

Abstract
Autophagy is a physiological cellular mechanism that degrades and recycles proteins and other molecules to maintain an adequate amino acid level during nutritional starvation of the cell. Autophagy is involved in cellular homeostasis and differentiation, as well as in tissue remodeling, aging, cancer, and other diseases. Under particular environmental conditions, autophagy can also be a contributor to programmed cell death, or can act as a defense mechanism for the elimination of intracellular bacteria and viruses. According to recent experimental data, autophagy may be implicated in autoimmunity by promotion of major histocompatibility complex (MHC) class II presentation of cytosolic antigens and control of T lymphocyte homeostasis, and its induction by Th1 cytokines and perhaps by specific serum autoantibodies. We review herein the role of autophagy in immune function and its possible contribution to breakdown of tolerance and development of autoimmunity.
Keywords: Apoptosis, autoimmunity, autophagy
1. INTRODUCTION
Autophagy (from the Greek for ‘self-digestion’) can be defined as the physiological mechanism that promotes the turnover of cell macromolecules and organelles via the lysosomal degradative pathway; as such, autophagy is involved in cell homeostasis, differentiation, and tissue remodeling. Three main types of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy [1-3]. During chaperone-mediated autophagy, proteins are directly delivered to the lysosome without formation of intermediate membranes [2], whereas during microautophagy proteins are locally taken up by the lysosome. Macroautophagy (hereafter referred to as autophagy) is the most active form of autophagy and is characterized by isolated membranes enveloping a fraction of the cytosol to contain non-specific cytosolic components, selectively targeted toxic protein aggregates [4], intracellular pathogens [5], or organelles such as mitochondria [6, 7]. Autophagy is no longer considered a mere demolition pathway producing substrates for energy production upon cell starvation; it is now well established that autophagy is constitutively essential for the maintenance of the overall cellular organization, with crucial connotations for survival and extension of the life span of the cell. Accordingly, autophagy has been implicated in several pathophysiological processes such as aging [8], neoplasia [9], myocardial ischemia [10], neurodegenerative disorders [4], responses to oxidative injury [11] and programmed cell death (PCD) under specific conditions. Autophagy can prevent apoptosis by isolating and removing damaged organelles while excessive autophagy ultimately leads to non-apoptotic PCD, as discussed below. The connection between autophagy and immunity should be emphasized in that autophagy contributes to the defense against microbial agents [5, 12], promotes antigen presentation through MHC class II [13, 14], is induced by cytokines [5, 15, 16], may regulate T lymphocyte survival and function [17], and may be stimulated by serum autoantibodies [18].
This review article seeks to provide a critical overview of current knowledge on autophagy with a focus on its proposed role in microbial immunity and its relevance to breakdown of tolerance.
2. SELF-DIGESTION FOR SURVIVAL
Autophagy is fundamentally a physiological process that plays an important role in the turnover of cellular proteins and other macromolecules. Moreover, it is to be considered the major catabolic route that eukaryotic cells will take to generate essential elements and preserve the need of the cell to maintain an adequate amino acid level and sustain protein synthesis during nutritional deprivation [19]. The concept that autophagy favors survival during nutritional stress is based on observations on mammalian hepatocytes, wherein both protein turnover and the need for endogenous amino acids are enhanced. In this setting, autophagy is highly activated and closely regulated by complex amino acid feedback and hormonal mechanisms. Autophagy is a dynamic process, and its regulation during normal development is based on different factors, only partially understood. Paradigmatic mechanisms leading to activation of autophagy are uniquely observed during growth and development. In fact, a significant increase in autophagy during the neonatal period, when the trans-placental nutrient supply is suddenly interrupted, has been reported [19]. There is a subsequent decline in autophagy during aging due to a reduced formation of autophagic vacuoles combined with a delay of fusion of autophagic vacuoles with lysosomes [8]. Recently, numerous studies have investigated the regulation of autophagy by nutrients, in particular amino acids as well as glucose and vitamins, and the signaling pathways underlying such regulation. Whereas amino acids are established regulators of autophagy and control autophagosome formation over its full range without hormonal assistance, the number of amino acids that perform these functions are limited, and varies among different cells and tissues. The induction classically begins after amino acid starvation, in particular leucine deprivation [20]. Other important signals are the absence of growth factors and the presence of specific compounds such as rapamycin [21].
2.1 Signaling pathways
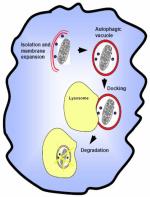
Following induction, autophagy begins with the formation of double membrane structures in the cytoplasm [1]. Importantly, it has been hypothesized that these structures are either derived from the smooth endoplasmic reticulum (SER), the Golgi complex, or are assembled de novo. In fact, molecular genetic studies in yeasts have shown that genes necessary for traffic within the SER are also required for autophagy [22, 23]. The nascent autophagosomal structure, after elongation and sequestration of cellular organelles, fuses with the vacuole/lysosome and ultimately produces degradation of the cellular structures mediated by the hydrolytic enzymes contained within the lysosomes.
The intracellular pathways of autophagy are mediated by phosphatidylinositol 3-kinases (PI3K) and the protein kinase, mTOR [21, 24]. The latter is the target of rapamycin and is a conserved Ser-Thr kinase that regulates cell growth and metabolism in response to environmental stimuli; its activation suppresses autophagy [24]. Importantly, TOR integrates inputs implicated in cell growth [21] that include growth factors, nutrients, energy, and environmental stress such as hypoxia. Nutrient starvation or hypoxia cause TOR signaling inhibition and consequently autophagy is activated; in fact insulin is well understood to activate the TOR pathway [25]. The molecular details of this process have been best characterized in the yeast Saccharomyces cerevisiae, and protein products of a group of genes known as autophagy genes (atg), which are evolutionarily conserved from yeasts to humans, have been found to regulate autophagy [3]. The yeast Atg1 protein initiates autophagy upon TOR signalling. Elongation is characterized by membrane bending and increase in size of the phagophore which wraps around cytoplasmic components, such as organelles, intracellular bacteria or just the cytosol, for degradation of macromolecules (e.g. stable proteins) [3]. Maturation involves fusion of autophagosomes with lysosomes, lumenal acidification and lysosomal hydrolysis leading to degradation of the ingested components. Different autophagy proteins (Atg) have specific functions from initiation to elongation to termination of the process. Briefly, some of the most important factors are Atg5 through Atg12 (further complexed with Atg16), and yeast Atg8 (LC3 in mammals) associated with the elongation of the phagophore and the formation of the autophagosome. Beclin1 (mammalian homolog of Atg6) and class III PI3K are positive regulators of the autophagic vacuole formation, whereas Atg12 and Atg16 are physically associated with the isolation membrane [3]. In mammals LC3 is currently the best marker of autophagy, a process that can be scored by following its translocation from the cytosol to punctate structures (Figure 1).
Figure 1. Signalling pathways activating (green) and inhibiting autophagy (orange).

The mTOR pathway responds to growth factors via the class I PI3K pathway. Inductive signals inhibit the mTOR kinase, which stimulates a partial dephosphorylation of Atg13. This allows the assembly of Atg13 with Atg 1 and Atg 17 (Atg-1 kinase complex) and the activity of the complex activates the autophagy cascade. Atg12 and LC3 are ubiquitin-like proteins; during autophagosome formation Atg12 is conjugated to Atg5 and LC3 binds phosphatidylethanolamine (PE), Atg7 and Atg3 to form LC3-II. Atg16 binds a pair of Atg12-Atg5 conjugates and this complex participates in autophagosomal membrane formation. The (Atg12-Atg5)2-Atg16 complex is required for the conjugation of LC3-II in the autophagosomal membrane. After completion of autophagosome formation LC3-II is reverted in LC3-I, which is released from the membrane. Proposed cytokine regulation checkpoints within this pathway are illustrated.
2.2 Autophagy and infections
It is well established that autophagy acts as a mechanism for the elimination of intracellular bacteria and viruses, first based on the role of autophagy in innate defense against bacterial pathogens eliminated by autophagosomes. However, some bacteria such as Shigella, Listeria monocytogenes and Rickettsia conorii can escape from phagosomes into the cytoplasm [12] while others often modulate their phagosome abode to establish an intracellular niche for survival and replication. Importantly, it was demonstrated that Mycobacterium tubercolosis, an intracellular organism which can survive within phagosomes, can be digested after pharmacological stimulation with IFN-γ which induces autophagy in macrophages [5]. These reports suggest that autophagy participates in the adaptive immune process and extends the role of autophagy beyond innate defenses. It might also suggest a role of autophagy in molecular mimicry, a widely proposed mechanism for the development of autoimmunity [26-28], but further studies are requested.
3. PROGRAMMED CELL DEATH: A FINE LINE BETWEEN AUTOPHAGY AND APOPTOSIS
The existence of non-apoptotic forms of programmed cell death (PCD) is currently accepted, after many years during which PCD had been equated with apoptosis. In fact, three different forms of PCD are recognized: type I, also known as nuclear or apoptotic; type II, also known as autophagic; and type III, also known as cytoplasmic. At present, only apoptosis and autophagy are generally accepted as being “legitimate” forms of PCD and their characteristics are summarized in table 1. Compared with apoptosis, relatively little is known about autophagic PCD and it is not clear whether autophagy directly contributes to death or is a failed effort to preserve cell viability. However, increasing evidence suggests that autophagic PCD is required in some tissues, and that apoptosis is not essential to limit the autonomous survival of the cell. Recently Levine proposed autophagy as one of the mechanism that contribute to the breakdown of cell grown control in cancer [9]. Studies on mice with double knock-out of pro-apoptotic family members [29] have demonstrated that following growth factor withdrawal, Bax−/− Bak−/− cells activated autophagy and progressive atrophy. Autophagy can lead to the degradation of catalase, a key enzymatic scavenger for intracellular reactive oxygen species (ROS); the resulting accumulation of ROS in the cell leads to membrane peroxidation with loss of membrane integrity and ultimately cell death [30]. Complex relationships exist between autophagy and apoptosis, and regulators of apoptosis activation often act as autophagy activators as well (Table 1). One of the most important points of confluence is the interaction between the antiapoptotic protein Bcl-2 and Atg6, part of the PI3K complex that leads to phagosome formation. Pattingre and colleagues have recently reported that Bcl-2 inhibits autophagy in vitro and in vivo via an interaction with Atg6 and proposed a model for understanding the relationship between autophagy, cell survival, and cell death [31]. According to this model, autophagy is necessary as an adaptive response to nutrient deprivation and other forms of cellular stress; the absence of autophagy increases susceptibility to death when cells are confronted by stressful stimuli. Another point of confluence between autophagy and apoptosis is the Atg5 protein that couples IFN-γ activated cell death to autophagic death and extrinsic apoptotic pathways via the interaction with FADD and possibly caspase 8 [16]. Atg5 was first described as an apoptosis specific protein [32], and was found to be upregulated in neurons during apoptosis activated by proteasomal inhibition. Finally, p53 has been also demonstrated to be involved in autophagy by three new target genes that cumulatively reveal unexpected functions for this tumor suppressor and apoptosis inducer in autophagy. Among these, DRAM (damage-regulated autophagy modulator), a gene targeted by p53 encoding a lysosomal protein that induces autophagy, was shown to be an effector of p53-mediated cell death. Overall, the discovery of DRAM unravels a novel link in the pathway by which p53 modulates autophagy, and suggests that induction of autophagy by p53 via DRAM also contributes to apoptotic cell death [33].
Table 1.
Comparison of apoptosis and autophagy
| Apoptosis | Autophagy | |
|---|---|---|
 |
 |
|
| Morphology | chromatin condensation nuclear fragmentation apoptotic bodies |
autophagic vacuoles |
| Triggers | extrinsic pathway → death receptors (fasL) | amino acid starvation |
| intrinsic pathway → viral infections, DNA damage, mitochondrial release of cytochrome c |
grown factor withdrawal energy withdrawal environmental stress (e.g. intracellular reactive oxygen species) |
|
| Mediators | extrinsic pathway → caspases 8, 10 | Atg 1, 5, 6, 8, 12, 17, 13 |
| intrinsic pathway → caspase 9 | ||
| Inhibitors | caspase inhibitors | Bcl-2? |
| anabolic metabolism (class I PI3K) |
In conclusion, how autophagy actually participates in cell death remains enigmatic despite the numerous hypotheses. Moreover, it is difficult to clearly ascertain the regulatory mechanisms that distinguish between cell death and protection, and the relationships between autophagy and apoptosis in cells destined for death. Finally, the effect of apoptosis in immunity has been largely studied [34, 35] while the importance of autophagy in the immune system is largely unknown.
4. AUTOPHAGY AND (AUTO)IMMUNITY
Recent studies have implicated autophagy in crucial immune mechanisms such as MHC class II presentation of intracellular antigens and, consequently, activation and regulation of CD4+ T cells by antigen-presenting cells (APC). A major review of the specific role of autophagy in immunity was pubblished in 2006 by Deretic who emphasized its role in adaptive immunity, mostly based on the strong influence that autophagy can have on MHC II presentation, and on the regulatory function of cytokines on autophagy [36].
Some recent reports have demonstrated the importance of autophagy on the development of autoinflammatory or autoimmune diseases including inflammatory bowel disease [37] or type I diabetes [38]. However, the importance of the autophagy pathway on immune function is still undetermined, as is how autophagy might favour the development of autoimmune diseases. In table 2 we indicate how autophagy might contribute to the development and perpetuation of autoimmunity.
Table 2.
Potential role of autophagy in autoimmunity
| Autophagy actions | Proposed autoimmunity-relevant effects | |
|---|---|---|
| elimination of intracellular bacteria by autophagic digestion [5, 12] |
→ | generation of autoantigens |
| → | molecular mimicry | |
| MHC class II presentation of cytosolic antigens [13] |
→ | presentation of intracellular autoantigens |
| → | regulation of APC | |
| → | impact on CD4+ T cell regulation | |
| inductive role of Th1 cytokines on autophagy [5, 15] |
→ | cytokine mediated inflammation |
| → | macrophage activation | |
| stimulation of autophagy by autoantibodies [18] | → | perpetuation of autoimmunity |
4.1 MHC class II presentation of antigens
It is known that both MHC class I and MHC class II molecules can present intracellular antigens, either foreign or intrinsic although in the past it was thought that this function was exclusive to MHC class I. Many reports suggest that autophagy is important for generation of cytosolic antigens and/or presentation through the MHC class II pathway, although the regulatory mechanisms as well as the consequences of this pathway in the immune system remain unknown [39]. In particular, how products of autophagy gain access to the MHC class II pathway remains to be elucidated. Recently Menendez-Benito proposed that MHC class II molecules could sample cytosolic antigenic peptides during the autophagosome fusion process [40]. After completion of autophagosome formation, the single-membrane autophagic body is dissolved by esterases, the cytosolic content is released into the late endosomal lumen and degraded by endosomal proteases, and proteolytic products are loaded onto MHC class II molecules. Paludan and colleagues have demonstrated the role of autophagy in MHC class II presentation of endogenously synthesized viral protein; after inhibition of lysosomal acidification, EBNA1 which is the dominant CD4+ T cell antigen of latent Epstein-Barr virus infection slowly accumulated in cytosolic autophagosomes, and inhibition of autophagy decreased recognition by EBNA1-specific CD4+ T cell clones [14]. This report asserts that the presentation of intracellular antigens on MHC class II is a “diffuse” mechanism with a larger impact on CD4+ T cell regulation than originally believed. There is another important report by Dengjel and colleagues who performed a detailed characterization of the MHC class II-bound ligand presented after increased autophagy, and demonstrated that the number of intracellular antigens presented by MHC class II was surprisingly enhanced compared with physiological condition [13]. Moreover, chaperone-mediated autophagy, another type of autophagy, is important for processing and delivery of antigens to MHC class II; it is accepted that chaperone-mediated autophagy imports individual cytosolic proteins into the lysosome and contributes to the endogenous pathway of cytoplasmic antigen presentation via MHC class II molecules [41].
In the context of APCs, MHC-mediated antigen presentation and T-cell activation, Schmid et al [42] confirmed the importance of autophagy by describing constitutive autophagosome formation in MHC class II-positive cells, including dendritic cells, B cells, monocytes, and certain epithelial cells: autophagy constitutively and efficiently delivers cytosolic antigens for MHC class II presentation in APCs that thereafter initiate CD4+ T cell activation. These experimental data strongly suggest a role for autophagy in conferring autoantigenic potential on intracellular constituents, and call for further studies on how antigens (and autoantigens) are processed for MHC class II presentation, and whether modulation of autophagy within APCs might regulate antigen presentation during pathological processes, thereby strengthening the concept of autophagy in the pathogenesis of autoimmune conditions.
4.2 The role of cytokines in the regulation of autophagy
Our understanding of the regulation of autophagy by cytokines is based on rather limited data accessed from mammalian cells. Among the first reports, as mentioned above, was the induction of autophagy in macrophages by exogenous IFN-γ [5]. Subsequently, it was found that cells exposed to IFN-γ underwent autophagic cell death [16]. Study of Th2 cytokines was encouraged by the induction of autophagy by Th1 cytokines (IFN-γ); intriguingly, Th2 cytokines act as suppressors of autophagy since IL-4 and IL-13 stimulate type I PI3K [43] which in turn activates the TOR cascade, although data are not conclusive on this issue. Jia and colleagues examined the effect of TNF-α and insulin-like growth factor-1 (IGF-1) on autophagy in vascular smooth muscle cells (VSMC) from atherosclerotic plaques, and reported that the expression of autophagy genes in VSMC was controlled by IGF-1 and TNF-α, and TNF-α could induce autophagy in VSMC [15]. The suppression of IGF-1 signaling further induces massive apoptosis while the administration of IGF-1 suppresses apoptosis in vitro and in vivo [44] thus suggesting a novel mechanism by which IGF-1 can induce cell survival by inhibiting autophagy in plaque-associated VSMC. Taken altogether, these lines of evidence support the role of inflammatory cytokines in the regulation of autophagy and may even indicate that the pro-inflammatory cytokine patterns usually observed in chronic autoimmune disorders ultimately disrupt the “balance” of autophagy in the affected organs. Moreover, it is currently accepted that the gene expression of cytokines in autoimmune diseases is stringently regulated both at the transcriptional and the post-transcriptional levels [45] and a relationship between autophagy and cytokine gene expression cannot be excluded. However, more solid experimental data to support these theses are needed and the development of specific animal models might be encouraged [46].
4.3 Autophagy and T cells
Human T lymphocytes stimulated and cultured in vitro form autophagosomes identifiable by transmission electron microscopy [47]. Also, thymocytes are capable of inducing autophagy [48] but there are not data regarding thymic cellular subtypes [49]. Interestingly, Li et al reported that autophagy is observed in activated CD4+ T cells whereas naive CD4+ T cells lack detectable autophagosomes and, in the same study, they found that T-cell receptor (TCR) stimulation, cytokine culturing, and prolonged serum starvation were the major determinants of autophagy in activated CD4+ T cells [17]. Lu et al further demonstrated that autophagy is induced in both Th1 and Th2 cells, and that autophagy is augmented by TCR signaling and IL-2 [50]. Hara and colleagues developed an atg5−/− mouse model [51] and afterwards demonstrated reduced lymphocyte counts, massive CD8+ cell apoptosis, and inability to undergo TCR-induced T-cell proliferation. These results strongly suggest that atg5 has multiple roles in lymphocyte development and function, and ultimately that autophagy is critical for T lymphocyte development. However, the massive apoptosis observed in atg5−/− CD8+ T cells suggests that the capacity to induce autophagy in these cells is actually essential for their survival in vivo [52].
4.4 Autophagy and B cells
Despite the important role of B cells in autoimmunity [53, 54], to the best of our knowledge, there is only one report suggesting that autoantibodies can stimulate autophagy [18]. This study purported to demonstrate that sera from type 2 diabetic patients with neuropathy were associated with increased levels of autophagosomes, and that this effect was mediated by increased levels of autoantibodies. The same laboratory group had previously reported that sera from patients with type 2 diabetes (T2D) with neuropathy contained an autoimmune immunoglobulin of undetermined specificity that induced apoptosis in neuronal cells [55]. These data might allow the hypothesis that autoimmune diseases correlate with unbalanced autophagy but, since T2D is not a classical autoimmune disease, further experimental studies based on well-characterized autoimmune antisera are needed.
4.5 Autophagy as innate immunity mechanism
There is solid evidence to suggest that autophagy is a key component of innate immunity, particularly in interactions against microbial pathogens [5, 12, 56] given that innate immune cells, and notably macrophages, use autophagy to clear infecting pathogens. Since macrophages also have been reported to utilize molecular mechanisms for the clearance of apoptotic fragments, autophagy may have a role here as well.
5. CONCLUSIONS
Autophagy, initially seen as a “cell-survival” response to adverse conditions, is now recognized as a multipurpose process involved in the normal physiology of the cell. In the context of immunity, there is clear evidence for participation of autophagy in intracellular defense against infectious agents and also perhaps, in disposal of unwanted e.g. misfolded self proteins, although there is no evidence yet for an ensuing inflammatory response to such disposal. Whatever the case, peptides from autophagic vacuoles can be delivered via lysosomes to MHC molecules and thereby access the cell surface; “immunophagy” has been coined to designate this [36]. Thus autophagy could be relevant to induction, or loss, of self-tolerance to intracellular molecules according to circumstances. There are parallels with apoptosis wherein intracellular molecules that are cleaved by caspases [57] can generate cell-surface expressed autoantigens that may jeopardize self tolerance. Students of autoimmunity have long been intrigued by the fact that many potent autoantigenic molecules/epitopes are located intracellularly, posing the question of how the protective effects of such localization are circumvented. As one example we can take the mitocondrially located PDC-E2 enzyme, which could be released when autophagy involves mitocondria (mitophagy). Other interesting questions include the correlation between autophagy and cytokine fluxes, the regulatory mechanisms for expression of autophagy genes, and the like utility of transgenic mice as a means to study the processes involved in autophagy.
In conclusion, autophagy is a physiological mechanism utilized by eukaryotic cells for the turnover of protein and other molecules to preserve their nutritional requirements during cellular starvation; under specific conditions autophagy may also determine programmed cell death. It participates in microbial immunity and may contribute to development of autoimmune diseases, but the mechanisms are yet to be clarified. Autophagy might act as a source of autoantigens derived from intracellular protein digestion, and in this way participate in the initiation or perpetuation of autoimmunity.
Finally, as suggested [39], research on autophagy may disclose potential therapeutic targets for diseases of protein misfolding, cancer and autoimmunity.
ABBREVIATIONS
- PCD
programmed cell death
- SER
smooth endoplasmic reticulum
- PI3K
phosphatidylinositol 3-kinases
- atg
autophagy genes
- Atg
autophagy proteins
- IGF-1
insulin-like growth factor-1
- APC
antigen-presenting cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 2.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 5.Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 6.Mijaljica D, Prescott M, Devenish RJ. Different fates of mitochondria: alternative ways for degradation? Autophagy. 2007;3:4–9. doi: 10.4161/auto.3011. [DOI] [PubMed] [Google Scholar]
- 7.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007 doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol. 2000;35:119–131. doi: 10.1016/s0531-5565(00)00075-9. [DOI] [PubMed] [Google Scholar]
- 9.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 10.Hamacher-Brady A, Brady NR, Gottlieb RA. The Interplay between Pro-Death and Pro-Survival Signaling Pathways in Myocardial Ischemia/Reperfusion Injury: Apoptosis Meets Autophagy. Cardiovasc Drugs Ther. 2006;20:445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 11.Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005;120:159–162. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Ogawa M, Yoshimori T, Suzuki T, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 13.Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 15.Jia G, Cheng G, Gangahar DM, et al. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 16.Pyo JO, Jang MH, Kwon YK, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Capan E, Zhao Y, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 18.Towns R, Kabeya Y, Yoshimori T, et al. Sera from patients with type 2 diabetes and neuropathy induce autophagy and colocalization with mitochondria in SY5Y cells. Autophagy. 2005;1:163–170. doi: 10.4161/auto.1.3.2068. [DOI] [PubMed] [Google Scholar]
- 19.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 20.Mordier S, Deval C, Bechet D, et al. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J Biol Chem. 2000;275:29900–29906. doi: 10.1074/jbc.M003633200. [DOI] [PubMed] [Google Scholar]
- 21.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Hamasaki M, Noda T, Baba M, et al. Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic. 2005;6:56–65. doi: 10.1111/j.1600-0854.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 23.Bernales S, McDonald KL, Walter P. Autophagy Counterbalances Endoplasmic Reticulum Expansion during the Unfolded Protein Response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 25.Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene. 2006;25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 26.Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padgett KA, Selmi C, Kenny TP, et al. Phylogenetic and immunological definition of four lipoylated proteins from Novosphingobium aromaticivorans, implications for primary biliary cirrhosis. J Autoimmun. 2005;24:209–219. doi: 10.1016/j.jaut.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Rieger R, Leung PS, Jeddeloh MR, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L, Wan F, Dutta S, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Grand RJ, Milner AE, Mustoe T, et al. A novel protein expressed in mammalian cells undergoing apoptosis. Exp Cell Res. 1995;218:439–451. doi: 10.1006/excr.1995.1177. [DOI] [PubMed] [Google Scholar]
- 33.Crighton D, Wilkinson S, O'Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 34.Rovere-Querini P, Manfredi AA, Sabbadini MG. Environmental adjuvants, apoptosis and the censorship over autoimmunity. Autoimmun Rev. 2005;4:555–560. doi: 10.1016/j.autrev.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 35.Gaipl US, Munoz LE, Grossmayer G, et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–382. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 37.Prescott NJ, Fisher SA, Franke A, et al. A Nonsynonymous SNP in ATG16L1 Predisposes to Ileal Crohn's Disease and Is Independent of CARD15 and IBD5. Gastroenterology. 2007;132:1665–1671. doi: 10.1053/j.gastro.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 38.Sooparb S, Price SR, Shaoguang J, et al. Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int. 2004;65:2135–2144. doi: 10.1111/j.1523-1755.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 39.Strawbridge AB, Blum JS. Autophagy in MHC class II antigen processing. Curr Opin Immunol. 2007;19:87–92. doi: 10.1016/j.coi.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Menendez-Benito V, Neefjes J. Autophagy in MHC class II presentation: sampling from within. Immunity. 2007;26:1–3. doi: 10.1016/j.immuni.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Zhou D, Li P, Lin Y, et al. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity. 2005;22:571–581. doi: 10.1016/j.immuni.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright K, Ward SG, Kolios G, et al. Activation of phosphatidylinositol 3-kinase by interleukin-13. An inhibitory signal for inducible nitric-oxide synthase expression in epithelial cell line HT-29. J Biol Chem. 1997;272:12626–12633. doi: 10.1074/jbc.272.19.12626. [DOI] [PubMed] [Google Scholar]
- 44.Hall JL, Gibbons GH, Chatham JC. IGF-I promotes a shift in metabolic flux in vascular smooth muscle cells. Am J Physiol Endocrinol Metab. 2002;283:E465–471. doi: 10.1152/ajpendo.00072.2002. [DOI] [PubMed] [Google Scholar]
- 45.Seko Y, Cole S, Kasprzak W, et al. The role of cytokine mRNA stability in the pathogenesis of autoimmune disease. Autoimmun Rev. 2006;5:299–305. doi: 10.1016/j.autrev.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Abbas AK, Lohr J, Knoechel B. Balancing autoaggressive and protective T cell responses. J Autoimmun. 2007;28:59–61. doi: 10.1016/j.jaut.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006;116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 49.Okada T, Inaba M, Naiki M, et al. Comparative immunobiology of thymic DC mRNA in autoimmune-prone mice. J Autoimmun. 2007;28:41–45. doi: 10.1016/j.jaut.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 50.Lu B, Capan E, Li C. Autophagy Induction and Autophagic Cell Death in Effector T Cells. Autophagy. 2007;3 doi: 10.4161/auto.3637. [DOI] [PubMed] [Google Scholar]
- 51.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 52.Pua HH, Dzhagalov I, Chuck M, et al. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2006 doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moritoki Y, Lian ZX, Ohsugi Y, et al. B cells and autoimmune liver diseases. Autoimmun Rev. 2006;5:449–457. doi: 10.1016/j.autrev.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 54.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–408. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Srinivasan S, Stevens MJ, Sheng H, et al. Serum from patients with type 2 diabetes with neuropathy induces complement-independent, calcium-dependent apoptosis in cultured neuronal cells. J Clin Invest. 1998;102:1454–1462. doi: 10.1172/JCI2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 57.Allina J, Hu B, Sullivan DM, et al. T cell targeting and phagocytosis of apoptotic biliary epithelial cells in primary biliary cirrhosis. J Autoimmun. 2006;27:232–241. doi: 10.1016/j.jaut.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]