

Abstract
Mapping the pathways that give rise to metastasis is one of the key challenges of breast cancer research. Recently, several large-scale studies have shed light on this problem through analysis of gene expression profiles to identify markers correlated with metastasis. Here, we apply a protein-network-based approach that identifies markers not as individual genes but as subnetworks extracted from protein interaction databases. The resulting subnetworks provide novel hypotheses for pathways involved in tumor progression. Although genes with known breast cancer mutations are typically not detected through analysis of differential expression, they play a central role in the protein network by interconnecting many differentially expressed genes. We find that the subnetwork markers are more reproducible than individual marker genes selected without network information, and that they achieve higher accuracy in the classification of metastatic versus non-metastatic tumors.
Keywords: breast cancer metastasis, classification, protein networks, pathways, microarrays
Introduction
Distant metastases are the main cause of death among breast cancer patients (Weigelt et al, 2005). Clinical and pathological risk factors, such as patient age, tumor size, and steroid receptor status, are commonly used to assess the likelihood of metastasis development. When metastasis is likely, aggressive adjuvant therapy can be prescribed which has led to significant decreases in breast cancer mortality rates (Weigelt et al, 2005). However, for the majority of patients with intermediate-risk breast cancer, the traditional factors are not strongly predictive (Wang et al, 2005). Accordingly, approximately 70–80% of lymph node-negative patients may undergo adjuvant chemotherapy when it is in fact unnecessary (van ‘t Veer et al, 2002). Moreover, it is believed that many of the current risk factors are likely to be secondary manifestations rather than primary mechanisms of disease. An ongoing challenge is to identify new prognostic markers that are more directly related to disease and that can more accurately predict the risk of metastasis in individual patients.
In the recent years, an increasing number of disease markers have been identified through analysis of genome-wide expression profiles (Golub et al, 1999; Alizadeh et al, 2000; Ben-Dor et al, 2000; Ramaswamy et al, 2003). Marker sets are selected by scoring each individual gene for how well its expression pattern can discriminate between different classes of disease. In breast cancer, two large-scale expression studies by van ‘t Veer et al (2002) and Wang et al (2005) each identified a set of ∼70 gene markers that were 60–70% accurate for prediction of metastasis, rivaling the performance of clinical criteria. Strangely, however, these marker sets shared only three genes in common, with the first set of markers predicting metastasis less successfully when scoring patients from the second study, and vice versa (Ein-Dor et al, 2006). One possible explanation for the different marker sets is that changes in expression of the relatively few genes governing metastatic potential may be subtle compared to those of the downstream effectors, which may vary considerably from patient to patient (Symmans et al, 1995; Ein-Dor et al, 2005; Tomlins et al, 2005).
Due to these types of difficulties, many groups have hypothesized that a more effective means of marker identification may be to combine gene expression measurements over groups of genes that fall within common pathways. Several approaches have been proposed to score known pathways by the coherency of expression changes among their member genes (Pavlidis et al, 2002, 2004; Doniger et al, 2003; Draghici et al, 2003; Subramanian et al, 2005; Tian et al, 2005; Wei and Li, 2007). Known pathways are drawn from sources such as the Gene Ontology (GO) (Harris et al, 2004) and KEGG (Kanehisa et al, 2004) databases. Recently, pathway-based analysis has been extended to perform classification of expression profiles and applied to discriminate irradiated from non-irradiated yeast cells (Rapaport et al, 2007). However, a remaining hurdle to pathway-based analysis is that the majority of human genes have not yet been assigned to a definitive pathway.
The recent availability of large protein networks provides one means to at least partially address these challenges. Using protein–protein interaction networks derived from literature, the yeast two-hybrid system, or mass spectrometry (reviewed by Mendelsohn and Brent, 1999), a number of approaches have been demonstrated for extracting relevant subnetworks based on coherent expression patterns of their genes (Ideker et al, 2002; Chen and Yuan, 2006) or on conservation of subnetworks across multiple species (Sharan et al, 2005). Each subnetwork is suggestive of a distinct functional pathway or complex, yielding many known and novel pathway hypotheses in organisms for which sufficient protein interaction data have been measured. Large protein networks have only recently become available for human (Peri et al, 2003; Ramani et al, 2005; Rual et al, 2005; Stelzl et al, 2005), enabling new opportunities for elucidating pathways involved in major diseases and pathologies (Calvano et al, 2005).
Here, we pursue a protein-network-based approach for identifying markers of metastasis within gene expression profiles, which can be used to identify genetic alterations and to predict the likelihood of metastasis in unknown samples. The markers in question are not encoded as individual genes or proteins, but as subnetworks of interacting proteins within a larger human protein–protein interaction network. We find that the network-based method has several advantages over previous analyses of differential expression. First, the resulting subnetworks provide models of the molecular mechanisms underlying metastasis. Second, although genes with known breast cancer mutations are typically not detected through analysis of differential expression, such as P53, KRAS, HRAS, HER-2/neu, and PIK3CA, they play a central role in the protein network by interconnecting many expression-responsive genes. Third, the identified subnetworks are significantly more reproducible between different breast cancer cohorts than individual marker genes selected without network information. Finally, network-based classification achieves higher accuracy in prediction, as ascertained by selecting markers from one data set and applying them to a second independent validation data set.
Results and discussion
Overview of subnetwork marker identification
We applied a protein-network-based approach to analyze the expression profiles of the two cohorts of breast cancer patients previously reported by van de Vijver et al (2002) and Wang et al (2005). Both sets of expression profiles had been obtained from primary breast tumors but hybridized to different microarray platforms (Agilent oligonucleotide Hu25K microarrays and Affymetrix HG-U133a GeneChips, respectively). We restricted our analysis to the 8141 genes present in both data sets. For 78 patients in van de Vijver et al (2002) and 106 in Wang et al (2005), metastasis had been detected during follow-up visits within 5 years of surgery. Profiles for these patients were assigned to the class ‘metastatic,' whereas profiles for the remaining 217 and 180 patients were labeled ‘non-metastatic.' To obtain a corresponding human protein–protein interaction network, we assembled a pooled data set comprising 57 235 interactions among 11 203 proteins, integrated from yeast two-hybrid experiments (Rual et al, 2005; Stelzl et al, 2005), predicted interactions via orthology and co-citation (Ramani et al, 2005), and curation of the literature (Peri et al, 2003; Alfarano et al, 2005; Joshi-Tope et al, 2005).
To integrate the expression and network data sets, we overlaid the expression values of each gene on its corresponding protein in the network and searched for subnetworks whose activities across the patients were highly discriminative of metastasis. This process involved several scoring and search steps, as illustrated in Box 1 and described further in Materials and methods. Briefly, a candidate subnetwork was first scored to assess its activity in each patient, defined by averaging its normalized gene expression values. This step yielded 295 and 286 activity scores per subnetwork, corresponding to the number of breast cancer patients in the two data sets, respectively. Second, the discriminative potential of a candidate subnetwork was computed based on the mutual information between its activity score and the metastatic/non-metastatic disease status over all patients. Significantly discriminative subnetworks were identified by comparing their discriminative potentials to those of random networks.
Schematic overview of subnetwork identification.
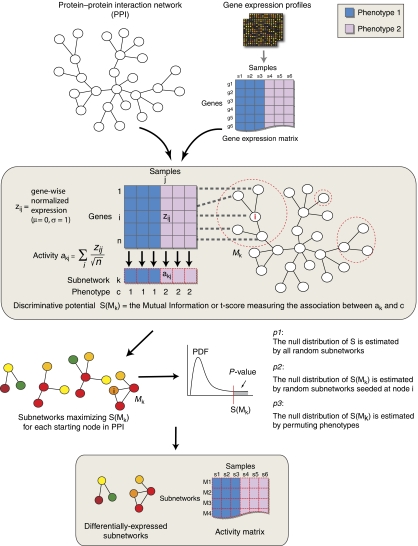
Protein–protein interaction networks are used to assign sets of genes to discrete subnetworks. Gene expression profiles of tissue samples drawn from each type of cancer (i.e., metastatic or non-metastatic) are transformed into a ‘subnetwork activity matrix'. For a given subnetwork Mk in the interaction network, the activity is a combined z-score derived from the expression of its individual genes. After overlaying the expression vector of each gene on its corresponding protein in the interaction network, subnetworks with discriminative activities are found via a greedy search. Significant subnetworks are selected based on null distributions estimated from permuted subnetworks (see Materials and methods). Subnetworks are then used to identify disease genes, and the subnetwork activity matrix is also used to train a classifier.
Subnetwork markers correspond to the hallmarks of cancer
A total of 149 and 243 discriminative subnetworks were identified in van de Vijver et al (2002) and Wang et al (2005) data sets (consisting of 618 and 906 genes, respectively, and based on a panel of three separate tests for statistical significance—see Materials and methods). A compendium including all of these subnetworks is available online via the Cell Circuits database (Mak et al, 2007) (www.cellcircuits.org), which provides each subnetwork in graphical (GIF) and machine-readable (SIF) formats. Each significant subnetwork may be viewed as a putative marker for breast cancer metastasis, which is not based on a single gene but rather on the aggregate behavior of genes connected in a functional network. This feature is a significant departure from conventional expression-alone analysis, which does not provide functional insight into the identified markers.
In all, 47.3% (van de Vijver et al, 2002) and 65.4% (Wang et al, 2005) of the discriminative subnetworks were enriched for proteins functioning in a common biological process as annotated by GO (hypergeometric test with a false discovery rate of 5%). To test whether this functional enrichment might be solely due to network topology, we extracted 1000 random subnetworks of the same size as the identified discriminative subnetworks, but without regard to the expression profiles. In the two sets of random subnetworks, 25.4±0.6 and 26.5±0.1% (mean±s.d.) were enriched for proteins with a common biological process. Our higher rate suggests that integrating protein networks with cancer expression profiles is able to identify proteins coordinately functioning in pathways. Among the discriminative subnetworks, 66 identified from van de Vijver et al (2002) and 153 identified from Wang et al (2005) corresponded to signaling of cell growth and survival, cell proliferation and replication, apoptosis, cell and tissue remodeling, circulation and coagulation, or metabolism (see Figure 1 for some example subnetworks; see CellCircuits database for all functional annotations). Together, these processes contribute to the major events that have been implicated in the progression of cancer (Hanahan and Weinberg, 2000). Many extracellular matrix and inflammatory proteins related to tumor aggression, such as matrix metallopeptidase 9 (MMP9 in Figure 1D) and interleukins (Figure 1H), were also included in the identified subnetworks. Approximately 88% of the 149 subnetworks identified from van de Vijver et al (2002) had higher activity levels in metastatic breast tumors than in non-metastatic ones, whereas the 243 subnetworks identified from Wang et al (2005) were split roughly equally in their direction of activity change (124 versus 119).
Figure 1.

Subnetworks enriched for the hallmarks of cancer. Example discriminative subnetworks from van de Vijver et al (2002) are shown in (A–E), whereas those from Wang et al (2005) are shown in (F–K). Nodes and links represent human proteins and protein interactions, respectively. The color of each node scales with the change in expression of the corresponding gene for metastatic versus non-metastatic cancer. The shape of each node indicates whether its gene is significantly differentially expressed (diamond; P<0.05 from a two-tailed t-test) or not (circle). The predominant cellular functions from Supplementary Figure 1 are indicated next to each module. Known breast cancer susceptibility genes are marked by a blue asterisk.
Subnetwork markers have increased reproducibility across data sets
Next, we examined the agreement between markers identified from the two breast cancer cohorts using our network-based approach. As shown in Figure 2A, the subnetwork markers were significantly more reproducible between data sets than were individual marker genes selected without network information (12.7 versus 1.3%). In terms of biological function, extracellular signal-regulated kinase 1 (MAPK3) was reproducible as a central node in subnetworks identified from both data sets (Figure 2C versus 2D). Figure 2E and F illustrate two other subnetworks that were discriminative in both data sets, although there was less consistency in the expression levels of genes comprising these subnetworks. For instance, PKMYT1 is significantly differentially expressed in van de Vijver et al (2002) but not in Wang et al (2005) (Figure 2E; diamond versus circle), whereas CD44 is significantly differentially expressed in Wang et al (2005) but not in van de Vijver et al (2002) (Figure 2F). However, by aggregating the expression ratios of these genes with their network neighbors, the subnetworks containing these genes are found to be significant in both data sets.
Figure 2.

Marker reproducibility and metastasis prediction performance. (A) Agreement in markers selected from the van de Vijver et al (2002) data set versus those selected from Wang et al (2005). Blue bars chart the magnitude of overlap on the left axis; the red line charts the hypergeometric P-values of overlap on the right axis. The first ‘single-gene' analysis was performed by using the same number of top discriminative genes as the number of genes covered by subnetwork markers. The second ‘single-gene' analysis was performed by using the same number of top discriminative genes as those in the gene signatures published in van de Vijver et al (2002) and Wang et al (2005). (B) AUC classification performance of subnetworks, individual genes, or modules from GO or MSigDB. The blue line charts the performance of markers selected based on the Wang et al (2005) data set and tested on the van de Vijver et al (2002) data set; the pink line represents the reciprocal test. The performance of the 1000 random subnetworks is denoted by its mean±s.d. (C and D) Erk1 (MAPK3) subnetworks in van de Vijver et al (2002) and Wang et al (2005). (E and F) Example network motifs shared between subnetworks selected from the two cohorts. The left-hand side motif is from van de Vijver et al (2002) and the right-hand side is from Wang et al (2005). (G and H) Examples of highly predictive subnetwork markers from Wang et al (2005). (I and J) Examples of highly predictive subnetwork markers from van de Vijver et al (2002).
One concern is that the increased overlap between subnetwork markers might be expected, given that the number of all possible subnetworks is smaller than the number of gene sets (selected irrespective of the network). However, the observed overlap between subnetworks was also significantly greater than that achieved among 1000 same-size sets of connected subnetworks chosen at random (P<0.002). Another question is why, even using subnetworks, the percentage overlap is not larger. One reason may be the difference in clinical design of the two data sets. While all of patients in Wang et al (2005) had lymph node-negative breast cancer, approximately half of the patients in van de Vijver et al (2002) were lymph node-positive and underwent adjuvant therapy before expression profiling. Another explanation may be the difference in microarray platforms or the incompleteness of the protein–protein interaction network, which covered only ∼40% of the gene expression levels measured in either study.
Subnetwork markers increase the classification accuracy of metastasis
We next tested the predictive performance of subnetwork markers during classification of a new expression profile as metastatic or non-metastatic. To use the subnetworks for classification, the expression levels of the genes in each subnetwork were averaged to compute a subnetwork activity score, in the same way the activity score was computed in identifying the subnetwork markers originally (see above). These activity scores were then used as feature values by a classifier based on logistic regression. At a fixed sensitivity of 90%, the subnetwork markers achieved 70.1% (van de Vijver et al, 2002) and 72.2% (Wang et al, 2005) accuracy, measured as the percentage of correct classifications using the technique of five-fold cross-validation within each data set. This accuracy compares favorably with those reported in the original studies (van de Vijver et al, 2002; Wang et al, 2005) (62 and 63%; see Supplementary Table S1).
In the above five-fold cross-validation, one-fifth of the samples were designated as ‘test' data and withheld during classifier training (in which the relative weights of each subnetwork feature are determined). However, the subnetwork features themselves were identified using all microarray samples before classification, which introduces possible circularity into the validation procedure. To achieve an unbiased evaluation of subnetwork performance, we further tested the subnetwork markers selected from one cohort of breast cancer patients as predictors of metastasis on the other cohort. This same cross-data set analysis was also run using individual marker genes according to the conventional method (controlled for size by providing the classifier with the set of 618 or 906 top discriminative genes in van de Vijver et al (2002) or Wang et al (2005), respectively, which is the same number of genes covered by the subnetwork markers; see Materials and methods). Similar to the procedure for the subnetwork markers, five-fold cross-validation was performed on one data set using the genes selected from the other data set.
At 90% sensitivity, the subnetwork markers from van de Vijver et al (2002) achieved 48.8% accuracy in classifying samples in Wang et al (2005), and 55.8% accuracy for the reciprocal test. The single-gene markers achieved 45.3 and 41.5% accuracies, respectively. Although all marker sets have decreased performance in predicting metastasis in an independent data set, the accuracies remain significantly higher than random guesses (31.2 and 39.7%, respectively). To show that the better performance was not dependent on the chosen classification algorithm, we evaluated the markers by support vector machines (SVM) (Chang and Lin, 2001), which led to the same trends (Supplementary Figure S1).
To capture performance over the entire range of sensitivity/specificity values, we also analyzed the classifiers using the AUC metric (area under ROC curve). As shown in Figure 2B and Supplementary Figure S2, the subnetwork markers significantly outperformed the single-gene markers in both data sets. Subnetwork classification performance was also higher than classifiers built on random subnetworks (P=0.046 and 0.012 against 1000 sets of same-sized random subnetworks on van de Vijver et al (2002) and Wang et al (2005), respectively); strangely, performance of the conventional classifiers was not (P=0.124 and 0.174, respectively).
Finally, we compared the classification performance of the subnetwork markers with markers based on predefined groups of functionally related genes (Figure 2B). These included 1446 sets of functionally related genes extracted from GO and 522 from the Molecular Signatures Database (MSigDB) (v1.0). Neither of these functionally related groupings performed as well as either the subnetwork markers or individual genes. This finding might indicate that some of the functional groupings relevant to breast cancer metastasis have not yet been curated in the current pathway databases.
Beyond achieving better performance, the discriminative subnetworks lend insight into the biological basis for why samples are classified as metastatic or non-metastatic. For instance, a single cell cycle-related subnetwork was identified from Wang et al (2005), which could be used to predict the metastatic outcome of ∼60% of patients in van de Vijver et al (2002) (Figure 2G). Thioredoxin (TXN), which was not differentially expressed, mediated interconnections among many cell mobility and DNA replication proteins that were differentially expressed in Wang et al (2005), forming subnetworks that were informative for metastasis in van de Vijver et al (2002) (see Figure 2H for the TXN core motif shared in multiple subnetworks). Conversely, several subnetworks identified from van de Vijver et al (2002), such as the RAD54L-related proteasome (Figure 2I) and a Ras-related subnetwork (RAB1A and RAB11A; Figure 2J), were predictive for patients in Wang et al (2005).
Subnetwork markers are informative of non-discriminative disease genes
Unlike conventional expression clustering or classification methods, network-based analyses can implicate proteins with low discriminative potential (e.g., those that are not differentially expressed), if such proteins participate in a subnetwork whose overall activity is discriminative. Such proteins can arise within a significant subnetwork if they are essential for maintaining its integrity, that is they are required to interconnect many higher scoring proteins. This property is important for the discovery of disease-causing genes, because the phenotypic changes most indicative of breast cancer metastasis need not be regulated at the level of expression (Turner et al, 2004).
Overall, 85.9 and 96.7% of the significant subnetworks contained at least one protein that was not significantly differentially expressed in metastasis (P>0.05 from a two-tailed t-test). Many well-established prognostic markers of breast cancer disease outcome, such as HER-2/neu (ERBB2), Myc, and cyclin D1, were not present in gene signatures from conventional expression-alone analysis (van ‘t Veer et al, 2002), but played a central role in the discriminative subnetworks by interconnecting many expression-responsive genes (see Figure 1C and J for examples and Supplementary Figure S3 for all). Other examples are the SMAD family and the phosphoinositide-3-kinase catalytic subunit (PIK3CA) (Figure 1E, F, I, and K): changes in SMAD phosphorylation have been linked to breast cancer metastasis (Kang et al, 2005), and somatic mutations in PIK3CA are associated with constitutive upregulation of kinase activity in ∼30% of breast cancers (Bachman et al, 2004; Campbell et al, 2004).
To evaluate the power of a network-based method to uncover disease genes, we assembled a list of 60 breast cancer susceptibility genes that had been reported as such in previous literature and were also represented in our expression data sets (de Jong et al, 2002; Lymberis et al, 2004; Online Mendelian Inheritance in Man (OMIM)) (the complete list is provided in Supplementary Table S2). We found that 32 out of 149 discriminative subnetworks from van de Vijver et al (2002) and 27 out of 243 from Wang et al (2005) contained at least one known cancer susceptibility gene (seven and five subnetworks, respectively, contained two or more known susceptibility genes). Some notable examples are RAD51 and TP53 shown in Figure 1A; ESR1 and TP53 in Figure 1B; ERBB2 in Figure 1C; BRCA1 in Figure 1F; ESR1, BRCA1, and CYP1A1 in Figure 1G; PIK3CA and HRAS in Figure 1I; GSTT1 in Figure 1J; and KRAS and PIK3CA in Figure 1K.
We compared these levels of enrichment to a conventional expression-alone analysis, which did not incorporate information on pathway structure. As shown in Figure 3A and B, subnetworks were significantly enriched with cancer susceptibility genes, in contrast to genes identified by a conventional analysis. Disease genes that can be only detected using network information include TP53, KRAS, HRAS, ERBB2, and PIK3CA.
Figure 3.

Detection of 60 known disease genes in breast cancer. The enrichment of disease genes is shown for subnetworks or individual genes selected from van de Vijver et al (2002) (A) or Wang et al (2005) (B). Blue bars chart the percentage of disease genes among all genes covered in the markers on the left axis; the red line charts the hypergeometric P-values of enrichment on the right axis. Numbers above the bars are the recovery rates of the known susceptibility genes in each marker set. (C–E) Example discriminative subnetworks containing genes with breast cancer mutations listed in Sjoblom et al. Mutation genes are marked by a plus sign.
Finally, we also examined the enrichment of the discriminative subnetworks for a recently published list of 122 genes with somatic mutations associated with breast cancer (Sjoblom et al, 2006) (71 of these were represented in the expression data sets we examined). Genes in this list were determined by DNA sequencing to have mutations in at least 1 of the 11 breast cancer cell lines, with no cancer cell line having more than six mutant genes in common with any other cancer. A total of 11 mutations mapped to proteins in the discriminative subnetworks (see Figure 3C–E for examples). Although still higher than the conventional method in van de Vijver et al (2002) (Supplementary Figure S4a), this enrichment was not significant by either approach (P=0.434 for subnetwork markers and 0.914 for single-gene markers). One explanation could be that the cancer cell lines capture a different disease state than that found in the population of patients surveyed by microarray profiling. Only two genes (p53 and BRCA1) reported in the sequencing study were linked with breast cancer in Online Mendelian Inheritance in Man, perhaps because the newly discovered mutations are rare or not genetically transmissible.
Conclusions
Human interaction databases are growing dramatically through systematic yeast two-hybrid and transcriptional interaction screens (Kim et al, 2005). Increased coverage, quality, and variety of human protein interaction data will, in turn, enable further opportunities for molecular characterization of human disease. Integrating other types of genome-wide data, such as sequence, transcription factor binding, gene and protein expression, or phenotypic information, holds further promise for determining cause and effect relationships within and between the network modules. At present, the success of network-based pathway identification and classification supports the notion that cancer is indeed a ‘disease of pathways' (Hanahan and Weinberg, 2000; Petricoin et al, 2005), and that the keys for understanding at least some of these pathways are encoded in the protein network.
Materials and methods
Scoring subnetworks
A subnetwork is defined as a gene set that induces a single connected component in the protein–protein interaction network. Given a particular subnetwork M, let a represent its vector of activity scores over the tumor samples, and let c represent the corresponding vector of class labels (metastatic or non-metastatic). To derive a, expression values gij are normalized to z-transformed scores zij which for each gene i has mean μ=0 and s.d. σ=1 over all samples j (Box 1). The individual zij of each member gene in the subnetwork are averaged into a combined z-score, which is designated the activity aj. Many types of statistic, such as the t or Wilcoxon score, could be used to score the relationship between a and c. In this study, we define the discriminative score S(M) as MI(a′,c), the mutual information MI between a′, a discretized form of a, and c
 |
where x and y enumerate values of a and c, respectively, p(x, y) is the joint probability density function (pdf) of a′ and c, and p(x) and p(y) are the marginal pdf's of a′ and c. To derive a′ from a, activity levels are discretized into ⌊log 2(# of samples) + 1⌋ = 9 equally spaced bins (Tourassi et al, 2001). A rationale for using MI in cancer classification is to capture potential heterogeneity of expression in cancer patients (Tomlins et al, 2005), that is, differences not only in the mean but in the variance of expression. For examples of the computation of MI see Supplementary Figure S5. The particular gene set maximizing S(M) is regarded as optimal for classification.
Searching for significant subnetworks
Given the discriminative score function S, a greedy search is performed to identify subnetworks within the protein–protein interaction network for which the scores are locally maximal. Candidate subnetworks are seeded with a single protein and iteratively expanded. At each iteration, the search considers addition of a protein from the neighbors of proteins in the current subnetwork and within a specified network distance d from the seed. The addition that yields the maximal score increase is adopted; the search stops when no addition increases the score over a specified improvement rate r. Given that the median distance between any two proteins in the human protein–protein interaction network is five, we set d=2 to provide a sufficient number of neighbors while keeping the search local. The parameter r is chosen as 0.05 to avoid over-fitting to the expression data used. The majority of searches terminate due to the constraint on r; increasing the value of d has only marginal effect on the results (data not shown).
To assess the significance of the identified subnetworks, three tests of significance are performed. For the first test, we perform the same search procedure over 100 random trials in which the expression vectors of individual genes are randomly permuted on the network. Such permutation disrupts the correlation between expression and interaction. The score of each real subnetwork is indexed on the ‘global' null distribution of all random subnetwork scores. The second test indexes each real subnetwork score on a ‘local' null distribution, estimated from the scores of 100 random subnetworks initialized from the same seed protein as the real subnetwork (the distribution is assumed to be gamma-distributed; Goebel et al, 2005). Third, we test whether the mutual information with the disease class is stronger than that obtained with random assignments of classes to patients (Tian et al, 2005). For the random model, these assignments are permuted in 20 000 trials, yielding a null distribution of mutual information scores for each trial; the real score of each subnetwork is indexed on this null distribution. In this study, significant subnetworks are selected that satisfy all three tests with P1<0.05, P2<0.05, and P3<0.00005, according to the three different null distributions of S.
Classification evaluation
Logistic regression models (Agresti, 1990) are trained on the subnetwork activity matrix (significant subnetworks versus patient samples) and the original gene expression matrix (i.e., conventional classification). Subnetwork markers or individual gene markers are selected using the whole first data set (van de Vijver et al, 2002) and then tested on the second data set (Wang et al, 2005); or vice versa. To measure unbiased classification performance, the patient samples in the second data set are divided into five subsets of equal size: three subsets are used as the training set to build the classifier using markers from the first data set, one subset is used as the validation set and the other subset is used as the test set. The P-value of discriminative power to classify training samples (P3) is used to rank markers (subnetworks or genes), after which the logistic regression model is built by adding markers sequentially in increasing order of P-value. The number of markers used in the classifier is optimized by evaluating its area under ROC curve (AUC; see Swets et al, 2000 for details) on the validation set. The final classification performance is reported as the AUC on the test set using the optimized classifier. Each of the five patient subsets in the second data set is evaluated in turn as the test set, with the other four sets providing training and validation. The averaged AUC values among the five test sets are reported as a final classification performance for each marker set.
Supplementary Material
Supplementary Figures and Tables
Supplementary Information
Acknowledgments
Funding for this work was provided by the National Institute of Environmental Health Sciences (ES14811-01) and Unilever, PLC. TI was additionally supported by a David and Lucille Packard Fellowship. EL and DL were supported by the Korean National Research Laboratory Grant (2005-01450) from the Ministry of Science and Technology.
References
- Agresti A (1990) Categorical Data Analysis. New York: Wiley [Google Scholar]
- Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, Buzadzija K, Cavero R, D'Abreo C, Donaldson I, Dorairajoo D, Dumontier MJ, Dumontier MR, Earles V, Farrall R, Feldman H et al. (2005) The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Res 33: D418–D424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403: 503–511 [DOI] [PubMed] [Google Scholar]
- Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, Peters BA, Velculescu VE, Park BH (2004) The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 3: 772–775 [DOI] [PubMed] [Google Scholar]
- Ben-Dor A, Bruhn L, Friedman N, Nachman I, Schummer M, Yakhini Z (2000) Tissue classification with gene expression profiles. J Comput Biol 7: 559–583 [DOI] [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF (2005) A network-based analysis of systemic inflammation in humans. Nature 437: 1032–1037 [DOI] [PubMed] [Google Scholar]
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA (2004) Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 64: 7678–7681 [DOI] [PubMed] [Google Scholar]
- Chang C-C, Lin C-J (2001) LIBSVM: a library for support vector machines. Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm
- Chen J, Yuan B (2006) Detecting functional modules in the yeast protein–protein interaction network. Bioinformatics 22: 2283–2290 [DOI] [PubMed] [Google Scholar]
- de Jong MM, Nolte IM, te Meerman GJ, van der Graaf WT, Oosterwijk JC, Kleibeuker JH, Schaapveld M, de Vries EG (2002) Genes other than BRCA1 and BRCA2 involved in breast cancer susceptibility. J Med Genet 39: 225–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR (2003) MAPPFinder: using gene ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol 4: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA (2003) Global functional profiling of gene expression. Genomics 81: 98–104 [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, Kela I, Getz G, Givol D, Domany E (2005) Outcome signature genes in breast cancer: is there a unique set? Bioinformatics 21: 171–178 [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, Zuk O, Domany E (2006) Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA 103: 5923–5928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel BDZ, Dawy Z, Hagenauer J, Mueller JC (2005) An approximation to the distribution of finite sample size mutual information estimates. In IEEE International Conference on Communications, Seoul, South Korea Vol. 2, pp 1102–1106 [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286: 531–537 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M et al. , Gene Ontology Consortium (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res 32 (database issue): D258–D261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Ozier O, Schwikowski B, Siegel AF (2002) Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18 (Suppl 1): S233–S240 [DOI] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, Lewis S, Birney E, Stein L (2005) Reactome: a knowledgebase of biological pathways. Nucleic Acids Res 33: D428–D432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277–D280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massague J (2005) Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA 102: 13909–13914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B (2005) A high-resolution map of active promoters in the human genome. Nature 436: 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymberis SC, Parhar PK, Katsoulakis E, Formenti SC (2004) Pharmacogenomics and breast cancer. Pharmacogenomics 5: 31–55 [DOI] [PubMed] [Google Scholar]
- Mak HC, Daly M, Gruebel B, Ideker T (2007) CellCircuits: a database of protein network models. Nucleic Acids Res 35: D538–D545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn AR, Brent R (1999) Protein interaction methods—toward an endgame. Science 284: 1948–1950 [DOI] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD) (accessed on 30 June 2006). World Wide Web: URL: http://www.ncbi.nlm.nih.gov/omim/
- Pavlidis P, Lewis DP, Noble WS (2002) Exploring gene expression data with class scores. Pac Symp Biocomput 474–485 [PubMed] [Google Scholar]
- Pavlidis P, Qin J, Arango V, Mann JJ, Sibille E (2004) Using the gene ontology for microarray data mining: a comparison of methods and application to age effects in human prefrontal cortex. Neurochem Res 29: 1213–1222 [DOI] [PubMed] [Google Scholar]
- Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC et al. (2003) Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res 13: 2363–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petricoin EF III, Bichsel VE, Calvert VS, Espina V, Winters M, Young L, Belluco C, Trock BJ, Lippman M, Fishman DA, Sgroi DC, Munson PJ, Esserman LJ, Liotta LA (2005) Mapping molecular networks using proteomics: a vision for patient-tailored combination therapy. J Clin Oncol 23: 3614–3621 [DOI] [PubMed] [Google Scholar]
- Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM (2005) Consolidating the set of known human protein–protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol 6: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33: 49–54 [DOI] [PubMed] [Google Scholar]
- Rapaport F, Zinovyev A, Dutreix M, Barillot E, Vert JP (2007) Classification of microarray data using gene networks. BMC Bioinformatics 8: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S et al. (2005) Towards a proteome-scale map of the human protein–protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T (2005) Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci USA 102: 1974–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber T, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314: 268–274 [DOI] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, Timm J, Mintzlaff S, Abraham C, Bock N, Kietzmann S, Goedde A, Toksoz E, Droege A, Krobitsch S, Korn B et al. (2005) A human protein–protein interaction network: a resource for annotating the proteome. Cell 122: 957–968 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swets JA, Dawes R, Monahan J (2000) Psychological science can improve diagnostic decisions. Psych Sci Public Interest 1: 1–26 [DOI] [PubMed] [Google Scholar]
- Symmans WF, Liu J, Knowles DM, Inghirami G (1995) Breast cancer heterogeneity: evaluation of clonality in primary and metastatic lesions. Hum Pathol 26: 210–216 [DOI] [PubMed] [Google Scholar]
- Tian L, Greenberg SA, Kong SW, Altschuler J, Kohane IS, Park PJ (2005) Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci USA 102: 13544–13549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, Pienta KJ, Rubin MA, Chinnaiyan AM (2005) Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310: 644–648 [DOI] [PubMed] [Google Scholar]
- Tourassi GD, Frederick ED, Markey MK, Carey E, Floyd J (2001) Application of the mutual information criterion for feature selection in computer-aided diagnosis. Med Phys 28: 2394–2402 [DOI] [PubMed] [Google Scholar]
- Turner N, Tutt A, Ashworth A (2004) Hallmarks of ‘BRCAness' in sporadic cancers. Nat Rev Cancer 4: 814–819 [DOI] [PubMed] [Google Scholar]
- van ‘t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415: 530–536 [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH et al. (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347: 1999–2009 [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA (2005) Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365: 671–679 [DOI] [PubMed] [Google Scholar]
- Wei Z, Li H (2007) A Markov random field model for network-based analysis of genomic data. Bioinformatics 23: 1537–1544 [DOI] [PubMed] [Google Scholar]
- Weigelt B, Peterse JL, van ‘t Veer LJ (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5: 591–602 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables
Supplementary Information