with TE buffer, pH 8.0. DNA-protein complexes were eluted from the agarose pellets with two sequential aliquots of 250 μl elution buffer (1% SDS, 0.1 M NaHCO3). The eluted samples were combined and protein-DNA crosslinking was reversed by incubation in 200 mM NaCl at 65°C for 4 h, followed by proteinase K (10 μg/ml) digestion at 45°C for 1 h. The ChIP DNA was extracted once with phenol/chloroform and once with chloroform and then precipitated in ethanol. Purified DNA was dissolved in 30 μl of H2O. One μl of DNA was amplified (56°C for annealing) in a 25 μl PCR reaction (forward primer: 5′-CGA ACC TTG AAT TAC GGG AAA; reverse primer: 5′-CCT GGT GTC AGA TGT TGC CT). PCR products (130 bp) were fractionated and visualized on a 1.2% agarose gel.
Reverse transcription and polymerase chain reaction (RT-PCR)
The general protocol has been described elsewhere (17). Specific primers and PCR conditions were as follows: SOD2 upstream primer: 5′-CTG GCC AAG GGA GAT GT-3′; downstream primer: 5′-GGC CTG TGG TTC CTT GC-3′ (27 cycles, producing a 309-bp product). β actin upstream primer: 5′-GTC CTC TGC CAT GTG GTT TTC-3′; downstream primer: 5′-GCT GCG CTC TCG TAA TTG TG-3′ (21 cycles, producing a 439-bp product). Annealing temperature for all reactions was 60°C. The cycle numbers and other conditions were optimized to achieve linear rates of amplification. All data depicted are representative of at least three experiments.
RESULTS
Sequence requirements for Sp-factors binding to κB sites
We previously determined that three different classes of transcription factor (Sp-factors, NFκB, and RBP-Jκ) can bind to the Ig/HIV-κB site (17), a sequence widely used as a probe in “NFκB” EMSA analyses. RBP-Jκ binding is conferred by a sequence located at the 3′ half of this site (5′-CCTGGGAAA-3′ in the complementary strand) and requires additional 3′ flanking sequences outside the core κB element. All these interactions between transcription factors and the Ig/HIV-κB sequence appeared to be sequence specific (17). We extended this investigation to establish the efficiency of the interaction between Sp-factors and this κB site. Nuclear extract containing Sp-factors, NFκB, and RBP-Jκ was prepared from LPS-activated N9 microglial cells; the Sp-factors in these cells consist primarily of Sp1 and Sp3 (5,6). EMSA analysis was performed using the Ig/HIV-κB sequence as a 32P-labeled probe and various sequences as unlabeled (“cold”) oligonucleotides in competition assays. When Ig/HIV-κB was used as an autologous competitor, binding was diminished for each of the three classes of transcription factors; but as a fraction of its baseline value, binding of the Sp-factors was the least sensitive among the three (Figure 1A). In contrast, a canonical Sp1 target sequence competed much more efficiently with the κB sequence for binding to Sp-factors; this sequence had no effect on the binding activity of NFκB or RBP-Jκ. No appreciable competition for any of the three transcription factors was observed when unrelated target sequences for transcription factors AP-1 or Oct1 was used. A similar competition pattern was observed when the Sp1 target sequence was used as a radiolabeled probe (data not shown). These results suggest that the affinity of Sp-factors for the Sp1 target sequence is approximately 30-fold greater than that for the Ig/HIV-κB sequence. However, a precise quantitation of this difference is confounded by a third-order competition for probe between the Sp-factors and the other transcription factors in these extracts, the quantities of which undoubtedly vary with cellular conditions.
Figure 1. GC-rich sequences required for the interaction of Sp-factors with Ig/HIV-κB sequence.
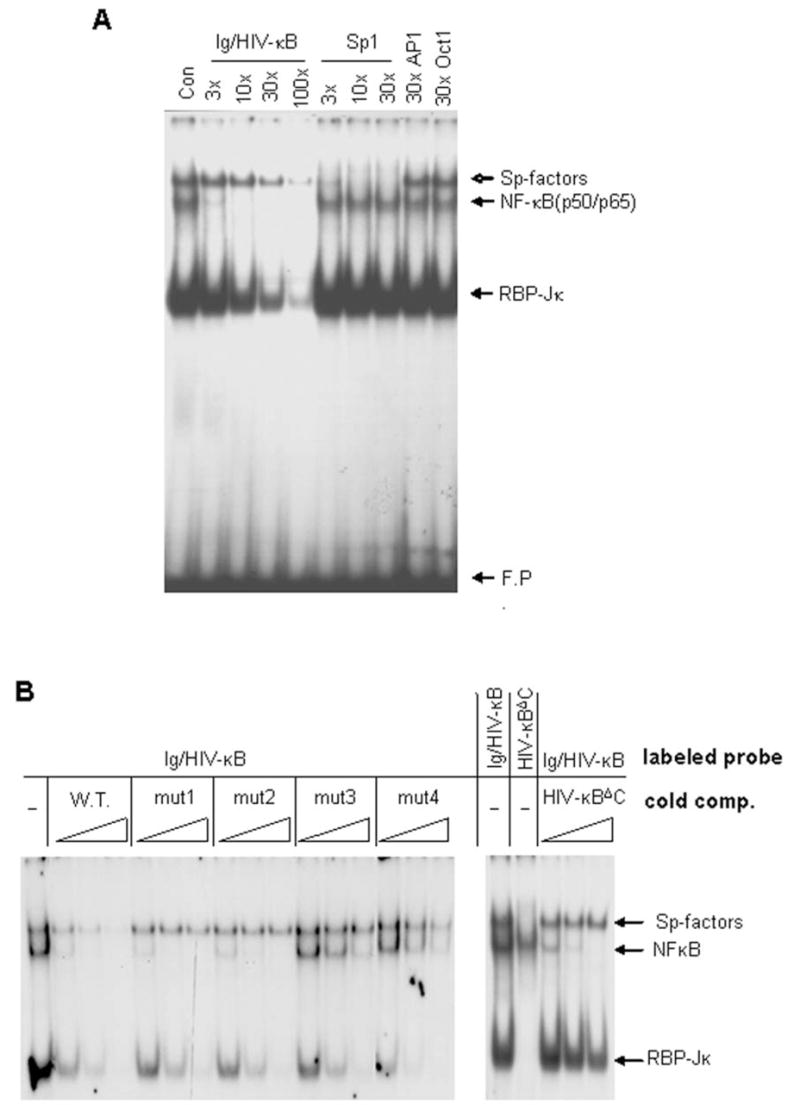
A. Nuclear extracts were prepared from N9 microglial cells activated with LPS and analyzed by EMSA. Three complexes [Sp-factors, NFκB (p50/p65) and RBP-Jκ] interacted with a radiolabeled Ig/HIV-κB probe. Cold competition was performed with a 3- to 100-fold molar excess of oligonucleotide containing the sequence of the probe (lanes 2–5), a sequence specific for Sp-factors (lanes 6–8), or sequences specific for AP-1 or Oct-1. B. In the left panel, Ig/HIV-κB and mutants thereof (see Table 1) were applied as cold competitors for the binding to radiolabeled Ig/HIV-κB probe. In the right panel, Ig/HIV-κBΔC was used as the radiolabeled probe for tests of direct binding (lane 2) or it was applied as a cold competitor of radiolabeled Ig/HIV-κB (lanes 3–5). In both panels, cold competitors were applied at 3-, 10-, or 30-fold molar excess.
Sp-factors bind to a subset of κB-elements (14), so it is important to determine the sequence requirements for this interaction. We made such determinations within the context of the widely used Ig/HIV-κB sequence. The 5′ end of the sequence contains four adjacent guanines (G4 motif). Each guanine in the G4 motif was individually mutated to cytosine (G→C) (κB-mut1-4, Table 1). To evaluate the binding efficiency, the mutated oligonucleotides were used as unlabeled competitors for Sp-factor binding to the 32P-labeled Ig/HIV-κB sequence. Again, activated N9 nuclear extracts were used to assay Sp-factors and NFκB in the same EMSA analysis. Mutation of any of the four guanines abolished Sp-factor binding, as substantial competition was not observed from any of the mutated sequences (Figure 1B). In contrast, none of the mutations interfered with RBP-Jκ binding, as the 3′-half of Ig/HIV-κB sequence was unchanged. The third and fourth guanines were required for NFκB binding, as expected from their location within the κB core sequence. But mutation of the first two guanines did not affect NFκB binding. The first guanine was predicted to be dispensible, as it lies outside the κB core sequence. NFκB binding after mutation of the second guanine can be explained by the retention of a κB-site (GGGAAAGTCC) in the complementary strand.
To test the sequence requirements of the 3′ end of the Ig/HIV-κB site for Sp1 binding, we made another mutation (κBΔC; Table 1) removing the distal cytosine from the three cytosines (C3 motif) present there. When κBΔC was used as a labeled probe, NFκB was the only prominent band and little Sp1 binding could be detected (Figure 1B). In competition assays, κBΔC could compete for NFκB binding with the same efficiency as the intact Ig/HIV-κB sequence (Figure 1B), while κBΔC was not an efficient competitor for RBP-Jκ or Sp-factors binding to the original Ig/HIV-κB probe. These data indicated that both the G4 motif and the C3 motif were essential for Sp-factors binding to the Ig/HIV-κB probe.
Together with our earlier work (19), the experiments presented here indicate that Sp-factor binding to the Ig/HIV-κB site requires four guanines at the 5′ end and three cytosines at the 3′ end of the same strand. For example, the IL6-κB site has three guanines at 5′, which makes it a poor site for Sp1 binding (17). Generally, the shorter the spacing between 5′ guanines and 3′ cytosines the more favorable is Sp1 binding; a heavy GC content in the spacer and flanking regions increased Sp1 binding efficiency to a κB site (data not shown).
Assessment of Sp-factor binding to gene-specific κB sites
Based on the sequence requirements noted above, we identified promising κB sites from other genes, focusing on genes with neuronal expression. These κB enhancer sequences were tested for binding by Sp-factors. Sequences were selected based on a high content of guanines and cytosines at respective ends with a low GC content between (the spacer region). The human amyloid precursor protein (APP) promoter contains two elements consisting of the same κB sequence (APPκB1) repeated; induction of APP by stimuli such as IL-1 reportedly depends on one or both of these elements (33). We found another candidate κB site (APPκB2) sandwiched between these two APPκB1 sites, apparent in the complementary strand. Functional κB sites have been reported in the following neuronally expressed genes: Bcl-x (34,35), COX-2 (36,37), MHC class I (38,39), IL-6 (40,41), and SOD2 (23,28). Oligonucleotides corresponding to the κB elements from these genes (Table 1) were used in competition assays with a 32P-labeled Ig/HIV-κB probe. All κB sequences except APPκB2 were able to compete for NFκB binding efficiently (Figure 2A). Only Sp-factors could be detected when APPκB2 site was used as a labeled probe in EMSA (data not shown). Of the remaining κB oligonucleotides, IL6-κB was unable to compete efficiently for Sp-factor binding, while APP-κB1 interacted with Sp-factors inefficiently (17). These data were consistent with our Ig/HIV-κB mutation results; specifically, the IL6-κB sequence contains only three guanines on the 5′ end, and the APP-κB1 sequence contains only two cytosines on the 3′ end. The COX2 and MHC class I sequences are identical in their core κB sequence, and neither could compete for Sp-factors efficiently. On the other hand, Bcl-x and rat SOD2 sequences indicated substantial Sp-factor binding.
Figure 2. Preference of Sp-factors for specific κB sequences.
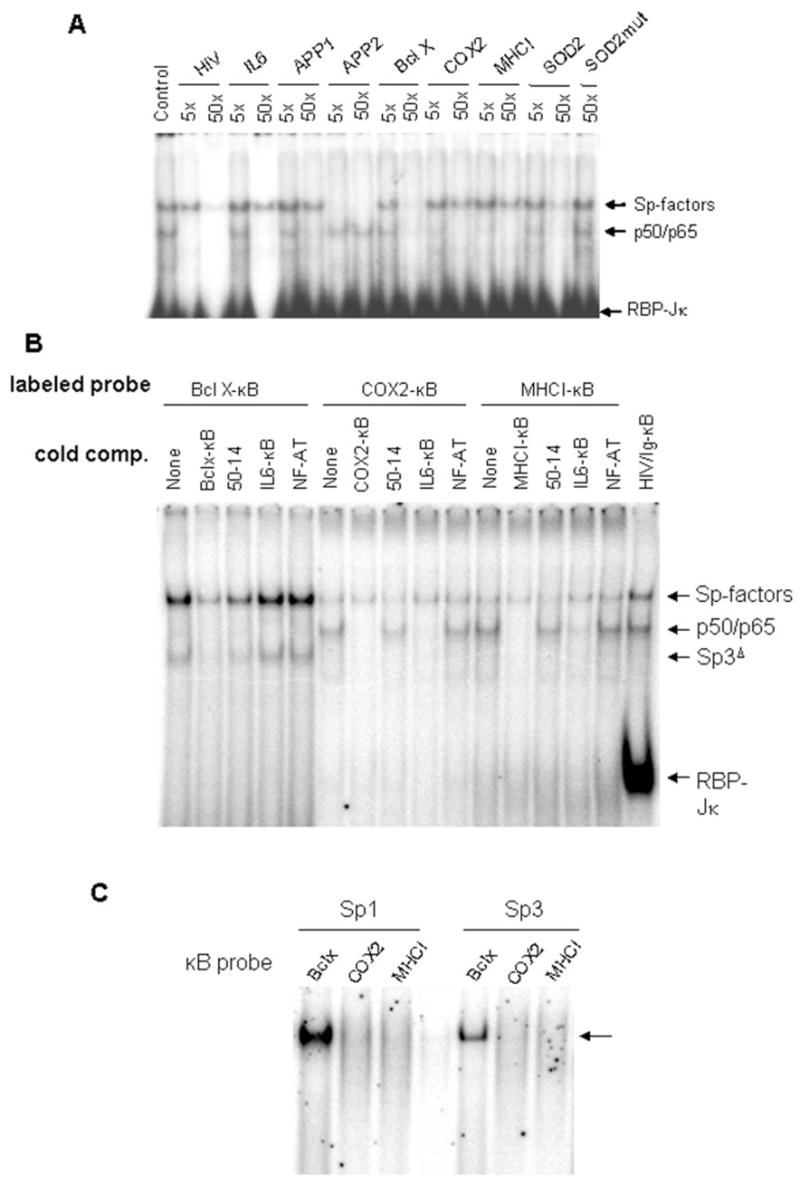
A. Nuclear extracts from activated N9 cells were prepared and an Ig/HIV-κB probe was used in EMSA. Excess (5- or 50-fold molar ratio) cold oligonucleotides were used to compete with the radiolabeled Ig/HIV-κB probe. Oligonucleotide sequences are listed in Table 1. B. BclX-κB, COX2-κB, and MHCI-κB sequences were radiolabeled to directly probe the binding activities in activated N9 nuclear extracts. The indicated cold oligonucleotides were added into some reactions to compete with these probes as a test of binding specificity. Cold oligonucleotides were at a 30-fold molar excess compared with probes. In the last lane, an Ig/HIV-κB probe was used as a positive control. C. Sp1 and Sp3 expressed in SL2 cells were used to show the interaction with different κB probes.
We labeled some of the oligonucleotides with 32P to test them in direct binding assays (Figure 2B). Interestingly, although the Bcl-x-κB sequence could compete with Ig/HIV-κB for both Sp-factors and NFκB, its prominent binding factors were of the Sp-family (Figure 2B), presumably because of the high GC content of the Bcl-x sequence. In contrast, COX2-κB and MHC I-κB were effective targets for NFκB but showed very poor Sp-factor binding. As competitors, the Sp-binding sequence 50-14 (19) and the NFκB-specific IL6-κB sequence had essentially reciprocal effects on active binding. It is noteworthy that the Bcl-x-κB sequence also showed substantial binding by Sp3Δ, an alternative translation product of the Sp3 mRNA that can be detected on a Sp1 consensus probe (e.g., ref. (17)).
We also tested recombinant Sp1 or Sp3 produced in SL2 cells. In a direct-binding EMSA, both proteins bound the Bcl-x-κB sequence but not COX2-κB or MHC I-κB sequences (Figure 2C), confirming that Sp-factors could not interact with COX2-κB and MHC I-κB efficiently. These data indicated that Bcl-x-κB was a Sp-favored site while COX2-κB and MHC I-κB were mainly NFκB binding sites.
Factors binding to SOD2-κB site in neurons and glia
The SOD2-κB site resides in the second intron of the gene and is reportedly essential for induction of SOD2 by multiple stimuli in human and mouse (23,42). The sequence of this site is highly conserved in human, rat, and mouse (Table 1). Although these differ slightly in the spacer region, all species contain a G4 motif at the 5′ end and a C3 motif at 3′ end. Also, the low GC content in the spacer region suggests that the SOD2-κB site could be a target sequence for both Sp-factors and NFκB. Indeed, consistent with the competition assays, both Sp-factors and NFκB were able to bind to the SOD2-κB site (Figure 3A). Two SOD2-κB-binding components could be detected in astrocytes that were not present in neurons, and these astrocytic complexes were further enhanced by TNF treatment (Figure 3B), suggesting these two bands were derived from Rel-family proteins related to NFκB. Consistent with our earlier results (17), the prominent neuronal κB-binding factors were Sp-factors that had activity diminished by toxic levels of glutamate when SOD2-κB, Ig/HIV-κB and Sp1 probes were surveyed (Figure 3B).
Figure 3. Rat SOD2 intronic κB site interaction with NFκB and Sp-factors.
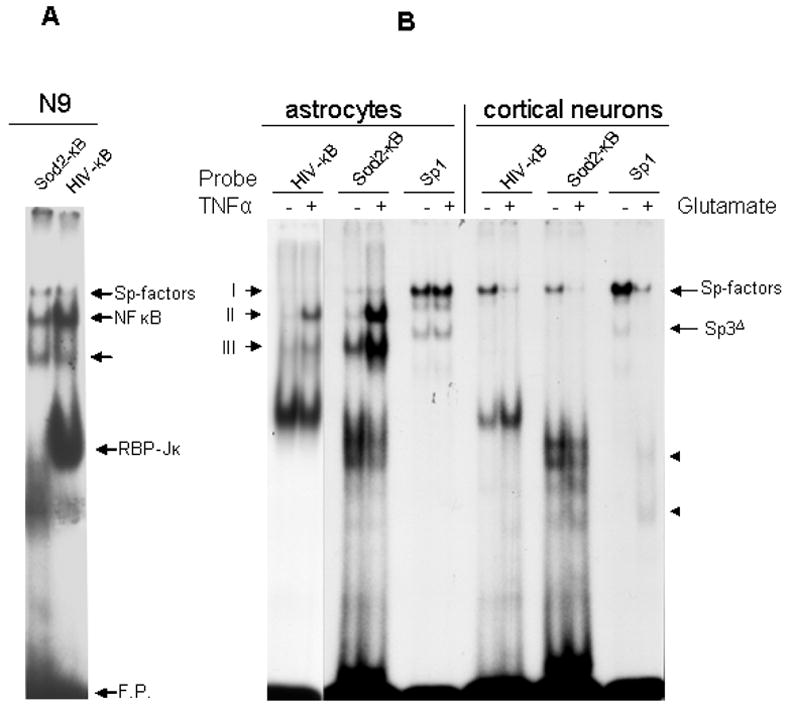
A. SOD2-κB and Ig/HIV-κB sequences were radiolabeled to probe the binding activities in nuclear extracts from activated N9 cells. Both Sp1-related factors and NFκB could be detected by each probe. B. Primary rat astrocytes were either left untreated or treated with TNF for 60 min prior to nuclear extraction. Rat primary cortical neurons were either left untreated or treated with glutamate for 60 min before nuclear extraction. All nuclear extracts were probed with Ig/HIV-κB, rat SOD2-κB, and Sp1 sequences in EMSA. The two fast-migrating bands (arrowheads) in the last lane are the DNA-binding remnants from calpain-cleaved Sp-factors.
We expanded our investigation of the SOD2 κB site to determine the effect of Sp-factors on the activity of the enhancer. First, we attempted to identify those factors binding to the SOD2-κB site in neurons and astrocytes. We focused on those bands with migration rates similar to known Sp-factors and NFκB (Complexes I, II, and III in Figures 3 and 4). We recently determined that Sp-factors are differentially expressed in neurons and glia: Sp3 is expressed in both glia and neurons, Sp1 is only detectable in glia, and Sp4 is highly enriched in neurons (5,6). These distinct expression patterns might have important consequences for cell function. Therefore, it is of interest to determine which of these Sp-factors and NFκB dimers bind to the SOD2-κB site in various cell types.
Figure 4. Characterization of protein complexes formed with rat SOD2 κB sequence: astrocytes vs. neurons.
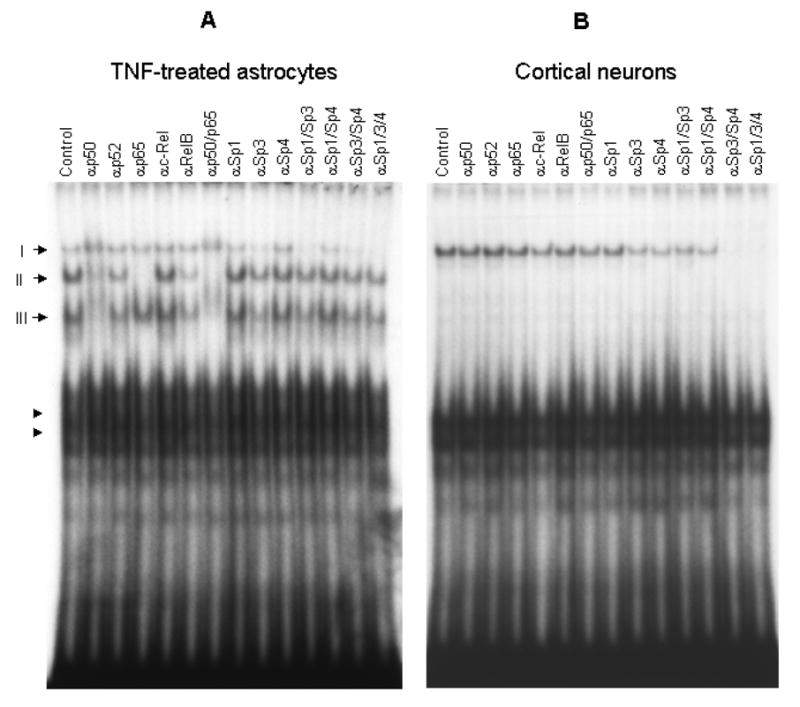
Nuclear extracts from TNFα activated astrocytes (A) and control cortical neurons (B) were used for EMSA when a rat SOD2-κB probe was used. Different antibodies against Rel- or Sp-factors were used to supershift the binding complexes. The total amount of antibodies used in each lane was 1.2 μg. An equal amount of each antibody was used when multiple antibodies were present.
Nuclear proteins from TNF-activated astrocytes or untreated cortical neurons were used in supershift assays. The top band (Complex I, Figure 4) was sensitive to Sp1 and Sp3 antibodies in astrocytes, whereas it was removed by Sp3 and Sp4 antibodies in neurons. Complex II, only present in astrocytes, was removed by antibodies against p50 or p65, indicating a p50/p65 heterodimer. Another astrocyte-specific species, Complex III, was sensitive to p50 antibodies but unaffected by p65 antibody, suggesting that it comprised p50 homodimer. Both Rel-family dimers were expressed in astrocytes under basal condition, and the level of p50/p50 was substantially higher than that of p50/p65 (Figure 3B). After TNF stimulation, the p50/p65 level surpassed that of p50/p50 (Figure 3B). Both astrocytes and neurons presented somewhat abundant, fast-migrating complexes (arrowheads, Figure 4), possibly produced by proteins binding to the flanking sequences. The levels of these uncharacterized complexes in astrocytes were diminished after activation of NFκB by TNF, suggesting that binding of the responsible proteins to the probe was competed by active NFκB, as seen with RBP-Jκ at the κB element in the IL-6 promoter (43).
To confirm that Sp-factors occupy the κB element from SOD2 intron 2 in situ, we performed chromatin immunoprecipitation (ChIP) assays. Because the available Sp-factor antibodies were generated against human proteins, it was necessary to utilize human cells for efficient immunoprecipition. Cells of the NT2 line were differentiated into neurons (31); the T98G astroglioma line was used as an astrocyte model and was treated with TNF to activate NFκB. NT2 cells showed prominent localization of Sp3 and Sp4 to a 130-bp sequence surrounding the κB element (Figure 5). In T98G cells, by contrast, the p65 and p50 components of NFκB were associated with this region more prominently. T98G cells also showed a paucity of Sp4 binding, consistent with data from primary astrocytes.
Figure 5. Association of Sp-factors with the SOD2 κB element in situ.
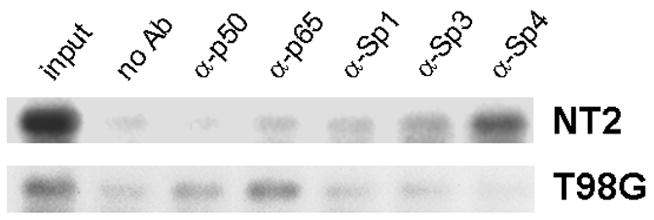
The NT2 cell line was differentiated into neuronal cells with retinoic acid, and the T98G astroglioma cell line was stimulated with 50 ng/ml TNF for 30 min. Cultures were then fixed in formaldehyde and processed for immunoprecipitation with antibody to p50, p65, Sp1, Sp3, or Sp4. A negative-control precipitation was also performed with protein A/G-agarose alone. Coprecipitating DNA was subjected to PCR with primers directed against the the κB region in the SOD2 second intron. From the blank precipitation, the extract from the boiled pellet (“no Ab”) and the initial supernatant (“input”) were also subjected to PCR as negative and positive controls, respectively.
The SOD2 intronic κB/Sp site is inhibitory in neurons
The κB site in the rat SOD2 intron (RSI) region was explored further for modulation by Sp-factors. The RSI, including approximately 400 bp upstream and 100 bp downstream from the κB site, was cloned into two different reporter vectors with different promoter activities (see Materials and Methods); a version in which the κB site was mutated was inserted in the same vectors. Reporter pGL3-promΔ has a very weak promoter, while pGL3-TK has a strong thymidine kinase promoter. When these reporters were transfected into primary neurons, their basal activities differed (Figure 6A & B). However, they both showed a similar pattern of inhibition by the RSI sequence and release from this inhibition by mutation of the κB site, indicating that the effect of the RIS was not dependent upon the basal promoter with which it was coordinated. Similar results were observed in the NT2 line (Figure 6C). While this repressive activity of the RSI in neuronal cells was surprising, the sequence had its expected stimulatory effect in rat primary astrocytes (Figure 6D). Mutation of the κB-site dramatically reduced the activation, though the remaining activity was significantly higher than promoter alone. This result is consistent with multiple sequences in the intron enhancing promoter activity in astrocytes, a substantial portion of which is mediated by the κB site. Together, these data indicate differential regulation of the SOD2 intronic element in neurons versus nonneuronal cells.
Figure 6. The SOD2 κB element is repressive in neurons but stimulatory in astrocytes.
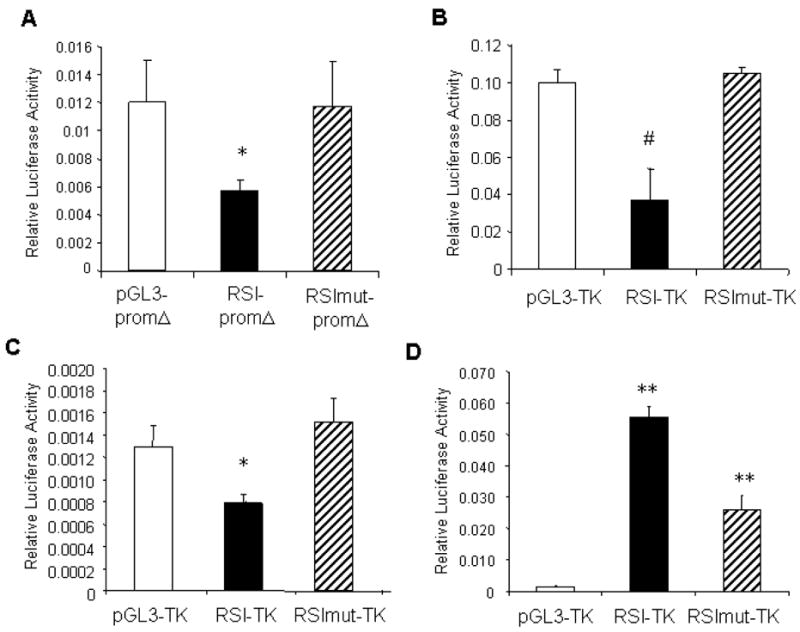
A portion of the second rat SOD2 intron (RSI), including a κB-element, was cloned into pGL3 reporters with different promoters (pGL3-promΔ and pGL3-TK). “RSImut” indicates constructs in which the κB sequence was mutated. The vector and reporter containing RSI or RSImut were transfected into highly enriched rat cortical neurons (A, B). pGL3-TK series were also transfected into neuronal NT2 cells (C) and rat primary astrocytes (D). In A, B, and C, *p<0.05 or #p<0.005 for RSI vs. vector or RSI vs. RSImut. In D, **p<0.001 between any two transfections.
It was important to confirm that the effects seen in reporter constructs were relevant to the actual SOD2 gene. Therefore, we performed RT-PCR to measure SOD2 mRNA levels in neurons treated with a double-stranded oligonucleotide containing a Sp-factor target sequence (“decoy”); an oligonucleotide of a scrambled sequence (“scram-con”) was applied as a control for the Sp decoy. When cortical neurons were exposed to Sp decoy for 20 h, steady-state levels of SOD2 mRNA appeared to rise (Figure 7). No such change occurred with application of the scrambled control oligonucleotide.
Figure 7. Elevation of expression of the endogenous SOD2 gene by Sp-factor inhibition.
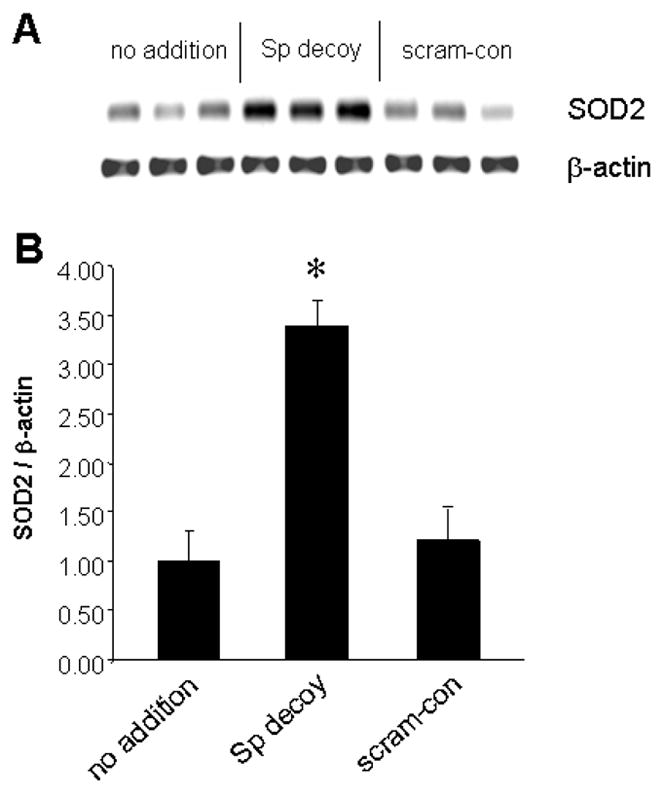
Highly enriched cultures of cortical neurons were left untreated or were transfected with double-stranded oligonucleotides containing a canonical binding site for Sp-factors (“Sp decoy”) or a scrambled control oligonucleotide (“scram-con”). After 20 h, RNA was prepared and subjected to RT-PCR with primers for SOD2 and β-actin. The triplicate samples depicted in A were quantified by densitometry for the graphical representation in B. *p<0.01, Student’s t-test
Rel- and Sp-factors differentially regulate the SOD2 intronic κB site
To explore the role of individual transcription factors in regulating the activity of the SOD2 intronic site, we used the Sp-factor decoy oligonucleotide and a plasmid encoding IκBα to repress the activities of Sp-factors and NFκB, respectively. In primary neurons, Sp1 decoy completely alleviated the inhibitory effect of RSI (Figure 8A). In contrast, scram-con appeared to be ineffective (p<0.01 for RSI-TK vs. RSI-TK/decoy; p<0.01 for RSI-TK/decoy vs. RSI-TK/scram-con; p>0.1 for comparisons between the results in any two decoy-treated conditions). Co-transfection of a plasmid encoding IκBα had no effect on reporter activities (p>0.1, RSI-TK vs. RSI-TK/IκBα; p>0.1, RSImut-TK vs. RSImut-TK/IκBα). We also co-transfected a plasmid encoding p65 with reporters. Such overexpression of p65 significantly enhanced RSI-TK promoter activity in neurons (p<0.001, RSI-TK vs. RSI-TK/p65). Surprisingly, p65 still increased expression moderately when the RSI was mutated (p<0.01, RSImut-TK vs. RSImut-TK/p65), suggesting that p65 might contribute to SOD2 intron-dependent transcription through a cryptic κB site or by activating other factors. The influence of p65 was completely blocked by IκBα co-transfection, consistent with our previous observation (32).
Figure 8. Sp-factors are inhibitory for RSI activity in neurons but stimulatory in astrocytes.
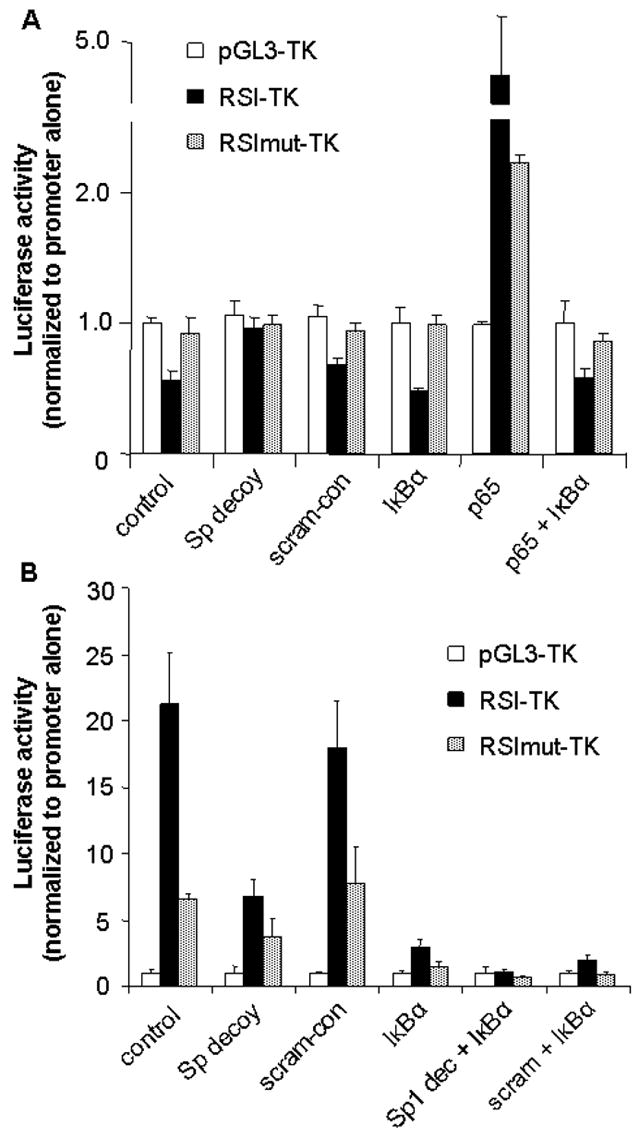
pGL3-TK, pGL3-RSI-TK, and pGL3-RSImut-TK were transfected in primary rat neurons (A) and astrocytes (B). Sp-factor decoy oligonucleotides (50 nM, final concentration) and IκBα were used to suppress the activity of Sp-factors and NFκB, respectively. In each set of transfection, the activity (ratio between firefly luciferase activity and Renilla luciferase activity) from pGL3-TK was arbitrarily set as 1.0. pCMV-RL was co-transfected to monitor the transfection efficiency. See text for statistical data.
Application of Sp1 decoy oligonucleotides and IκBα to primary astrocytes yielded results very different from those observed in neurons (Figure 8B). Sp1 decoy significantly reduced the activity of RSI-TK (p<0.01 for RSI-TK vs. RSI-TK/decoy); the scram-con was ineffective (p>0.1, RSI-TK vs. RSI-TK/scram-con). This result indicates that the Sp-factors in astrocytes (Sp1 and Sp3) act as activators on the κB site. Co-transfection of IκBα suppressed RSI-dependent transcription more effectively than Sp1 decoy (p<0.005 for RSI-TK vs. RSI-TK/IκBα; p<0.01 for RSI-TK/decoy vs. RSI-TK/IκBα). Interestingly, IκBα also significantly reduced expression when the RSI was mutated (p<0.01 for RSImut-TK vs. RSImut-TK/IκBα), consistent with the ability of p65 to activate the mutated intron in neurons. A combination of Sp1 decoy and IκBα co-transfection completely removed the activity derived from the SOD2 intron (p<0.005, RSI-TK/IκBα vs. RSI-TK/decoy/IκBα; p>0.05 for RSI-TK/decoy/IκBα vs. RSImut-TK/decoy/IκBα). Application of scram-con along with IκBα did not significantly alter the activity from transfection with IκBα alone. These data indicate that the dominant activator for the SOD2-κB in astrocytes was NFκB, while Sp1 and Sp3 also significantly enhanced the intron activity through the same site.
DISCUSSION
The transcriptional induction of SOD2 by NFκB in some cell types has been invoked in support of the broader hypothesis that this transcription factor participates in responses to oxidative stress. Our results indicate that the cis element most important to NFκB-dependent inductions of SOD2 is capable of interacting with either NFκB or Sp-factors, to a degree dependent on cell type-specific determinants. More surprisingly, this κB element participates in the suppression of SOD2 in CNS neurons, as a result of the specific Sp-factors they express and the recalcitrance of their NFκB. We have further delineated the sequence requirements for Sp-factors binding to κB-sites; low GC content in the spacer sequences seems to be required. However, a very high overall GC content in the sequence will favor Sp-factor over NFκB, as seen in the APP2 sequence (and less dramatically in the Bcl-x sequence), effectively creating a Sp-dependent element from a sequence that might otherwise share binding and regulation by NFκB. Conversely, low GC content discourages the interaction with Sp-factors, as seen in the IL6 sequence. Hence, DNA sequences exist with overlapping binding affinities for NFκB and Sp-factors, and this relationship can lead to surprising regulatory consequences.
Sp-factors are abundantly expressed and constitutively active in most cell types, and NFκB activity is undetectable in many cells under basal conditions. In addition, Sp1 is not sensitive to CpG methylation but NFκB is. Therefore, the interaction between Sp-factors and κB sites could play an important role in homeostatic expression of certain genes through their κB element. The situation in CNS neurons appears to be an extreme example. Evidence indicates that DNA-binding activity from bona fide NFκB is severely restricted in CNS neurons [(17,19,20,32); and Figure 3B], making the role of Sp-factors in these cells much more critical. In contrast to neurons, astrocytes were found to exhibit varying degrees of NFκB activity under basal and TNF-stimulated conditions. Adding to this contrast between cell types, we have recently determined that Sp-factors are differentially expressed in neurons and astrocytes (5,6). Sp1 is much more highly expressed in glial cells than in neurons, while Sp4 is conversely enriched in neurons.
The above relationships of transcription factors to specific cell types appear to create remarkable consequences for expression of SOD2. In neurons, the prominent proteins interacting with SOD2-κB site were Sp3 and Sp4. Reporter assays indicated these neuronal Sp-factors repressed promoter activity through the intronic κB site, because the inhibition was relieved by either mutation of the κB site or by application of Sp-decoy. Co-transfection of IκBα suppressed the RSI promoter activity in astrocytes (but not in neurons), permitting detection and manipulation of residual activity that resulted from the Sp1 and/or Sp3 expressed abundantly in astrocytes. The role of Sp3 notwithstanding, this contrast is consistent with prior observations by ourselves (17) and others (44–46) with regard to the transactivating potential of Sp1 and Sp4. The latter appears to act more commonly as a transrepressor, though its behavior is not completely understood and may depend on the context of the binding sequence. In a separate but analogous situation, mutation of the κB element in the RSI left a residual promoter activity in astrocytes that was still sensitive to IκBα. This is consistent with earlier reports analyzing promoter activity from intron-2 of the mouse and human SOD2 genes, wherein the identified κB site was shown to be only partially responsible for inductions by TNF and interleukin-1 (23,28,42).
Several Sp-binding κB sites have been reported so far, and consistent with our data, all these sequences have high GC content. For the classical Ig/HIV-κB site, the affinity for Sp-factors was approximately 30-fold lower than that of NFκB (p50/p65). The four Gs at the 5′ end and three Cs at the 3′ end were all required for Sp-factors to interact with this site. Our data are largely consistent with an earlier report using HeLa cell extracts (14). However, that study found the IL6-κB sequence to be a good target for Sp1, but we were unable to detect a substantial interaction between Sp1 and the IL6-κB in either direct binding assays or competition assays; it is unclear what causes this discrepancy. Sp1 interacts with a κB site (GGGGGTGACCCC) located in the P-selectin promoter, where it confers constitutive basal activity (14). In that system, Sp-binding activity can be replaced by higher affinity NFκB (p50/p50) factors to turn off the promoter activity. A similar competitive situation has been observed in the human Fas promoter (15), where Sp1 is bound to a κB site (GGGCGTTCCC) under unstimulated conditions then replaced by NFκB after activation. Our results from primary neurons are analogous to these systems in that transfected p65 was able to relieve repression caused by Sp-factors.
By contrast to these instances of physical and functional competition between NFκB and Sp-factors, there are other systems in which the two classes of factors appear to function simultaneously in the same cell or population of cells. For instance, a body of data assembled by Liu et al. (18) document Sp-binding at a κB site (GGGACTGGCC) in the mouse NMDA receptor 1 (NR1) promoter in the P19 neuroblast cell line. Though NFκB activity is present in those cells, Sp-factors drive most of the activity of the NR1 κB element therein. Interestingly, P19 cells express the Sp-factors (Sp1/Sp3) we observed in astrocytes. And it was in astrocytes that we found NFκB and Sp-factors to co-exist and both transactivate the RSI. Consistent with those functional data, EMSA indicated that TNF-mediated activation of NFκB in astrocytes did not alter Sp-factor binding to the SOD2-κB.
We found the repressive effect of neuronal Sp-factors to be promoter-specific (Fig. 7). This is consistent with the identification of the neuronal Sp-factors as Sp3 and Sp4, as these factors are more equivocal in their tendency to activate or repress than is Sp1. For instance, Ahlgren et al. (47) found that Sp3 inhibits transcription from a G-C/T box in an artificial reporter construct but not one in the promoter for cytochrome P450. We previously showed that two κB sites in the human APP promoter can interact with Sp-factors (17). No effect on reporter gene expression was detected when these two sites were mutated within the context of the entire promoter (our unpublished results). Also, when APP-κB2 sites were cloned into pGL3-promΔ, these sites did not repress expression in primary neurons. Thus, binding of an Sp-factor to a DNA sequence is not tantamount to a transcriptional effect, and this is true even for repression.
Oxidative stress typically induces SOD2 expression, and excitotoxic levels of glutamate produce superoxide and other reactive oxygen species (48,49). We previously documented a rapid attenuation of the DNA-binding and transactivation activity of neuronal Sp-factors upon challenge of cerebral neurons with glutamate (17,21). Severe cellular oxidation after initiation of the Fenton reaction in neurons attenuates Sp-factor activity, as well (21). Thus, under conditions of stress, the tonic repression of the SOD2 gene by Sp3 and Sp4 may be alleviated by removal of these factors. We recently found that most of the effect of glutamate is explained by the activation of calpain, which converts Sp3 and Sp4 into fragments that retain DNA-binding activity (6). Although these fragments lack the transactivation domain, we and others (50) have determined that the zinc-finger domain that remains after proteolysis of Sp1 can serve as a transcriptional inducer, perhaps functioning as a co-factor of sorts. Therefore, it cannot be presumed that transcriptional repression by Sp-factors is necessarily attenuated by this limited proteolysis. Nevertheless, glutamate causes a substantial reduction of total DNA binding by Sp-factors and their fragments (as does oxidation), suggesting that the repression of SOD2 expression by Sp-factors is indeed relieved under these conditions.
In conclusion, we have demonstrated for the first time an inhibitory effect of Sp-factors mediated through a κB site. This effect is a direct result of the transcription factor milieu in neurons, particularly the relative dominance of Sp4 over Sp1 and the paucity of NFκB activity. The same κB site was regulated in an opposite fashion in astrocytes, which contained Sp1 and active NFκB. Transfection of Sp1 was unable to reverse the repressive effect of the RSI in neurons (our unpublished results). This is consistent with the dominant-repressive effect that Sp4 had over Sp1 in our earlier experiments (17). Further, other laboratories have found that Sp4 and/or Sp3 can exert a dominant-repressive effect even when Sp1 is present (44,51). In addition to providing insights about the regulation of SOD2, these data bolster the conclusion supported by several other approaches (5,32) that NFκB is relatively unimportant for transcription in cerebral neurons.
Abbreviations used are
- APP
amyloid precursor protein
- CMV
cytomegalovirus
- COX2
cyclooxygenase-2
- EGFP
enhanced green fluorescent protein
- EMSA
electrophoretic mobility shift assay
- FBS
fetal bovine serum
- HIV-LTR
human immunodeficiency virus long terminal repeat
- IL-6
interleukin-6
- LPS
lipopolysaccharide
- MEM
minimal essential medium (Earle’s salts)
- MHC
major histocompatibility complex
- NR1
N-methyl D-aspartate receptor-1 subunit
- ROS
reactive oxygen species
- RSI
rat SOD2 intron
- SOD2
superoxide dismutase-2
- RT-PCR
reverse transcription and polymerase chain reaction
- TK
thymidine kinase
- TNF
tumor necrosis factor
Footnotes
The authors appreciate the technical services of Mandy Porter. This work was supported by funds from the National Institute of Neurological Disorders and Stroke (R01NS046439).
References
- 1.Marin M, Karis A, Visser P, Grosveld F, Philipsen S. Cell. 1997;89:619–628. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- 2.Bouwman P, Gollner H, Elsasser HP, Eckhoff G, Karis A, Grosveld F, Philipsen S, Suske G. Embo J. 2000;19:655–661. doi: 10.1093/emboj/19.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Supp DM, Witte DP, Branford WW, Smith EP, Potter SS. Dev Biol. 1996;176:284–299. doi: 10.1006/dbio.1996.0134. [DOI] [PubMed] [Google Scholar]
- 4.Zhou X, Long JM, Geyer MA, Masliah E, Kelsoe JR, Wynshaw-Boris A, Chien KR. Mol Psychiatry. 2005;10:393–406. doi: 10.1038/sj.mp.4001621. [DOI] [PubMed] [Google Scholar]
- 5.Massa PE, Aleyasin H, Park DS, Mao X, Barger SW. J Neurochem. 2006;97:607–618. doi: 10.1111/j.1471-4159.2006.03810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao X, Yang S-H, Simpkins JW, Barger SW. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.04297.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouwman P, Philipsen S. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 8.Suske G. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 9.Lee JA, Suh DC, Kang JE, Kim MH, Park H, Lee MN, Kim JM, Jeon BN, Roh HE, Yu MY, Choi KY, Kim KY, Hur MW. J Biol Chem. 2005;280:28061–28071. doi: 10.1074/jbc.M414134200. [DOI] [PubMed] [Google Scholar]
- 10.Sapetschnig A, Koch F, Rischitor G, Mennenga T, Suske G. J Biol Chem. 2004;279:42095–42105. doi: 10.1074/jbc.M404989200. [DOI] [PubMed] [Google Scholar]
- 11.Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. Science. 2004;303:383–387. doi: 10.1126/science.1088679. [DOI] [PubMed] [Google Scholar]
- 12.Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 13.Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ. Mol Cell Biol. 2002;22:1277–1287. doi: 10.1128/mcb.22.5.1277-1287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I, Scheidereit C. Mol Cell Biol. 1998;18:1266–1274. doi: 10.1128/mcb.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan H, Bartos DP, Owen-Schaub LB. Mol Cell Biol. 1999;19:2098–2108. doi: 10.1128/mcb.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saito T, Takahashi Y, Hashimoto H, Kamataki T. J Biol Chem. 2001;276:38010–38022. doi: 10.1074/jbc.M106130200. [DOI] [PubMed] [Google Scholar]
- 17.Mao X, Moerman AM, Barger SW. J Biol Chem. 2002;277:44911–44919. doi: 10.1074/jbc.M204292200. [DOI] [PubMed] [Google Scholar]
- 18.Liu A, Hoffman PW, Lu W, Bai G. J Biol Chem. 2004;279:17449–17458. doi: 10.1074/jbc.M311267200. [DOI] [PubMed] [Google Scholar]
- 19.Moerman AM, Mao X, Lucas MM, Barger SW. Mol Brain Res. 1999;67:303–315. doi: 10.1016/s0169-328x(99)00091-1. [DOI] [PubMed] [Google Scholar]
- 20.Jarosinski KW, Whitney LW, Massa PT. Lab Invest. 2001;81:1275–1288. doi: 10.1038/labinvest.3780341. [DOI] [PubMed] [Google Scholar]
- 21.Mao X, Moerman AM, Lucas MM, Barger SW. J Neurochem. 1999;73:1851–1858. [PubMed] [Google Scholar]
- 22.Saha RN, Pahan K. J Neurochem. 2005;94:S127. [Google Scholar]
- 23.Jones PL, Ping D, Boss JM. Mol Cell Biol. 1997;17:6970–6981. doi: 10.1128/mcb.17.12.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kokoszka JE, Coskun P, Esposito LA, Wallace DC. Proc Natl Acad Sci U S A. 2001;98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duttaroy A, Paul A, Kundu M, Belton A. Genetics. 2003;165:2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu Y, Porntadavity S, St Clair DK. Biochem J. 2002;362:401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kiningham KK, Xu Y, Daosukho C, Popova B, St Clair DK. Biochem J. 2001;353:147–156. [PMC free article] [PubMed] [Google Scholar]
- 30.Barger SW, Harmon AD. Nature. 1997;388:878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 31.Cheung WM, Fu WY, Hui WS, Ip NY. Biotechniques. 1999;26:946–948. 950–942, 954. doi: 10.2144/99265rr04. [DOI] [PubMed] [Google Scholar]
- 32.Barger SW, Moerman AM, Mao X. Curr Pharm Des. 2005;11:985–998. doi: 10.2174/1381612053381594. [DOI] [PubMed] [Google Scholar]
- 33.Grilli M, Goffi F, Memo M, Spano P. J Biol Chem. 1996;271:15002–15007. doi: 10.1074/jbc.271.25.15002. [DOI] [PubMed] [Google Scholar]
- 34.Chen C, Edelstein LC, Gelinas C. Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaltschmidt B, Linker RA, Deng J, Kaltschmidt C. BMC Mol Biol. 2002;3:16. doi: 10.1186/1471-2199-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukiw WJ, Bazan NG. J Neurosci Res. 1998;53:583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Segars JH, Nagata T, Bours V, Medin JA, Franzoso G, Blanco JC, Drew PD, Becker KG, An J, Tang T, et al. Mol Cell Biol. 1993;13:6157–6169. doi: 10.1128/mcb.13.10.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Barger SW, Liu L, Mrak RE, Griffin WST. J Neurochem. 2000;74:143–150. doi: 10.1046/j.1471-4159.2000.0740143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juttler E, Potrovita I, Tarabin V, Prinz S, Dong-Si T, Fink G, Schwaninger M. Neuropharmacology. 2004;47:580–592. doi: 10.1016/j.neuropharm.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Xu Y, Kiningham KK, Devalaraja MN, Yeh CC, Majima H, Kasarskis EJ, St Clair DK. DNA Cell Biol. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- 43.Kannabiran C, Zeng X, Vales LD. Mol Cell Biol. 1997;17:1–9. doi: 10.1128/mcb.17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwon HS, Kim MS, Edenberg HJ, Hur MW. J Biol Chem. 1999;274:20–28. doi: 10.1074/jbc.274.1.20. [DOI] [PubMed] [Google Scholar]
- 45.Pan X, Solomon SS, Shah RJ, Palazzolo MR, Raghow RS. J Lab Clin Med. 2000;136:157–163. doi: 10.1067/mlc.2000.108149. [DOI] [PubMed] [Google Scholar]
- 46.Bai L, Collins JF, Xu H, Ghishan FK. Am J Physiol Cell Physiol. 2001;280:C1168–1175. doi: 10.1152/ajpcell.2001.280.5.C1168. [DOI] [PubMed] [Google Scholar]
- 47.Ahlgren R, Suske G, Waterman MR, Lund J. J Biol Chem. 1999;274:19422–19428. doi: 10.1074/jbc.274.27.19422. [DOI] [PubMed] [Google Scholar]
- 48.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 49.Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. J Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- 50.Pascal E, Tjian R. Genes Dev. 1991;5:1646–1656. doi: 10.1101/gad.5.9.1646. [DOI] [PubMed] [Google Scholar]
- 51.Choi HS, Hwang CK, Kim CS, Song KY, Law PY, Wei LN, Loh HH. Mol Pharmacol. 2005;67:1674–1683. doi: 10.1124/mol.104.008284. [DOI] [PubMed] [Google Scholar]