

Abstract
NUP98-HOXD13 (NHD13) fusions have been identified in patients with myelodysplastic syndrome (MDS), acute myelogenous leukemia (AML) and chronic myeloid leukemia blast crisis (CML-BC). We generated “knock-in” mouse embryonic stem (ES) cells that express a NHD13 fusion gene from the endogenous murine NUP98 promoter, and used an in vitro differentiation system to differentiate the ES cells to haematopoietic colonies. Replating assays demonstrated that the partially differentiated NHD13 ES cells were immortal, and two of these cultures were transferred to liquid culture. These cell lines are partially differentiated immature haematopoietic cells, as determined by morphology, immunophenotype and gene expression profile. Despite these characteristics, they were unable to differentiate when exposed to high concentrations of Epo, G-CSF, or M-CSF. The cell lines are incompletely transformed, as evidenced by their dependence on IL3, and their failure to initiate tumours when injected into immunodeficient mice. We attempted genetic complementation of the NHD13 gene using IL3 independence and tumorigenicity in immunodeficient mice as markers of transformation, and found that BCR-ABL successfully transformed the cell lines. These findings support the hypothesis that expression of a NHD13 fusion gene impairs haematopoietic differentiation, and that these cell lines present a model system to study the nature of this impaired differentiation.
Keywords: NUP98, HOXD13, ES Cells, leukemia, oncogene, BCR-ABL
INTRODUCTION
The NUP98-HOXD13 (hereafter NHD13) fusion gene is formed by the t(2;11)(q31;p15) translocation, which has been observed in patients with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [1]. The NUP98 gene is involved in numerous other translocations resulting in at least 20 different fusion genes, with nine of the known fusions involving homeodomain genes. NUP98 translocations have been recognised in patients with a wide array of haematological malignancy, including MDS, AML, CML and pre-T cell lymphoblastic leukemia/lymphoma (pre-T LBL) [2].
NUP98 is located at chromosome 11p15.5 and encodes a nucleoporin protein that normally mediates transport of RNA and protein across the nuclear membrane. GLFG repeats within the amino-terminal region of the NUP98 protein are thought to form a docking site for the nuclear transport signal receptor proteins known as karyopherins [3, 4]. A GLEBS motif, also within the amino terminal portion of the NUP98 protein, forms a binding site for the RAE1 mRNA export factor [5]. In addition, a recent report suggests that this interaction is important for proper mitotic division [6].
The NUP98 fusion genes invariably encode a fusion protein consisting of the amino terminal portion of the normal NUP98 protein fused to the carboxyl terminal portion of the partner gene. In the case of the homeobox partners, this portion includes the DNA-binding homeodomain. As the N-terminal portion of NUP98 has transactivation potential [7], these NUP98-HOX fusion proteins are suspected to act as aberrant transcription factors.
Vertebrate HOX genes are organised into four genomic clusters (A, B, C and D), each containing between 9 and 11 genes arranged in 13 paralogous groups. The HOX genes are transcription factors, featuring a common DNA-binding domain termed the homeodomain. The HOX genes are critical for developmental processes including haematopoiesis, during which genes of the A, B and C (but not D) clusters are expressed [8]. Several lines of evidence have implicated HOX gene deregulation in leukemic transformation. First, multiple HOX genes have been identified as fusion partners of NUP98 in chromosomal translocations associated with acute leukemia [2]. Second, gene expression profiling has shown that upregulation of several HOX genes occurs in leukemias of different origins [8, 9]. This overexpression has been demonstrated to have functional significance during leukemic transformation [10–12] and to be a marker of poor prognosis [13]. Finally, we and others have reported that expression of NUP98-HOX fusions leads to myeloid malignancies in transgenic and retroviral transduction model systems [14–17]. NHD13 transgenic mice developed a highly penetrant MDS which was either fatal or developed to a fatal acute leukemia [17].
To determine whether expression of a NHD13 fusion gene under control of endogenous NUP98 regulatory sequences is leukemogenic, we used homologous recombination to produce “knock-in” (hereafter KI) ES cells that expressed the NHD13 fusion gene from the endogenous NUP98 locus. Herein, we report that the NHD13 embryonic stem cells were not able to contribute to a live mouse. However, by subjecting the NHD13 ES cells to an in vitro differentiation system, we were able to establish a haematopoietic cell line model of NHD13 haematopoietic disease, offering a novel type of model system to investigate this disease.
MATERIALS AND METHODS
Generation of NHD13 ES Cells
The NHD13 KI vector was constructed by ligation of the 3’ end of the NHD13 fusion gene derived from the index patient to exon 12 of the murine NUP98 using the ApaI site common to human and murine NUP98 exon 12 (Figure 1a). A floxed neomycin resistance cassette was inserted for selection of the targeted ES cells [18].
Figure 1. Insertion and expression of NHD13 allele into NUP98 loci.
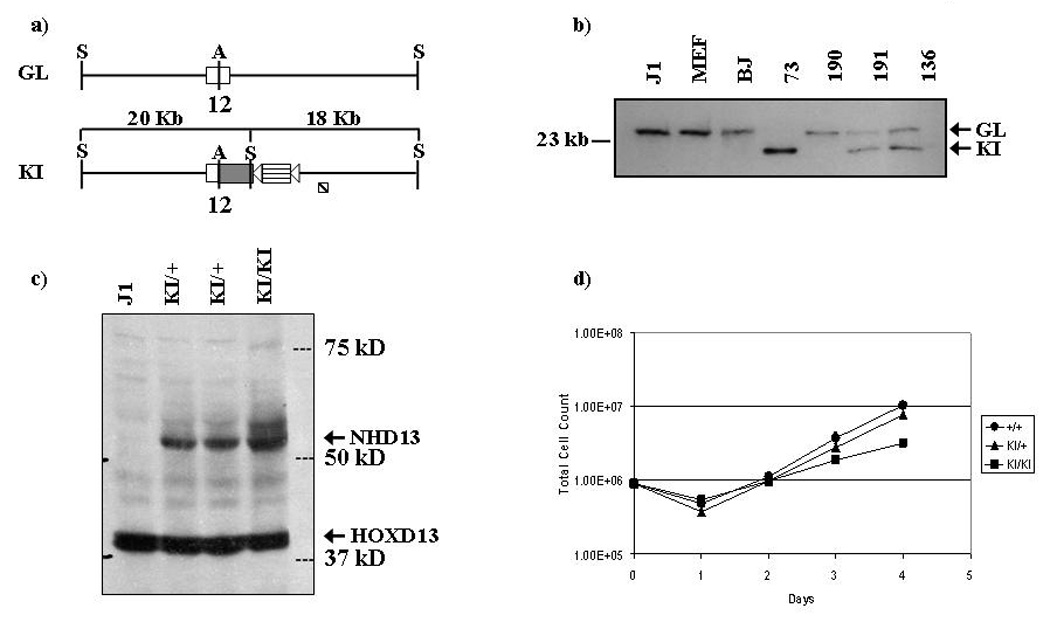
a) Schematic comparing NUP98 allele with NHD13 KI allele. S = SstI, A = ApaI. 12 denotes exon 12 of NUP98 gene. Open boxes represent mouse sequence (endogenous NUP98); grey boxes represent human sequence (inserted portion of NHD13). Triangles represent loxP sites. A G418 resistance cassette is indicated as a horizontal striped box‥ The diagonally striped box represents the position of the probe used for Southern blot. b) Southern blot of parental and NHD13 ES cells. J1 indicates the parental ES cell, BJ indicates ES cells targeted with an unrelated construct, and. MEF indicates murine embryonic fibroblasts. 73, 190, 191, and 136 are independent ES clones. Sizes of germline (GL) and NHD13 knock-in (KI) alleles are shown. c) Western blot showing expression of NHD13 allele in parental, heterozygous and homozygous targeted clones. The endogenous HOXD13 and NHD13 proteins are indicated with arrows. d) Growth curves of parental and heterozygous and homozygous NHD13 ES cells in culture.
Blastocyst injection
To generate chimeric mice, day e3.5 blastocysts were harvested and injected with 10–12 ES cells. Up to 10 injected blastocysts per uterine horn were transferred to pseudopregnant recipient mice through a dorsal medial incision, and the pregnancies proceeded to term [19]. For some experiments, the pseudopregnant recipient mice were euthanized at pre-term time points, and the embryos dissected to remove the placenta, yolk sac, and fetus.
In vitro differentiation of ES cell lines
ES cell lines were cultured in ES Maintenance Media (15% FBS, 1mM sodium pyruvate, 100 U/ml penicillin, 100 ug/ml streptomycin, 2mM L-glutamine, 0.1 mM Non-Essential Amino Acids, 10 ng/ml mLIF and 100 uM monothioglycerol (MTG) in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose (Stem Cell Technologies)) on a layer of primary embryonic fibroblasts. Cell lines were cultured under standard conditions in Iscove’s Modified Dulbecco’s Medium (IMDM) with 15% fetal bovine serum, 2mM L-glutamine, 100 U/ml penicillin and 100 ug/ml streptomycin.
Differentiation of ES cells to hematopoietic colonies was accomplished using Stem Cell Technologies protocols and reagents [20]. Briefly, 2 × 10³ ES cells were plated in 35 mm non-adherent petri dishes in “Primary Differentiation Medium” (IMDM in 0.9% methylcellulose plus 15% FCS, 150 uM MTG, and 40 ng/ml mSCF) to induce embryoid body (EB) formation. At day 7, the EBs were given 1.0 ml of “Feed Medium” (IMDM plus 15% FCS, 150 µM MTG, 160 ng/ml mSCF, 30 ng/ml mIL-3, 30 ng/ml hIL-6, and 3 U/ml hEpo). After 12 to 13 days of primary differentiation, EBs were harvested, disrupted with collagenase, and plated in “Hematopoietic Medium” [0.9% methylcellulose, 15% FCS, 150 µM MTG, BIT 9500 (1% BSA, 10 ug/ml Insulin, 200 ug/ml Transferrin), 3 U/ml hEpo, 160 ng/ml mSCF, 30 ng/ml mIL-3 and 30 ng/ml hIL-6]. Colonies were evaluated at day 10 post secondary differentiation.
Treatment with differentiating agents
The cytokine-induced differentiation assay was performed using methylcellulose (Methocult M3120, StemCell Technologies Inc.) with 15% FBS, along with a low concentration of IL3 (1 ng/ml) to support growth and 1x or 10x cytokine concentrations for the cytokine under investigation. The 1x concentrations used in the forced differentiation assay were IL6 (10ng/ml), SCF (160 ng/ml), EPO (3U/ml) granulocyte colony stimulating factor (G-CSF) (10ng/ml), macrophage colony stimulating factor (M-CSF) (10ng/ml) and granulocyte macrophage colony stimulating factor (GM-CSF) (10ng/ml). For treatment of cells in liquid culture, 1X concentrations of the individual cytokines were used.
Treatment with decitabine (Sigma) and trichostatin A (TSA; Sigma) was performed in IMDM with 15% FBS, along with a low concentration of IL3 (1 ng/ml) to support growth. In the first protocol, cells were split the day before treatment, and were then treated with decitabine alone (3d at 1 uM or 100nM), TSA alone (1d at 50 nM or 5 nM), or decitabine followed by TSA (decitabine, 3d at 1 uM followed by TSA, 1d at 50 nM, or decitabine 3d at 100 nM followed by TSA, 1d at 5 nM). Following the treatment period, drugs were washed out and cells were allowed to recover for three days before being assayed by FACS. In the second protocol, cells were split the day before treatment and were then treated with decitabine alone (2d at 100 nM), TSA alone (16 h at 5 nM), or decitabine followed by TSA (decitabine, 2d at 100 nM followed by TSA, 16 h at 5 nM). Following the treatment period, drugs were washed out and cells were allowed to recover in normal growth medium for one day before FACS analysis.
Retroviral transduction
Infectious retroviral particles was constructed by cotransfection of 293T cells with pVpack-GP, pVpack-Eco (Stratagene) and plasmids prepared from the Human Fetal Liver Plasmid cDNA Library (Stratagene) according to the manufacturer’s instructions. Virus containing media (VCM) was harvested 48 hours post-transfection. Infection of the 188D and 189L2 cell lines was performed by resuspending 5×105 to 5×106 cells with VCM containing 5ug/ml protamine sulfate and 10 ng/ml IL3. VCM was replaced with fresh medium after 24 hours.
Cell Transfection
Transfections of the cell lines were performed using DMRIE-C transfection agent (Invitrogen), according to the manufacturer’s instructions. Plasmids used were pMIG-BCR-ABL (gift from Dr Warren Pear), pEGFP-C2 (Clontech) and pTRE2hyg (Clontech). Selection was performed in 500ug/ml G418 (Invitrogen) or 500ug/ml hygromycin (Invitrogen).
Flow cytometry
A total of 106 cells were washed in phosphate-buffered saline (PBS) and then resuspended in 100 ul 2% FBS in Hanks’ Buffered Saline Solution (HBBS). 5ul of appropriate antibodies were added and the mixture incubated at 4°C for 30 min. The following antibodies were used as PE conjugates: IgG (as isotype control; 0.4 ug/ml); CD34 (1.25 ug/ml); CD3 (0.4 ug/ml); Sca (0.4 ug/ml); Mac-1 (0.2 ug/ml); IgM (0.4 ug/ml); CD41 (1.0 ug/ml). The following antibodies were used as FITC conjugates: IgG (as isotype control; 0.4 ug/ml); Gr-1 (0.1 ug/ml); Ter119 (0.4 ug/ml); Kit (1.0 ug/ml); B220 (1.0 ug/ml) (all antibodies were purchased from BD Biosciences). Cells were then washed twice in PBS and resuspended in 500 ul of 1% propidium iodide in 2% FBS in HBBS. Fluorescence was detected and analysed using a FACSort flow cytometer (Beckman Coulter) with CellQuest data analysis software.
Microarray analysis
Total RNA was isolated from cultured cells using TRIZOL reagent (Invitrogen). Control RNA was Universal Murine Reference RNA (Stratagene). Fifteen micrograms of total RNA was reverse transcribed using oligo (dT) primer and Stratascript reverse transcriptase (Stratagene) for 1 hour at 42°C. Cell line and reference cDNA were labeled with Cy3 or Cy5 (Amersham Biosciences) and then combined and hybridized to 9984-feature murine cDNA arrays (NCI-Frederick), for 16 hours at 42°C. The hybridized slides were scanned using a GenePix 4000A Microarray Scanner (Axon). Image analysis was performed using the GenePix 5 software (Axon).
Ligation-mediated PCR
Genomic DNA was digested with NlaIII or MseI and ligated to linkers constructed by annealing the oligonucleotides 5’-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGACCATG-3’ and 5’-Phos-GTCCCTTAAGCGGAG-C3spacer-3’ for NlaIII-digested DNA, and 5’-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC-3’ and 5’-Phos-TAGTCCCTTAAGCGGAG-C3spacer-3’ for MseI-digested DNA (Integrated DNA Technologies, Coralville, IA). Primary PCR was performed using primers designed to the linkers and the LTR of the pFB retroviral sequence. Secondary PCR was performed using nested primers after 1:50 dilution of the primary PCR product. Products were ligated into pGEM-T Easy (Promega) and transformed into DH5α cells (Invitrogen). Plasmid DNA was isolated (Qiagen) and sequencing was performed using an SP6 primer with BigDye Terminator and analysed on a 3730 DNA Analyzer (Applied Biosystems).
Immunodeficient mouse xenotransplant tumour assays
Nude and Scid mice were obtained from Charles River Laboratories. Cells were washed and resuspended in PBS at a concentration of 2.5 × 107/ml. Subsequently, aliquots of 200 µL (5 × 106 cells) were injected either subcutaneously or intraperitoneally into nude or Scid recipient mice. Mice were monitored daily and euthanised when tumour formation became apparent. Mice in which tumours did not form were monitored for six months.
RESULTS
Generation of NHD13 ES Cells
The NHD13 targeting construct is shown in Figure 1a. Proper targeting of the murine NUP98 locus will generate a chimeric mouse-human transcript in which the first 11 exons are derived entirely from the endogenous murine Nup98, exon 12 is a mouse/human hybrid exon, and exon 2 of human HOXD13 is fused in frame to NUP98 exon 12 (Figure 1a). This fusion recapitulates the NHD13 fusion seen in patients with the t(2;11)(q31;p15). The NHD13 vector was electroporated into J1 ES cells, which were then selected with G418 and ganciclovir counter-selection. Southern blot analysis was performed to identify those clones in which homologous recombination had occurred (Figure 1b). An extraordinarily high targeting frequency was obtained; 185/228 (81%) of the ES cells had been properly targeted. Indeed, one clone, designated 73, was found to have no germline allele, suggesting that the clone had either undergone two recombination events, a gene conversion, or a non-disjunction event (Figure 1b).
To verify that the targeted locus was properly transcribed, RNA was extracted from four heterozygous clones and assayed by RT-PCR for expression of the fusion transcript. All expressed similar levels of the transcript (data not shown). To test whether the transcript is correctly translated within the ES cells, three clones (two with one allele targeted and one with two alleles targeted) were subjected to western blot analysis using an anti-HOXD13 antisera to detect expression of the NHD13 fusion protein. The expected size of the NHD13 protein is approximately 50 kD. The blot confirmed that a protein of slightly more than 50 kD was expressed at high levels (Figure 1c). Growth curves indicated that the heterozygous NHD13 cells had growth characteristics similar to the parental cells, and that the homozygous NHD13 cells expanded at a reduced rate (Figure 1d).
Inability to generate chimeric mice with NHD13 cells
In an attempt to generate NHD13 mice, several independent NHD13 clones were microinjected into wild-type blastocysts, and the blastocysts transferred to pseudo-pregnant mothers. Several experiments were performed, and a total of 254 blastocysts were injected with four independent NHD13 ES clones, by two experienced microinjectionists. A total of 26 pups were born; seven died in the neonatal period, and 19 developed into adulthood. None of these 19 mice were positive for the NHD13 allele, as assessed by coat color as well as PCR analysis of tail biopsy DNA or peripheral blood DNA. Reasoning that these mice might still be chimeric in the germ tissue, several of the 19 surviving mice were crossed with wild-type mice; none of 32 offspring of these mice were positive for the NHD13 allele.
We considered that the inability to generate chimeric mice may have been due to technical problems with these ES clones. Therefore we repeated the targeting in a different facility with an independent strain of ES cells (CJ7). We again noted a very high targeting efficiency (35/76 or 46%), and obtained a large number of successfully targeted clones. Both a pool of 12 individual clones as well as single clones were injected into blastocysts. In sum, a total of 85 blastocysts were injected and transferred; again, none of 19 pups born were chimeric by coat color.
To assess whether we could detect the NHD13 allele in the developing embryo, we euthanized mice implanted with blastocysts between e10.5 and e14.5 and analyzed contribution of the J1 NHD13 cells by PCR and/or Southern blot (Table 1). The implantation and resorption rates of 76% and 13%, respectively, were within an expected range. The NHD13 allele was consistently detected in a fraction of embryos at all gestational periods by PCR. However, Southern blot analysis failed to show any contribution from the NHD13 ES cells in three litters that were analyzed. This indicated that the contribution was below the level of sensitivity of the Southern blot assay, and suggested that the contribution of the NHD13 ES cells to the developing embryo was minimal.
We considered the possibility that the neomycin resistance cassette and PGK promoter used for selection of the NHD13 ES cells might effect the expression of nearby genes, and lead to a lack of chimeric animals through an unexpected positional effect [21–23]. Therefore, the floxed PGK-Neo resistance cassette was removed using Cre recombinase. The resultant PGK-Neo-deleted NHD13 ES cells were microinjected into blastocysts to generate chimeric mice. Again, the microinjections resulted in very few liveborn pups, and none were chimeric. Taken together, these findings suggest that NHD13 ES cells are impaired in their ability to differentiate and contribute to an adult animal.
In vitro differentiation of ES cells
The NHD13 fusion gene has been shown to impair megakaryocytic differentiation of K562 cells [17], and impaired differentiation was considered to be the likely cause for failure of the NHD13 ES cells to contribute to the adult mouse. To explore the effects of the NHD13 knock in on the differentiation potential of the targeted cells, we performed a series of in vitro differentiation experiments, using an established ES cell differentiation protocol [20]. In this procedure, ES cells are first differentiated to embryoid bodies (EBs; primary differentiation), which are then disaggregated and the single cells plated in methylcellulose containing SCF, IL3, IL6, and EPO (secondary differentiation). Although the primary differentiation of the parental J1 as well as two independent NHD13 ES clones (designated 188 and 189) produced typical-appearing EBs, the secondary differentiation showed clear differences between the J1 and NHD13 derived cells. Grossly, the methylcellulose dishes from the parental J1 cells showed primarily loose aggregates of cells typical of CFU-GM colonies(Figure 2a–Figure 2b), whereas the NHD13 dishes had a preponderance of dense clusters of cells, similar to BL-CFCs (Figure 2d–Figure 2e and Figure 2g). Wright-Giemsa stained cytospin slides showed that these cells had a very primitive morphology, with a high nuclear/cytoplasmic ratio, prominent nucleoli, and uncondensed chromatin (Figure 2f, compare with Figure 2c). Replating of the methylcellulose cultures resulting from the secondary differentiation demonstrated that the partially differentiated NHD13 cells were effectively immortal in this system, surviving more than 12 replatings, whereas cells derived from the parental J1 ES stem cell line survived only 3 replatings. The entire differentiation procedure was repeated 3 times, with similar results for each experiment.
Figure 2. In vitro differentiation of J1 parental and NHD13 clones.
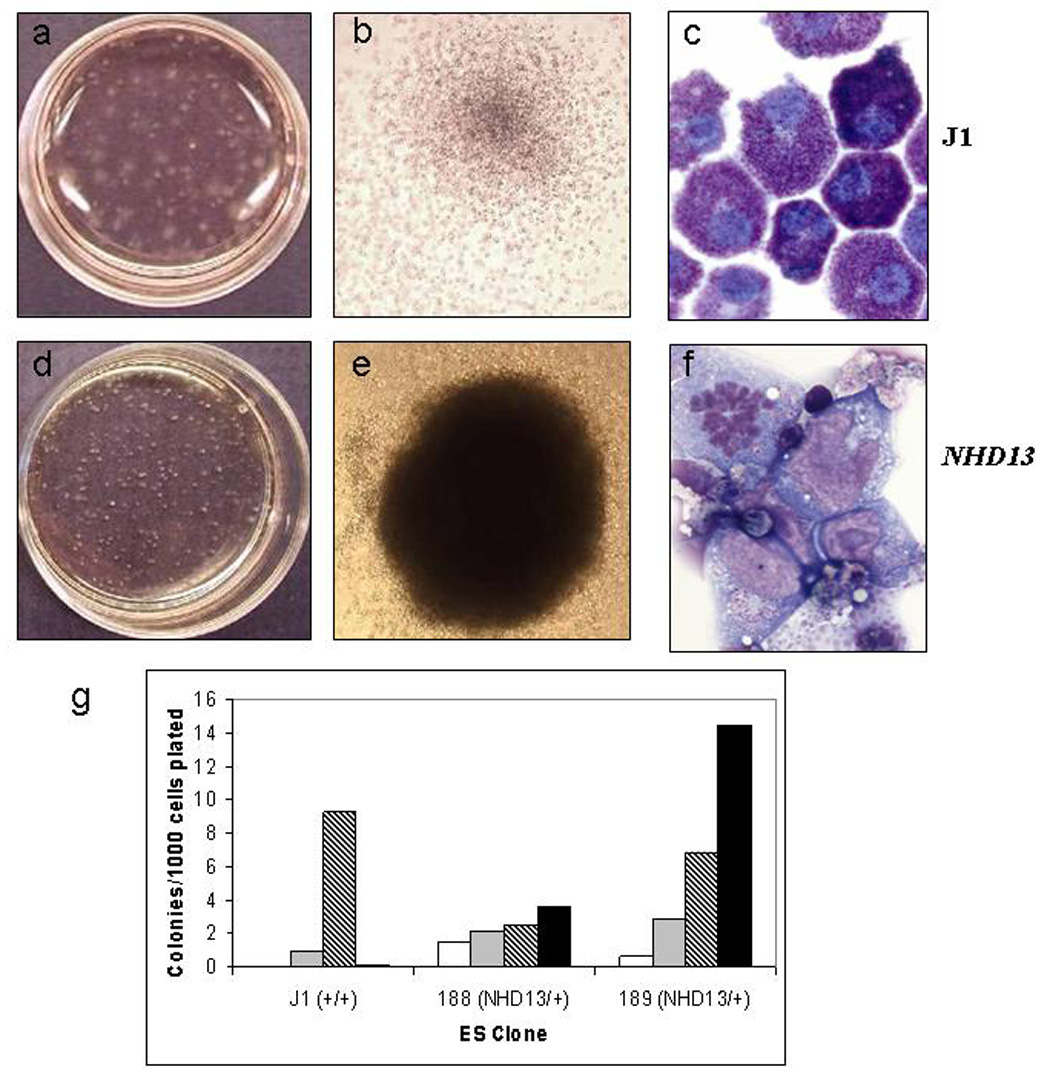
Entire dish (a,d) single colony (b,e) , and Wright-Giemsa stained single cell (c,f) images of the secondary differentiation (a–c, parental J1 cells; d–f, NHD13 knock-in cells). Note the denser, more compact colony formation of the NHD13 clones. g) Counts of colony types formed during secondary differentiation. Open bars represent GEMM colony number; grey bars represent BFU-E colonies; diagonally striped bars represent GM colonies; black bars represent BL-CFC colonies.
Generation of IL3 dependent cell lines
After the cells derived from NHD13 clones 188 and 189 cells had been replated 12 times in methylcellulose with IL3, IL6, SCF and Epo, they were transferred to liquid culture to determine whether they could grow in suspension, and to define which cytokines were necessary for survival. Successive withdrawal of each of the cytokines demonstrated that IL3 was necessary and sufficient for the cells to survive and proliferate in liquid culture (Figure 3a). We established cell lines from three separate differentiation experiments; these cell lines were designated 188D, 189D, 188L1, 189L1, 188L2, and 189L2. We have focused our more detailed studies on one IL3 dependent cell line derived from NHD13 ES clone 188 (188D), and one derived from NHD13 ES clone 189 (189L2). These cell lines have now been in continuous liquid culture for over three years.
Figure 3. Growth of differentiated NHD13 cells in liquid culture.
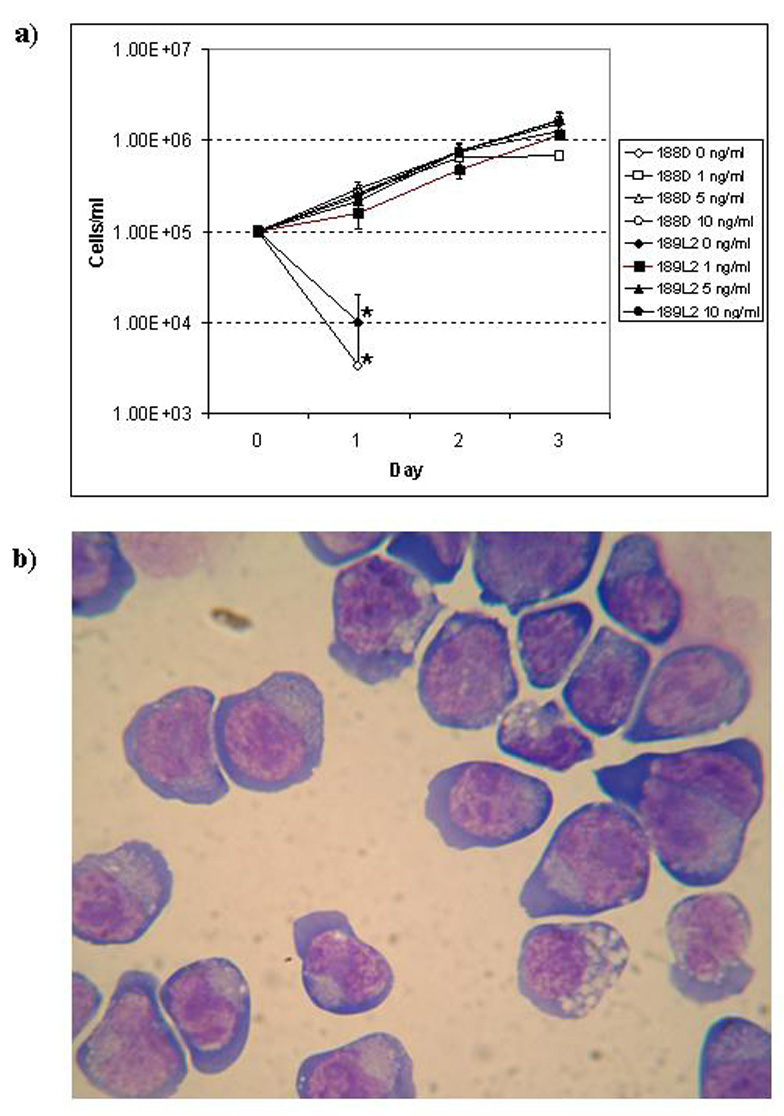
a) Growth curve of 188D and 189L2 cell lines in various concentrations of IL3. Asterisks indicate the last data point that could be plotted, as no viable cells were identified under these conditions after day 1. b) Wright-Giemsa stained cytospin of 188D cell line. Note deep basophilic cytoplasm, perinuclear clearing, and nuclear blebbing.
Both the morphology and FACS profile of the 188D and 189L2 cell lines were similar. The morphologic appearance of these cells was reminiscent of an erythroid or megakaryoblastic leukemia, as the cells displayed deeply basophilic cytoplasm, cytoplasmic blebbing, perinuclear clearing, prominent nucleoli, and variable vacuolisation (Figure 3b). FACS analysis showed that the 188D and 189L2 cells stained positively for markers of haematopoietic progenitor cells, including c-kit, CD31, and CD34. The cells stained weakly for CD41, and were negative for markers of more differentiated haematopoietic cells, including Mac1, Gr1, ter119, CD3 and B220 (Figure 4a and Figure 4b).
Figure 4. Immunophenotype of 188D and 189L2 cell lines.
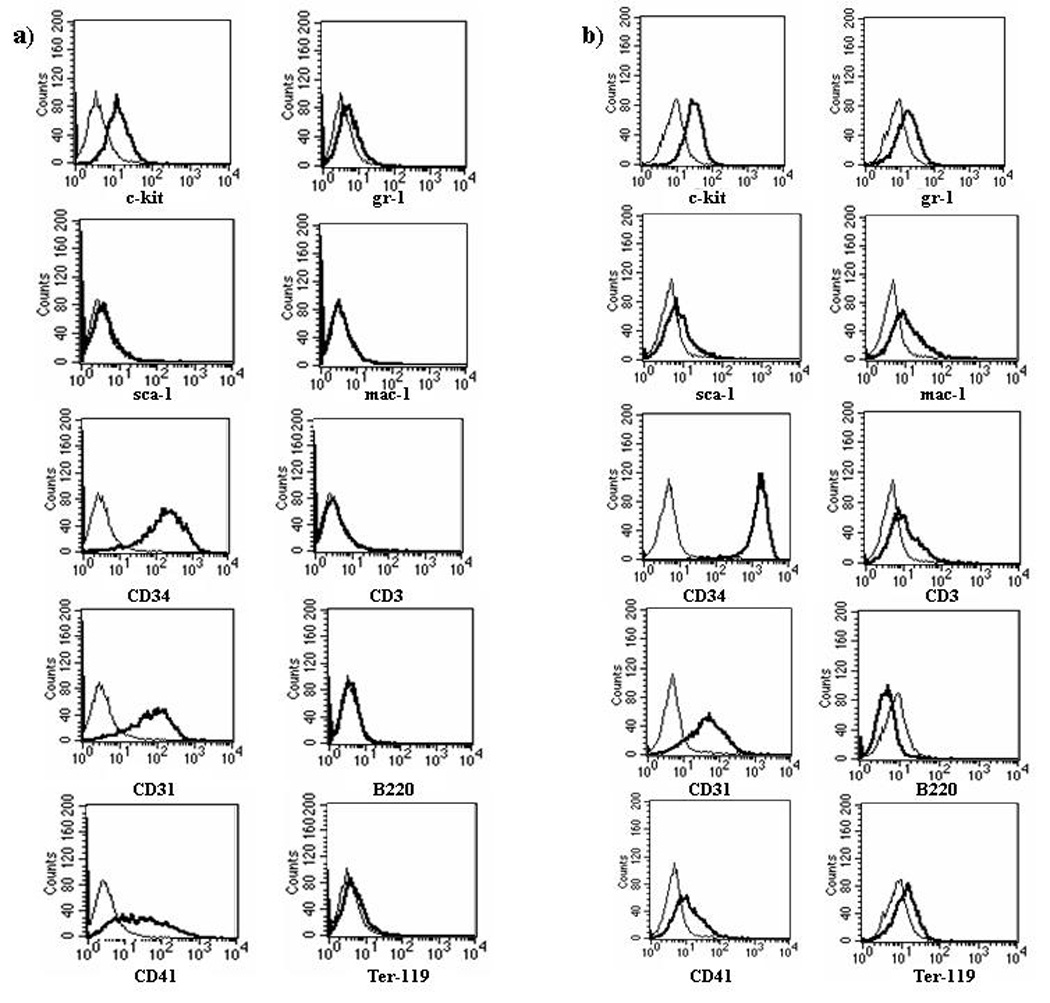
a) 188D immunophenotype. b) 189L2 immunophenotype. Thin lines represent isotype control; heavy lines represent CD antigen.
The mRNA expression profile of the 188D and 189L2 cell lines was determined by comparing the expression profile of the NHD13 cell lines with that of a universal reference RNA consisting of RNA pooled from 11 different cell lines (Stratagene). The two genes most highly expressed relative to the reference RNA were Hbb (haemoglobin beta) and Csf2rb2 (IL3 receptor), consistent with the IL3 dependence and erythroid appearance of the cells. In addition, several other highly expressed genes (haemoglobin alpha, Eraf (erythroid associated factor), and Nfe2 (nuclear factor, erythroid derived 2)), are known to be specifically involved in haematopoiesis, especially erythropoiesis (Table 2). Taken together, the morphology, FACS analysis, and gene expression profile support the assertion that these are primitive, incompletely differentiated haematopoietic cells.
Response to differentiating agents
We treated the 188D and 189L2 cell lines with single cytokines and combinations of cytokines to determine if they could differentiate into mature haematopoietic cells. 188D and 189L2 cells that were plated in methylcellulose supplemented with either IL6 (10 ng/ml), SCF (160 ng/ml), Epo (3 U/ml or 30 U/ml), G-CSF (10 ng/ml or 100 ng/ml), M-CSF (10 ng/ml or 100 ng/ml), or GM-CSF (10 ng/ml) alone did not form colonies. To determine if the cells would differentiate if survival was supported by IL3, the experiment was repeated in the presence of low (1 ng/ml) or high (10 ng/ml) concentrations of IL3. In this experiment, the cells survived, however, the number and morphology of CFU-GM, CFU-GEMM, and BFU-E colonies was not appreciably different between the IL3 only controls and the samples incubated in the presence of IL6, SCF, Epo, G-CSF, M-CSF, or GM-CSF (data not shown).
A similar experiment was performed in liquid culture to allow for analysis of the results by FACS. The 188D and 189L2 cells were incubated for five days in the presence of G-CSF (100 ng/ml), M-CSF (100 ng/ml), or Epo (30 U/ml). At the end of five days, all cells were dead. To determine if the cells would differentiate if survival was supported by IL3, the experiment was repeated in the presence of low (1 ng/ml) or high (10 ng/ml) concentrations of IL3; control cells were treated with low or high concentrations of IL3 alone. After five days incubation, cells were analyzed by May-Giemsa staining and FACS with Mac-1 and Gr-1. In no instance did the cells show evidence of differentiation by morphology or flow cytometry. This inability to respond to supraphysiological concentrations of cytokines is a measure of the robustness of the block in differentiation induced by the NHD13 gene.
We treated the 189L2 cell line with either a demethylating agent (decitabine) or a deacetylation agent (Trichostatin A; TSA) or both to determine whether the cells would differentiate under these conditions. The treatment protocols used were taken from published reports that treated hematopoietic cell lines with these agents [24, 25]. The initial conditions as described in methods were highly toxic to the cells (3–23% cell viability). Subsequent conditions using a shorter exposure to decitabine and TSA (see methods) were less toxic (52–69% cell viability). FACS analysis of cells following these treatments showed no change in the level of expression of c-kit, CD34, Gr-1, Mac-1, Ter119, B220 or CD3, indicating that no differentiation occurred under these treatment conditions.
Tumorigenicity in immunodeficient mice
To investigate the tumorigenic potential of these cells, nude mice were injected either intraperitoneally or subcutaneously with parental J1 ES cells and NHD13 ES cells (clone # 188). These mice developed teratomas within 2–6 weeks and were sacrificed (Table 3; Figure 5a and Figure 5b). When the IL3 dependent 188D and 189L2 cell lines were injected into nude mice, the mice survived six months without developing teratomas, subcutaneous tumors, or leukemia. These findings support the contention that the 188D and 189L2 cell lines have partially differentiated and are therefore no longer totipotent, and that they are also not fully transformed.
Figure 5. ES cells form teratomas, whereas 188D/bcr-abl cells form chloromas.
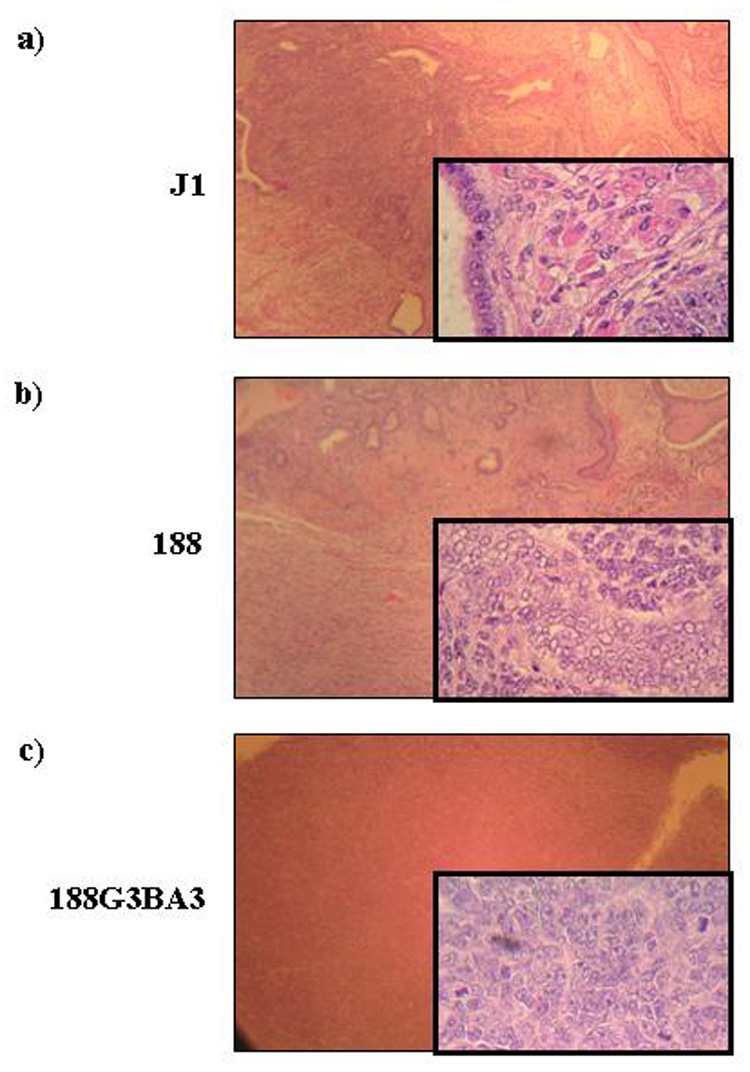
a) Haematoxylin and eosin (H&E) stained teratoma formed by injection of J1 ES cells. b) H&E stained teratoma formed by injection of 188 ES cells. c) H&E stained chloroma formed by injection of 188D cells expressing BCR-ABL. Magnifications shown are 100x, insets 400x.
Complementation with a cDNA Library
Dependence on IL3 and failure to induce tumours in nude mice are both indicative of cells that are not fully transformed. In an effort to identify genes which may complement the NHD13 fusion protein during leukemic transformation, we infected the 188D and 189L2 cell lines with a human fetal liver cDNA retroviral library and selected for clones that were able to survive and proliferate in the absence of IL3. This infection was performed three times. In all experiments, the infection efficiency was determined by GFP expression in a parallel infection, and a minimum transduction efficiency of 30% was achieved. In total, two clones, from one experiment only, grew in the absence of IL3. PCR using primers specific to the retroviral vector sequence amplified the cDNAs that had been integrated into the genomes of these two clones. We determined that these two clones were closely related, with one clone containing six insertions and the other containing the same six insertions as well as a seventh insertion. The seven cDNA inserts were identified as ARMC1 (Armadillo repeat containing 1), AHSG (Alpha-2-HS-glycoprotein), Transferrin, Beta actin, Angiotensinogen, GrpE-like 1 (chaperone protein) and Haemoglobin alpha 2. While none of these were considered likely candidates for complementation, they were nonetheless cloned into the pFB (Invitrogen) retroviral vector and used to infect 188D and 189L2 cells. However, none of these seven cDNAs conferred IL3 independence. We next considered whether the retroviral insertions may have upregulated a gene in the mouse genome and thereby made the cell IL3 independent. To address this, we used ligation-mediated PCR to identify retroviral insertion sites. We identified four insertion sites using this technique. One of these was an insertion in 11qB1.3, 5 kb 5’ of the IL3 gene. We conclude that upregulation of the IL3 gene is responsible for the IL3 independent status of the clone. Therefore, while our infection protocol and selection system were both successful, we were unable to identify any collaborating genes using this approach.
Complementation with an activated tyrosine kinase
Because we were unable to identify a complementary gene by retroviral infection, we sought instead to test a candidate gene for complementation as a proof of principle. An evolving paradigm suggests that AML patients have one mutation which impairs differentiation, and a complementary mutation, often involving tyrosine kinases, that increases cell proliferation and/or decreases apoptotic cell death [26]. Since the NHD13 protein seems to impair haematopoietic differentiation, we reasoned that the NHD13 cells could be complemented by an oncogenic tyrosine kinase such as BCR-ABL. To test this hypothesis, after removal of the floxed G418 cassette with a Cre expression vector, we co-transfected the 188D and 189L2 cell lines with a BCR-ABL expression vector and pEGFP-C2 or pTRE2hyg to allow G418 or hygromycin selection respectively. Stable transfectants were selected, and when IL3 was removed from the cultures, the BCR-ABL transfected cell lines survived and expanded at a rate similar to that of the parental cell lines in the presence IL3, demonstrating that BCR-ABL was able to effectively complement NHD13 in this system (Figure 6).
Figure 6. Expression of BCR-ABL in 188D cells eliminates the dependence on IL3.
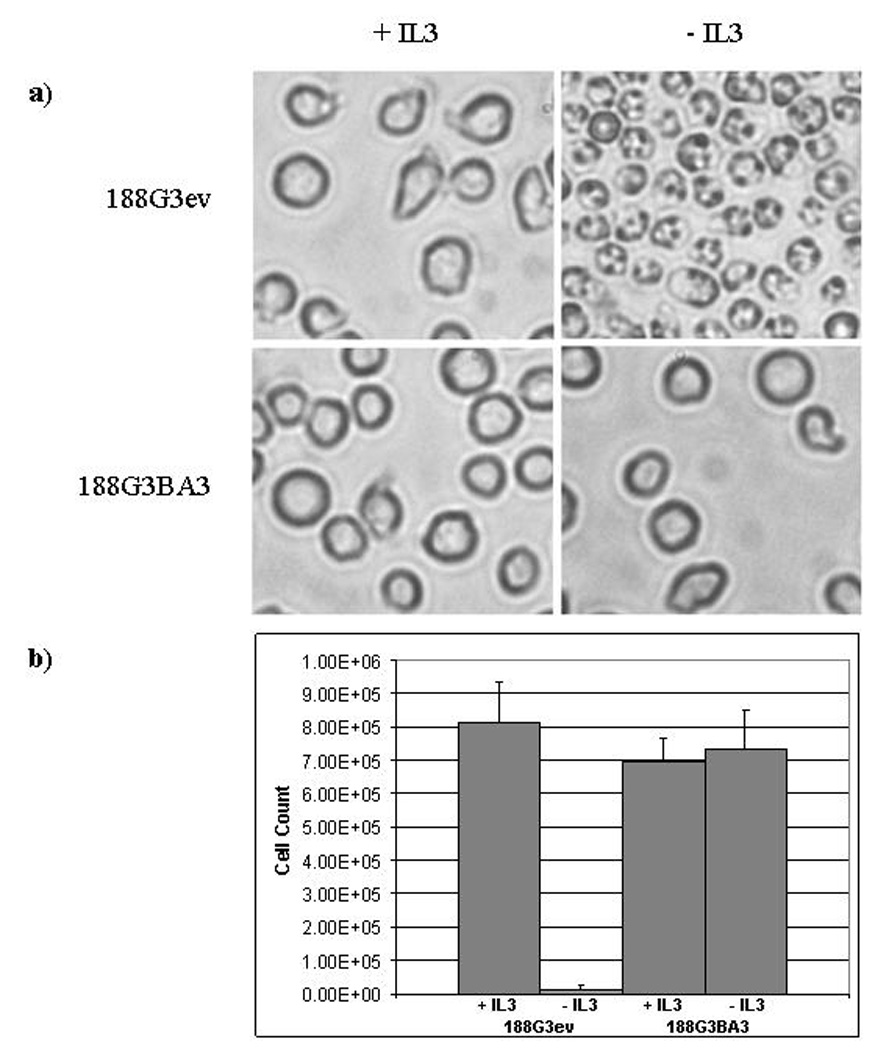
a) 188G3ev (empty vector transfectant) and 188G3BA3 (BCR-ABL transfectant) cells in culture in the presence and absence of IL3. Note the apoptotic appearance of 188G3ev (empty vector) cells and the round, refractile, and intact cell membranes of 188G3BA3 cells in the absence of IL3. b) Cell counts (in cells/ml) were performed 24 hours after the withdrawal of IL3 from the cultures. Cells were seeded at 5×105/ml.
To test the hypothesis that BCR-ABL expression had converted the non-malignant, IL3 dependent 188D cell line to fully transformed cells, one of the 188D transformants that expressed BCR-ABL (188G3BA3) was injected subcutaneously into Scid mice. These mice uniformly developed chloromas at the site of injection within two weeks, indicating that the cell line was fully transformed (Table 3, Figure 5c). Mice injected in parallel with PBS or parental 188D cells were healthy without tumours for at least six months. This reaffirms the complementary ability of NHD13 and BCR-ABL to fully transform cells. However, as these cell lines formed chloromas rather than teratomas, the partially differentiated NHD13 cells have lost the totipotency that the ES cells possessed. The chloroma cells stained negative for myeloperoxidase, and the BCR-ABL transfected cells were negative by FACS analysis for Gr-1 and Mac-1, similar to the untransfected cells. This finding is similar to the immunophenotype seen in mouse BCR-ABL/NUP98-HOXA9 leukemias [27].
DISCUSSION
We generated NHD13 KI ES cells, and demonstrated that the mRNA and protein is correctly expressed. Following blastocyst injection of these ES cells, however, the resultant mice showed no contribution of the NHD13 cells in any tissues examined. These observations suggest that as differentiation of the embryo proceeds, cells carrying the NHD13 allele are lost, consistent with the notion that the NHD13 fusion gene impairs differentiation. Consistent with the above findings, NHD13 ES cells did not differentiate normally using an in vitro methylcellulose assay, and produced predominantly BL-CFCs. However, some CFU-GM colonies were identified, indicating that the differentiation block is not absolute.
The morphology, FACS analysis, and gene expression profile support the assertion that the IL3 dependent cell lines 188D and 189L2 established through the in vitro differentiation protocol are primitive, incompletely differentiated haematopoietic cells. The contention that these immortalised cell lines were at least partially differentiated was further supported by the observation that these cells did not form teratomas in vivo, indicating that the cell lines were no longer totipotent. Of note, the 188D and 189L2 cell lines were unable to differentiate further following exposure to either demethylation or deacetylation agents, or supraphysiological concentrations of the cytokines GM-CSF, G-CSF, M-CSF or erythropoietin. These observations support the hypothesis that the NHD13 gene induces a strong, but not complete, differentiation block and are consistent with reports demonstrating transgenic mice expressing NHD13 develop a myelodysplastic syndrome with differentiation arrest that progresses to acute leukemia after a long latent period [17]. Taken together, these findings reaffirm the classification of the NHD13 fusion gene as a Class II type event according to the proposed two class model of leukemogenesis [28].
The partially differentiated, IL3-dependent, 188D and 189L2 cell lines were not fully malignant, since injection of these cell lines into immunodeficient mice did not produce tumours. As the cell lines were not fully malignant, it seemed that they might provide a useful system to test for complementation of the NHD13 fusion gene, which could be assayed by conversion to IL3 independence, or by tumour formation in immunodeficient mice. Our first effort to complement, using a retroviral cDNA library from human fetal liver, did not succeed. In those experiments, we were only able to produce one true IL3 independent clone, and it seems likely that this clone was converted to IL3 independence by upregulation of the IL3 gene, as the clone contained a retroviral integration immediately 5’ of the IL3 locus. As a complementary approach, we turned to a specific candidate gene, BCR-ABL, which provides a strong proliferation signal that might complement the NHD13 differentiation block. This approach was successful; expression of the BCR-ABL gene in either 188D cells or 189L2 cells converted them to IL3 independence, and a representative 188D BCR-ABL cell line was tumorigenic in nude mice. The reason we were unable to complement NHD13 with cDNA from a fetal liver library is unclear. It may be that no single gene, other than the potent BCR-ABL kinase, is capable of complementing NHD13 in this system.
Although it is not clear how NUP98-HOX fusion genes impair differentiation, it has been suggested that NUP98-HOX fusions act similar to overexpression of native HOX genes, with the NUP98 portion of the fusion providing a cryptic transcription activation domain, and the HOX portion providing DNA binding activity [29, 30]. However, relatively few bona fide targets for the abd-b HOX genes have been identified. Intriguingly, MYB, which has recently been shown to be upregulated in response to overexpression of the abd-b HOX gene HOXA9 [31], was shown to be highly expressed in the IL-3 dependent NHD13 cell line (Table 2). Additionally, MYB is well known to inhibit hematopoietic differentiation in a number of model systems [32, 33]. Thus, one can speculate that the NHD13 cell lines inhibit differentiation, at least in part, through overexpression of MYB.
In summary, we present a novel model system of haematopoietic disease caused by expression of a NHD13 fusion gene. A block in the normal hematopoietic differentiation scheme seems to occur at the haematopoietic progenitor cell stage. Although the impaired differentiation caused by the NHD13 fusion gene is insufficient for complete transformation of a cell, we demonstrate that the tyrosine kinase BCR-ABL is capable of complementing the NHD13 fusion gene to form a fully malignant cell.
ACKNOWLEDGEMENTS
We thank our colleagues Du H. Lam, Linda Lowe, Zhenhua Zhang, David Caudell and Stephanie Strahan for technical assistance and critical discussion. We thank R. Keith Humphries, Michael Kuehl, and Ilan Kirsch for their insight and advice. We thank Warren Pear for the gift of the bcr-abl plasmid. This research was supported by the Intramural Research Program of the NIH, NCI.
References
- 1.Raza-Egilmez SZ, Jani-Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. Nup98-HoxD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer Research. 1998;58(19):4269–4273. [PubMed] [Google Scholar]
- 2.Slape C, Aplan PD. The role of NUP98 gene fusions in hematologic malignancy. Leuk Lymphoma. 2004;45(7):1341–1350. doi: 10.1080/10428190310001659325. [DOI] [PubMed] [Google Scholar]
- 3.Radu A, Moore MS, Blobel G. The peptide repeat domain of nucleoporin Nup98 functions as a docking site in transport across the nuclear pore complex. Cell. 1995;81(2):215–222. doi: 10.1016/0092-8674(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 4.Bayliss R, Littlewood T, Strawn LA, Wente SR, Stewart M. GLFG and FxFG nucleoporins bind to overlapping sites on importin-beta. J Biol Chem. 2002;277(52):50597–50606. doi: 10.1074/jbc.M209037200. [DOI] [PubMed] [Google Scholar]
- 5.Pritchard CE, Fornerod M, Kasper LH, van Deursen JM. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. Journal of Cell Biology. 1999;145(2):237–254. doi: 10.1083/jcb.145.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438(7070):1036–1039. doi: 10.1038/nature04221. [DOI] [PubMed] [Google Scholar]
- 7.Kasper LH, Brindle PK, Schnabel CA, Pritchard CEJ, Cleary ML, van Deursen JMA. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Molecular & Cellular Biology. 1999;19(1):764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol. 2005;12(3):210–216. doi: 10.1097/01.moh.0000160737.52349.aa. [DOI] [PubMed] [Google Scholar]
- 9.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 10.Su X, Drabkin H, Clappier E, Morgado E, Busson M, Romana S, et al. Transforming potential of the T-cell acute lymphoblastic leukemia-associated homeobox genes HOXA13, TLX1, and TLX3. Genes Chromosomes Cancer. 2006;45(9):846–855. doi: 10.1002/gcc.20348. [DOI] [PubMed] [Google Scholar]
- 11.Beslu N, Krosl J, Laurin M, Mayotte N, Humphries KR, Sauvageau G. Molecular interactions involved in HOXB4-induced activation of HSC self-renewal. Blood. 2004;104(8):2307–2314. doi: 10.1182/blood-2004-04-1653. [DOI] [PubMed] [Google Scholar]
- 12.Fischbach NA, Rozenfeld S, Shen W, Fong S, Chrobak D, Ginzinger D, et al. HOXB6 overexpression in murine bone marrow immortalizes a myelomonocytic precursor in vitro and causes hematopoietic stem cell expansion and acute myeloid leukemia in vivo. Blood. 2005;105(4):1456–1466. doi: 10.1182/blood-2004-04-1583. [DOI] [PubMed] [Google Scholar]
- 13.Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16(2):186–195. doi: 10.1038/sj.leu.2402354. [DOI] [PubMed] [Google Scholar]
- 14.Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98-HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. Embo J. 2001;20(3):350–361. doi: 10.1093/emboj/20.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pineault N, Buske C, Feuring-Buske M, Abramovich C, Rosten P, Hogge DE, et al. Induction of acute myeloid leukemia in mice by the human leukemia-specific fusion gene NUP98-HOXD13 in concert with Meis1. Blood. 2003;101(11):4529–4538. doi: 10.1182/blood-2002-08-2484. [DOI] [PubMed] [Google Scholar]
- 16.Gurevich RM, Aplan PD, Humphries RK. NUP98-topoisomerase I acute myeloid leukemia-associated fusion gene has potent leukemogenic activities independent of an engineered catalytic site mutation. Blood. 2004;104(4):1127–1136. doi: 10.1182/blood-2003-10-3550. [DOI] [PubMed] [Google Scholar]
- 17.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106(1):287–295. doi: 10.1182/blood-2004-12-4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tessarollo L. Manipulating mouse embryonic stem cells. Methods Mol Biol. 2001;158:47–63. doi: 10.1385/1-59259-220-1:47. [DOI] [PubMed] [Google Scholar]
- 19.Bonin A, Reid SW, Tessarollo L. Isolation, microinjection, and transfer of mouse blastocysts. Methods Mol Biol. 2001;158:121–134. doi: 10.1385/1-59259-220-1:121. [DOI] [PubMed] [Google Scholar]
- 20.Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13(1):473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnold HH, Braun T. Targeted inactivation of myogenic factor genes reveals their role during mouse myogenesis: a review. Int J Dev Biol. 1996;40(1):345–353. [PubMed] [Google Scholar]
- 22.Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71(3):383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 23.Braun T, Rudnicki MA, Arnold HH, Jaenisch R. Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell. 1992;71(3):369–382. doi: 10.1016/0092-8674(92)90507-9. [DOI] [PubMed] [Google Scholar]
- 24.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 25.Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet. 2005;37(3):265–274. doi: 10.1038/ng1521. [DOI] [PubMed] [Google Scholar]
- 26.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 27.Dash AB, Williams IR, Kutok JL, Tomasson MH, Anastasiadou E, Lindahl K, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci U S A. 2002;99(11):7622–7627. doi: 10.1073/pnas.102583199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1(5):417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 29.Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19(1):764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmqvist L, Argiropoulos B, Pineault N, Abramovich C, Sly LM, Krystal G, et al. The Flt3 receptor tyrosine kinase collaborates with NUP98-HOX fusions in acute myeloid leukemia. Blood. 2006;108(3):1030–1036. doi: 10.1182/blood-2005-12-007005. [DOI] [PubMed] [Google Scholar]
- 31.Hess JL, Bittner CB, Zeisig DT, Bach C, Fuchs U, Borkhardt A, et al. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006;108(1):297–304. doi: 10.1182/blood-2005-12-5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakamoto H, Dai G, Tsujino K, Hashimoto K, Huang X, Fujimoto T, et al. Proper levels of c-Myb are discretely defined at distinct steps of hematopoietic cell development. Blood. 2006;108(3):896–903. doi: 10.1182/blood-2005-09-3846. [DOI] [PubMed] [Google Scholar]
- 33.McClinton D, Stafford J, Brents L, Bender TP, Kuehl WM. Differentiation of mouse erythroleukemia cells is blocked by late up-regulation of a c-myb transgene. Mol Cell Biol. 1990;10(2):705–710. doi: 10.1128/mcb.10.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]