

SUMMARY
Hedgehog (Hh) proteins signal by inhibiting the proteolytic processing of Ci/Gli family transcription factors and by increasing Ci/Gli specific activity. In the absence of Hh, phosphorylation of Ci/Gli triggers binding to SCF ubiquitin ligase complexes and consequent proteolysis. Here we define the principal SCFSlimb binding site in Ci as an extended variant of a canonical Slimb/β-TRCP binding motif that can be created by PKA-priming of five successive CK1 sites. GSK3 enhances binding primarily through a nearby region of Ci, which may contact an SCF component other than Slimb. Studies of Ci variants with altered CK1 and GSK3 sites suggest that the large number of phosphorylation sites that direct SCFSlimb binding confers a Hh response that is both sensitive and graded, and that in the Drosophila wing disc, morphogenetic responses involve changes in both the level and specific activity of Ci. We also show that when Ci proteolysis is compromised, its specific activity is limited principally by Su(fu) and not by Cos2 cytoplasmic tethering or PKA phosphorylation.
INTRODUCTION
Drosophila Hedgehog (Hh) and its vertebrate homologs are secreted signaling proteins that directly influence cell fate and cell proliferation. Accordingly, severe developmental abnormalities and multiple forms of cancer are associated with genetic aberrations that alter the generation and transduction of Hh signals (McMahon et al., 2003; Pasca di Magliano and Hebrok, 2003). Cells respond to Hh proteins primarily through altered transcription of specific “Hh target genes” that respond directly to Cubitus interruptus (Ci) in Drosophila and Gli 1−3 in vertebrates (Hooper and Scott, 2005). In Drosophila, full-length Ci (Ci-155) is proteolytically processed to a repressor form (Ci-75) in the absence of Hh. Exposure of cells to Hh inhibits Ci-155 processing, thereby increasing Ci-155 levels while eliminating Ci-75 repressor, and also converts Ci-155 into an effective transcriptional activator. In vertebrates only Gli2 and Gli3 are processed to shorter forms, the latter of which provides most repressor function, while Gli2 and Gli1 are the major sources of regulated activator function (Huangfu and Anderson, 2006).
Ci/Gli activator and repressor forms each have some unique roles. For example, repressor forms have essential roles in patterning vertebrate limb digits and in silencing decapentaplegic (dpp) and hh expression in anterior Drosophila wing disc cells, whereas induction of most Hh target genes in the vertebrate neural tube and Drosophila wing discs requires Ci/Gli activator function (Huangfu and Anderson, 2006; Methot and Basler, 2001). However, it is not known whether dose-dependent responses to Hh proteins acting as morphogens involve different degrees of Ci/Gli proteolysis, or if they depend on a collaboration between changes in Ci/Gli proteolysis and specific activity.
Proteolytic processing of Ci-155 to Ci-75 repressor is relatively well understood in outline (Figure 1A) (Jiang, 2006). Ci-155 is phosphorylated at three Protein Kinase A (PKA) sites. The resulting phosphoserines create recognition sites for Glycogen Synthase Kinase 3 (GSK3; consensus site, [S/T]xxx[Sp/Tp]) and Casein Kinase 1 (CK1; consensus site, [Sp/Tp]xx[S/T]), which propagate an extensive cascade of localized Ci-155 phosphorylation (Ilouz et al., 2006; Jiang, 2006; Kobe et al., 2005). This creates a binding site for Slimb, the homolog of vertebrate β-TRCP, which is the key substrate recognition component of an E3 SCF ubiquitin ligase complex. Consequent proteasome digestion of ubiquitinated Ci-155 appears to be arrested fairly efficiently by the stable folding of the Ci zinc finger domain coupled to the proteolytic resistance of the adjacent polypeptide region, thereby sparing the N-terminal Ci-75 polypeptide (Tian et al., 2005). Similar mechanisms apply to Gli2 and Gli3 proteins. In Drosophila, the Kinesin-related protein, Costal-2 (Cos2) binds Ci-155, PKA, CK1 and GSK3 to facilitate Ci-155 phosphorylation. Partial dissociation of those protein kinases from Cos2 is observed in response to Hh and likely underlies the accompanying inhibition of Ci-155 processing (Zhang et al., 2005).
Figure 1. Identification of a Slimb binding site on Ci between PKA sites 1 and 2.
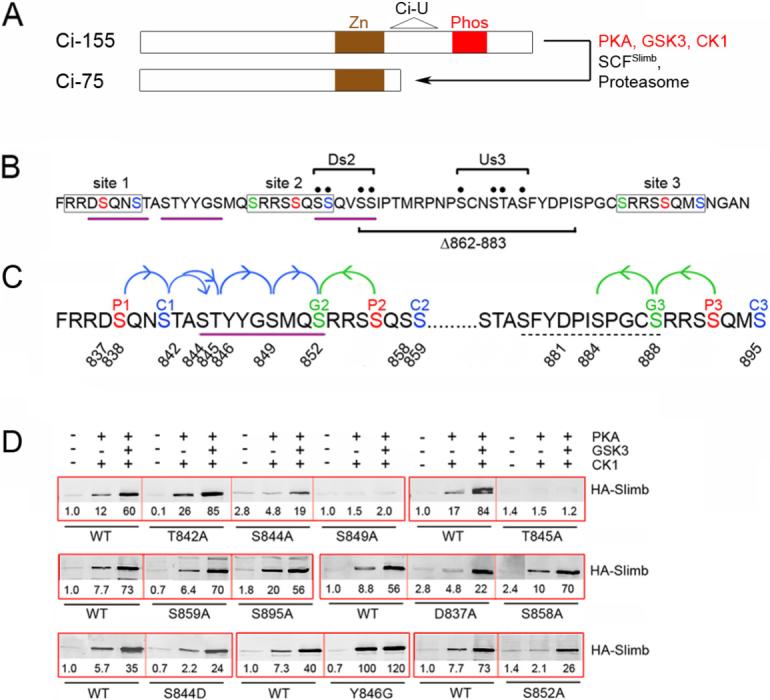
(A) Schematic showing the Zinc finger (Zn) DNA-binding domain shared by Ci-155 and Ci-75, the phosphorylated region (Phos) depicted in B and C, and the region of Ci deleted in the Ci-U transgene. (B) Amino acids 834−899 of Ci showing the three (lightly boxed) groups of PKA sites (red, P1-P3), PKA-primed GSK3 sites (green, G2 and G3) and PKA-primed CK1 sites (blue, C1-C3). Motifs resembling a consensus (DSGxxS) Slimb binding site are underlined in purple. The four Ala substitutions of Ci-Ds2 and Ci-Us3 (bracketed black dots) and residues deleted in Ci-Δ862−883 are indicated. (C) Single Ala substituents tested in this study are indicated by residue number. The extended Slimb binding motif defined here is underlined (purple) and arrows indicate critical consecutive CK1 (blue) and GSK3 (green) phosphorylations. Dashed underlining indicates residues of a potential weak Slimb binding motif. (D) GST-Ci proteins with the named substitutions were phosphorylated by the indicated protein kinases and incubated with extracts of Kc cells transfected with an HA/Flag-Slimb-Myc expression vector. Proteins brought down with glutathione beads were visualized on Western blots with HA antibody. In this and other Figures, thick red boxes group single experiments that test GST-Ci variants relative to each other and a wild-type (WT) control strictly in parallel. HA-Slimb band intensities were measured, corrected for background and expressed relative to GST-Ci-WT with no phosphorylation (fixed at 1.0) for that experiment. GST blots (not shown) confirm similar protein levels and similarly efficient phosphorylation, judged by mobility shifts.
Ci-155 specific activity is regulated by its binding partners. Ci-155 is found largely in complexes containing either Suppressor of fused (Su(fu)) or Cos2 (Hooper and Scott, 2005). These complexes are thought to restrain Ci-155 activity in the absence of Hh because excess Ci-155 can activate Hh target genes and both Cos2 and Su(fu) can limit ectopic Hh target gene induction (Ingham and McMahon, 2001). However, it is not clear how the burden of silencing Ci-155 is shared among these complexes. Su(fu) appears to restrict both the nuclear entry and nuclear activity of Ci-155, whereas Cos2 limits Ci-155 nuclear import and promotes PKA-dependent Ci-155 proteolysis (Methot and Basler, 2000; Sisson et al., 2006; Wang et al., 2000; Wang and Holmgren, 2000). Phosphorylation of Ci-155 at the PKA sites that promote processing to Ci-75 has also been suggested to reduce Ci-155 specific activity (Wang et al., 1999). Hh promotes nuclear entry of Ci-155, although the majority of Ci-155 still remains cytoplasmic. The protein kinase activity of Fused (Fu) is required to induce a full spectrum of Hh target genes, but only if Su(fu) is present. Hence, Fu kinase activity is required principally to counter inhibition of Ci-155 by Su(fu), even though Fu binds, and likely phosphorylates both Su(fu) and Cos2 (Hooper and Scott, 2005; Ingham and McMahon, 2001).
Here we look at the mechanisms that regulate Ci-155 phosphorylation, proteolysis and activity in greater detail through in vitro phosphorylation and binding assays, coupled with activity assays in wing discs. In the wing disc, Hh is produced in the posterior (P) compartment and is secreted to anterior (A) cells, which express the 12-pass transmembrane receptor, Patched (Ptc) and Ci. Hh binding to Ptc relieves an inhibition on the seven pass transmembrane protein, Smoothened (Smo), which is essential for signal propagation. Specific target genes respond to different concentrations of Hedgehog (Hooper and Scott, 2005). Thus, dpp expression is induced in a stripe of roughly a dozen cell widths extending from the site of Hh expression, whereas ptc is induced in a narrower domain and engrailed (en) only in a few cells exposed to the highest levels of Hh. Elevated Ci-155 levels are seen over the whole range of Hh signaling at the anterior-posterior (AP) border, suggesting that PKA-dependent processing is inhibited even by low levels of Hh. Within the AP border region, Ci-155 levels are in fact lowest in the cells responding to the highest levels of Hh because of a second proteolytic mechanism that uses Cullin3 (Cul3) and a Hh-inducible protein to target Ci for complete destruction (Jiang, 2006; Strigini and Cohen, 1997).
Previous studies established that binding of a Ci fragment to Slimb in vitro is stimulated by PKA, CK1 and GSK3 phosphorylation and depends on the primary PKA, CK1 and GSK3 sites that are required for Ci-155 processing in vivo (Jia et al., 2005; Smelkinson and Kalderon, 2006). We now define a Slimb-binding site in Ci generated by PKA and CK1 that differs from the canonical β-TRCP/Slimb motif. We define a second region phosphorylated by PKA and GSK3 that enhances binding only when Slimb assembles into an SCF complex, suggesting the possibility of direct interaction with complex components other than Slimb. We also uncover differing requirements for CK1 and GSK3 in regulating Ci-155 processing, provoking a model for sensitive, but potentially graded inhibition of Ci-155 proteolysis by Hh. Finally, we show that PKA phosphorylation does not influence Ci-155 activity beyond regulating its proteolysis, that cytoplasmic retention of Ci-155 by Cos2 contributes little to restraining Ci-155 activity and that regulation of Ci-155 activity by Su(fu) and Fu depends critically on whether Ci-155 is efficiently proteolyzed or not.
RESULTS
Ci residues directly recognized by Slimb
Ci-Slimb binding was assayed previously by measuring the amount of HA-tagged Slimb from Drosophila Kc cell extracts that associated with immobilized GST-Ci685−920 proteins phosphorylated by purified kinases in vitro (Smelkinson and Kalderon, 2006). Binding was induced only when GST-Ci was phosphorylated by both PKA and CK1, and was increased further by GSK3. Substitution of each of three PKA sites, both PKA-primed GSK3 sites together or all three PKA-primed CK1 sites together (red, green and blue, respectively, in Figure 1B and 1C) reduced binding significantly (Smelkinson and Kalderon, 2006). However, it was not established whether those phosphorylated residues bind to Slimb or are required just to prime further phosphorylation by CK1 or GSK3. To define which phosphorylated residues interact directly with Slimb, we tested Ci variants with single amino acid substitutions in each PKA-primed GSK3 and CK1 site and in several potential CK1-primed CK1 sites (indicated by numbered residues in Figure 1C).
Only two residues (T845 & S849) were found to be essential for binding to Slimb (Figure 1D). Substitution of S844 (adjacent to T845) also substantially reduced Slimb binding, particularly in response to phosphorylation by just PKA and CK1 (Figure 1D). If phosphorylated, S844, T845 and S849 would contribute to a sequence, SpTpYYGSp, that resembles the β-TRCP/Slimb binding site consensus DSpGxxSp with respect to the three negatively charged residues that are known to make key binding contacts (Wu et al., 2003). Replacing S844 with Asp in GST-Ci did not alter binding to Slimb significantly (Figure 1D), suggesting that S844 functions as a phosphorylated residue and that the Asp residue in the DSpGxxSp consensus can be substituted by phosphoserine or phosphothreonine, as suggested previously (Eide et al., 2005; Shirogane et al., 2005).
Any residue other than Gly at the third position of a consensus β-TRCP binding site is expected to disrupt binding substantially (Wu et al., 2003). When the Tyr residue at this position in GST-Ci was substituted by Gly (Y846G), the efficiency of phosphorylation of GST-Ci was unaltered but binding to Slimb was greatly increased in response to PKA and CK1 (Figure 1D and Figure S2). This indicates that the SpTpYYGSp motif of GST-Ci binds less well than a canonical DSpGxxSp peptide to Slimb and that PKA and CK1 suffice to phosphorylate this motif. S849 is essential for binding to Slimb but, as summarized in Figure 1C and justified below, S849 is not required to prime any additional phosphorylation events in the presence of PKA, CK1 and GSK3. We therefore conclude that Slimb-Ci association in vitro requires direct binding of Slimb to the SpTpYYGSp motif of Ci.
The function of this motif was tested in vivo using a transgene encoding full-length tagged Ci (Flag-HA-Ci-Myc) with the S849A substitution. Proteolytic processing of Ci variants to Ci-75 repressor can be assayed in posterior wing disc cells, where endogenous ci is not expressed, by preventing Hh signaling in smo mutant clones and looking for repression of a hh-lacZ reporter gene (Methot and Basler, 1999). In contrast to equivalently tagged Ci-WT, Ci-S849A failed to repress hh-lacZ in posterior smo mutant clones, indicating that it cannot be converted to the Ci-75 repressor form (Figure S1A and S1B).
PKA site 1 primes CK1 to generate a Slimb binding site
Substitution of the GSK3 site primed by PKA site 2 (S852; “G2”, Figure 1C) with Ala eliminated Slimb binding in response to just PKA and CK1 (S852A in Figure 1D). The observation that S844, T845, S849 and S852 (G2) are all required for Slimb binding stimulated by PKA and CK1 suggests that each of these residues can be phosphorylated by CK1 following PKA priming, even though S852 (G2) is clearly also a PKA-primed GSK3 site (Figure 1C). We confirmed this inference using an antibody raised against a Ci peptide centered on phosphorylated S852 (CGSMQSpRRSSQS). We found that S852 could indeed be phosphorylated either by GSK3 primed by PKA site 2 or by CK1 primed initially by PKA site 1 (Supplemental Data and Figure S2). Furthermore, S852 phosphorylation by CK1 required S849 (Figure S2), confirming the chain of successive phosphorylation shown in Figure 1C. S844 and S852 are consensus (Sp/Tp)xx(S/T) CK1 sites, whereas T845 and S849 fall within a variant CK1 consensus, (Sp/Tp)xxx(S/T), that is used almost as efficiently when assayed with peptide substrates in vitro (Bustos et al., 2005; Kobe et al., 2005).
Ci has an extended Slimb binding site
S852 contributes to Slimb binding stimulated by PKA and CK1, and might therefore be part of an extended Slimb-binding sequence (Figure 1). We tested this hypothesis by inserting five residues immediately preceding S852 (to form “VQANLS”; Figure 2A) to disrupt the motif, while preserving GSK3 phosphorylation of S852. We observed complete loss of Slimb binding stimulated by PKA and CK1 and greatly reduced binding in response to PKA, CK1 and GSK3 together. Both defects were largely remedied by replacing the first of the five inserted residues with Ser to restore the putative extended Slimb recognition motif, SpTpYYGSpMQSp (Figure 2A). Similarly robust binding to Slimb was retained when this motif was separated from PKA site 2 by as many as six non-polar residues (in “SQANLAFS”) and was not increased in this variant by introducing a Ser at a position analogous to PKA site 2 (“S852QANSAFS”) (Figure 2A). In contrast, substitution of S852 with Ala (to form “A852QANSAFS”) greatly reduced binding to Slimb (Figure 2A), confirming that S852 is part of an extended Slimb-binding motif that retains function even when its position is altered relative to PKA site 2 and other, more C-terminal phosphorylation sites.
Figure 2. The primary Slimb binding site of Ci includes S852 (G2).
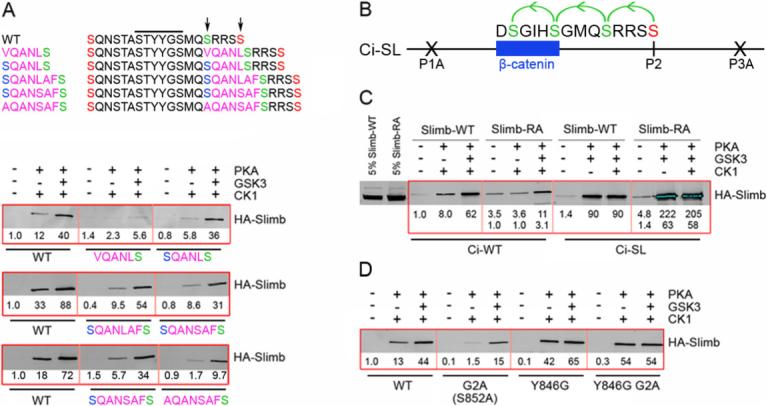
(A) Sequences of named GST-Ci variants (left) between PKA sites 1 and 2 (orange), highlighting inserted residues in pink, GSK3 sites primed by PKA site 2 in green and S852 phosphorylated only by CK1 in blue. The core Slimb binding motif (black line), S852 and PKA site 2 (arrows) are highlighted. (A, D) Flag/HA-Slimb-Myc binding to GST-Ci variants, as in Figure 1. (B) Ci-SL has altered PKA sites 1 and 3 (P1A, P3A). PKA site 2 (red) primes phosphorylation of three GSK3 sites (green) to create the Slimb/β-TRCP binding consensus of β-catenin. (C) Binding of phosphorylated GST-Ci-WT and GST-Ci-SL to Flag/HA-Slimb-Myc-WT (“Slimb-WT”) or Flag/HA-Slimb(R333A/R353A)-Myc (“Slimb-RA”). Numbers show binding relative to Slimb-WT (top row) or Slimb-RA (bottom row). 5% of the extract used for each binding reaction is also shown.
We used the crystal structure of β-TRCP1/Skp-1 bound to a dually phosphorylated β-catenin peptide (Wu et al., 2003) to identify residues that might provide a favorable electrostatic interaction with phosphorylated S852 and substituted two such residues at a time in tagged Slimb proteins used for GST-Ci binding experiments. The R333A/R353A substitutions (in “Slimb-RA”) eliminated stimulation of GST-Ci binding by phosphorylation with PKA and CK1 (Figure 2C), while the R310A/K356A substitutions had no effect (data not shown). For comparison we used a previously described GST-Ci-SL protein (Smelkinson and Kalderon, 2006), in which PKA site 2 primes successive GSK3 phosphorylation events to produce a prototypical β-TRCP/Slimb binding motif (DSpGIHSp) identical to that of β-catenin (Figure 2B). This GST-Ci-SL protein bound equally well to wild-type Slimb and Slimb-RA after phosphorylation by PKA and GSK3 (Figure 2C). Thus, binding to phosphorylated GST-Ci involves residues of Slimb that are not required to recognize the canonical β-TRCP/Slimb substrate, β-catenin. We suggest that one or more of the electropositive Slimb residues, R333 and R353, interacts directly with phosphorylated S852.
To determine which feature of the binding site in Ci allows interaction of an extended motif with Slimb we substituted S852 with Ala in the Y846G variant (to form STGYGSMQA852; “Y846G G2A”). This substitution had no effect on the strong Slimb binding afforded by phosphorylation with PKA and CK1 (Figure 2D). Thus, the presence of Tyr rather than Gly at the third position of the Slimb binding motif in Ci engenders a local peptide conformation that allows favorable contacts of an additional phosphoserine (S852 at position 9) with Slimb.
In summary, we have defined an essential, direct Slimb binding site on Ci on the basis of a single amino acid substitution (S849A) that abolishes binding without affecting further phosphorylation of Ci, and from the contributions of neighboring phosphorylated residues, which form a linear sequence motif with some resemblance to a canonical Slimb/β-TRCP binding site. A key difference from canonical motifs is the substitution of a critical Glycine residue with Tyrosine (Y846). This attenuates the interaction but allows Ci to make use of an additional phosphorylated residue (S852) within an extended binding motif. Full recognition of the extended motif depends on Slimb residues (R333/R353) that have not been implicated in binding to canonical substrates.
GSK3 enhances Slimb binding via GSK3 sites primed by PKA site 3
Loss of either PKA site 3 (“P3A”; S891A/S892A) or the preceding GSK3 site (“G3”; S888A) did not affect Slimb binding to GST-Ci phosphorylated only by PKA and CK1 but more or less eliminated further stimulation of binding by GSK3 (Figure 3A). Full-length Ci-G3A protein also accumulated to abnormally high levels in anterior wing disc cells, implying a significantly reduced rate of Ci-155 proteolysis (Figure 7; discussed later). Some processing to Ci-75 was evident from the substantial repression of hh-lacZ observed in posterior smo mutant clones of wing discs expressing Ci-G3A (Figure S1D). These properties of Ci-G3A are similar to those of wild-type Ci in cells lacking shaggy GSK3 activity, where some processing to Ci-75 persists (Figure S3A) despite a large increase in Ci-155 levels (Jia et al., 2002; Price and Kalderon, 2002). Thus, GSK3 acts principally through sites primed by PKA site 3 to enhance Ci binding to Slimb and ensure efficient Ci-155 proteolysis in the absence of Hh signaling.
Figure 3. GSK3 enhancement of Ci-Slimb binding requires GSK3 sites primed by PKA site 3 and SCF complex formation.

(A) GST-Ci variants binding to wild-type Flag/HA-Slimb-Myc probed with HA antibody. Also shown are relevant Ci and Gli-2/3 sequences. (B) Western blots for the indicated antigens (right) of extracts of Kc cells expressing transfected Myc-Slimb and the indicated Flag/HA-Slimb derivatives (top line) before (“Input”) or after immune precipitation with Flag antibody (“IP: Flag”). (C) Schematic of domains of Slimb and associated SCF complex proteins as a dimer bound to E2 enzymes. Folding of the two SCF complexes around the D-D axis toward each other likely gives a more accurate representation (Tang et al., 2007). (D, E) Binding of Flag/HA-Slimb-ΔD (D) or Flag/HA-Slimb-ΔF (E) expressed alone (upper panels) or together with Flag/HA-Slimb-WT to GST-Ci and GST-Ci-SL assayed with Flag antibody (D) or HA antibody (E). Corrected band intensities are shown relative to SL-WT binding to Ci-WT, and also for the lower panels relative to Sl-ΔD or SL-ΔF binding to Ci-WT (bottom line).
Figure 7. Ectopic Hh-target gene induction due to inefficient Ci-155 proteolysis.

C-terminal Myc epitope staining (red) of Ci variants expressed at 29C using C765-GAL4 was highest in posterior cells (right) only for Ci-Y846G and some Ci-Us3 and Ci-Ds2 lines. The inferred inefficient proteolysis of Ci-155 in other lines was accompanied in every case except for one Ci-Us3 line by ectopic induction of anterior ptc-lacZ (blue), assayed by β-galactosidase activity. “G2,3E” is S852E/S888E and “SRRAS” is S855A/S891A. Results of Ci-75 repressor assays (“hh-lacZ repression”) and Slimb binding assays (expressed as a percentage of wild-type Ci binding after phosphorylation with PKA, CK1 and GSK3) are also tabulated for each Ci variant.
How does GSK3 enhance Ci-Slimb binding? G3 (S888) phosphorylation can prime phosphorylation of another GSK3 site (S884) (Figure 1C). Alteration of that site (S884A) and a nearby acidic residue (D881A) reduced the enhancement of Slimb binding by GSK3 without altering the binding induced by PKA and CK1 alone (Figure 3A). The residue analogous to S884 in mammalian Gli2 and Gli3 proteins is the seventh residue of a potential, variant β-TRCP binding site (DSpYDPISp) (Figure 3A). The first Ser in this motif is required for normal Gli2 processing and the function of this motif in promoting Gli3 processing and β-TRCP association can be substituted by a canonical β-TRCP binding motif, suggesting that this Gli2/3 motif binds directly to β-TRCP (Pan et al., 2006; Tempe et al., 2006). The analogous sequence in Ci (SFYDPIS884) lacks an acidic or phosphorylatable residue at the second position and substitution of the first Ser (S878) together with the preceding three phosphorylatable residues (S872, S875 and T876) with Ala (to form “Us3; Upstream of site 3”; Figure 1B) only marginally reduced Slimb binding (Figure 3A). Moreover, S878 is not a consensus site for PKA, CK1 or GSK3 and is therefore not expected to be phosphorylated in our assays. Even removal of amino acids 862 to 883 from GST-Ci (Δ862−883, Figure 1B) did not prevent reasonably strong PKA/CK1-dependent binding to Slimb that was enhanced by GSK3 (Figure 3A). Likewise, alteration of four putative CK1 sites primed by PKA site 2 (“Ds2; Downstream of site 2”, Figure 1B) did not significantly impair Ci-Slimb binding (Figure S4A). Thus, none of the phosphorylatable residues between PKA site 2 and S884 (Figure 1) strongly promotes Ci binding to Slimb in vitro.
To ascertain whether the apparently minimal role of the S878FYDPIS884 motif in Ci resulted from a failure to phosphorylate S878 in vitro or from differences between the Ci and Gli2/3 motif, we converted S878 to a PKA site and we also changed the local Ci sequence to that of Gli-2/3 (DS879YDPIS884), while ensuring S879 phosphorylation as a PKA-primed CK1 site (Figure S4B). Neither change increased binding to Slimb, even though a consensus DSGIHS884 motif introduced at the same position created a strong Slimb binding site (Figure S4B). Thus, only the third of the three critical electronegative residues of the putative β-TRCP binding motif of Gli-2/3 (underlined in DSpYDPISp) contributes significantly to Ci/Slimb binding and provision of all three did not enhance binding. We therefore infer that GSK3 enhancement of Ci-Slimb binding is not mediated by a recognizable Slimb/β-TRCP binding motif and may not result from a direct interaction of the critical phosphorylated GSK3 sites with Slimb.
Strong Ci binding requires SCF complex assembly
We examined whether assembly of Slimb into SCF complexes affected Ci binding. Slimb and β-TRCP proteins can bind directly through their F-box domain to Skp1 (SkpA in Drosophila), which binds directly to Cullin1 (Cul1) (Willems et al., 2004). Cul1 can be modified by covalent linkage to Nedd8 and associates with an Rbx/Roc protein to form a complete SCF complex (Figure 3C). Several F-box proteins can also form oligomers to generate higher order SCF complexes. In particular, β-TRCP proteins dimerize through a “D-domain” preceding the F-box domain (Suzuki et al., 2000). We expressed Flag/HA-Slimb together with Myc-Slimb in Kc cells and observed robust co-immune precipitation of Myc-Slimb with antibodies to the Flag epitope, indicating abundant Slimb dimers or higher order oligomers (Figure 3B). Removal of the D-domain to form Flag/HASlimb-ΔD eliminated co-precipitation of Myc-Slimb but Cul1 binding was retained, indicating SCF complex formation (Figure 3B). Flag/HA-Slimb lacking the F-box domain co-precipitated Myc-Slimb as efficiently as observed for wild-type Slimb and also co-precipitated a small amount of Cul1, presumably indirectly via Myc-Slimb. Accordingly, Flag/HA-Slimb lacking both the F-box and D-domain did not bind Cul1 at all.
We tested the binding properties of SCF complexes using extracts from Kc cells co-transfected with tagged Slimb and tagged SkpA. SkpA bound to phosphorylated GST-Ci with the same characteristics as Slimb (Figure S5A), as did endogenous Cul1 (Figure S5B). To test if higher order SCF complexes bind to GST-Ci we first constructed a Slimb variant that could not associate directly with substrate. This variant (“Slimb-NB”) showed minimal phosphorylation-dependent binding to GST-Ci (Figure S5C). However, co-expression of Myc-Slimb allowed Flag/HA-Slimb-NB to associate with phosphorylated GST-Ci, presumably indirectly via Myc-Slimb (Figure S5C). Thus, higher order, likely dimeric, SCFSlimb complexes can bind to GST-Ci.
To examine whether the co-operative binding afforded by the CK1- and GSK3-phosphorylated regions of Ci depends on simultaneous contacts with a higher order SCF complex we used extracts expressing Slimb proteins deficient for dimerization (Slimb-ΔD) or SCF complex assembly (Slimb-ΔF). Slimb-ΔD bound as well as Slimb-WT to the canonical DSpGIHSp motif of GST-Ci-SL phosphorylated by PKA and GSK3 but (roughly 5-fold) less well than Slimb-WT to GST-Ci-WT (Figure 3D). Nevertheless, PKA/CK1-stimulated binding of Slimb-ΔD to GST-Ci was still enhanced by GSK3 (Figure 3D). Thus, the two phosphorylated regions of Ci can co-operate to bind Slimb even if dimeric or higher order SCF complexes cannot form.
Slimb-ΔF bound less efficiently than Slimb-WT to GST-Ci in response to PKA and CK1, consistent with a role for intact SCF dimers (or oligomers) in strong Ci binding. Strikingly, however, GSK3 phosphorylation of GST-Ci-WT did not enhance binding to Slimb-ΔF (Figure 3E). Thus, GSK3-stimulated binding is evident only for Slimb that is assembled into an SCF complex. This suggests that the primary mechanism by which GSK3 regulates Ci may be to create a binding site for another SCF component, not for Slimb itself.
Regulation of Ci-155 proteolysis via CK1 and GSK3
The association of PKA, CK1 and GSK3 with Cos2 facilitates Ci-155 phosphorylation and processing in Drosophila wing discs, and is reduced in response to Hh, implying that kinase dissociation from Cos2 may be instrumental in the stabilization of Ci-155 by Hh (Zhang et al., 2005). We used Ci variants to test if the regulation of Ci-155 processing by Hh requires the participation of both CK1 and GSK3. GST-Ci-G2,3E has Glu substituents at both PKA-primed GSK3 sites (S852E/S888E); its binding to Slimb in vitro is stimulated by PKA and CK1 but not by GSK3 (Smelkinson and Kalderon, 2006). Ci-G2,3E is processed to Ci-75 repressor in posterior smo mutant wing disc clones, where Hh signaling is blocked, but hh-lacZ is strongly expressed in surrounding tissue, indicating substantial inhibition of processing by Hh (Smelkinson and Kalderon, 2006). To test more sensitively for any residual processing in cells undergoing Hh signaling we expressed Ci-G2,3E and wild-type Ci (Ci-WT) in positively-marked posterior wing disc clones that retain normal Smo function. We did not observe any hh-lacZ repression in such clones (Figures 4A and 4B), implying that Hh inhibits Ci-155 proteolysis efficiently even when there is no regulation of Ci phosphorylation at its critical GSK3 sites.
Figure 4. Hh only fully blocks Ci-155 processing if CK1 regulates Slimb binding.
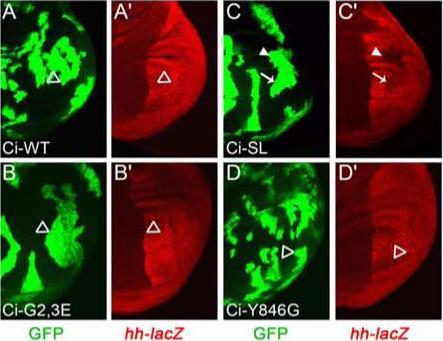
(A-D) Conversion of Ci-155 to Ci-75 repressor was assayed in positively marked wing disc clones that lack GAL80 and therefore express the indicated Ci variants together with GFP (green). Reduced expression of hh-lacZ (red) in posterior cells (right), indicating Ci-75 repressor, was seen for Ci-SL (C, arrow), especially outside the central wing blade primordia (C, arrowhead), but not (open arrowheads) for Ci-WT (A), Ci-G2,3E (B) or Ci-Y846G (D).
The binding of GST-Ci-SL to Slimb in vitro depends only on phosphorylation by PKA and GSK3 (Figures 2B and 2C) (Smelkinson and Kalderon, 2006). Ci-SL is processed to Ci-75 repressor in posterior smo mutant clones, whereas strong hh-lacZ expression was evident in surrounding posterior wing disc tissue, indicating substantial inhibition of Ci-155 processing by Hh signaling (Smelkinson and Kalderon, 2006). However, positively marked clones expressing Ci-SL in otherwise wild-type wing discs resulted in significant repression of hh-lacZ (Figure 4C). Thus, Hh signaling does not completely inhibit the processing of Ci-SL, which is driven by only PKA and GSK3 phosphorylation.
Since GST-Ci-SL binds slightly more strongly than GST-Ci-WT to Slimb in vitro we explored whether the failure of Hh to inhibit Ci-SL processing completely might be due to a stronger Ci-Slimb interaction rather than to the specific protein kinases driving proteolysis. Ci-Y846G binding to Slimb in vitro is even stronger than observed for Ci-WT or Ci-SL and depends on phosphorylation by only PKA and CK1 (Figure 1D and Figure 2D). However, no hh-lacZ repression was observed in clones that expressed Ci-Y846G in the presence of Hh (Figure 4D). This confirms the conclusion (from Ci-G2,3E) that GSK3 participation is not essential for Hh to block Ci-155 processing efficiently and indicates that the failure of Hh to block Ci-SL processing completely is because CK1 does not participate. Increased affinity of phosphorylated Ci for Slimb (as for Ci-Y846G) does not cause constitutive processing of Ci-155 to Ci-75 in the presence of Hh.
PKA-dependent proteolysis of Ci-U invalidates the prior conclusion that PKA sites 1−3 regulate Ci-155 specific activity
Having learned more about the mechanisms of Ci phosphorylation and proteolysis, we explored further how these regulatory influences affect Ci activity in vivo. Ci-155 levels are increased substantially and Ci-75 repressor activity is undetectable in both slimb and PKA mutant wing disc clones, but only the latter induce the Hh target gene reporter, ptc-lacZ and En (Ingham and McMahon, 2001) (Figure S6A-J) Furthermore, ptc-lacZ is induced in slimb mutant clones that express a PKA inhibitor (Wang et al., 1999) and in slimb PKA double mutant clones (Figure S6K-T), suggesting that the specific activity of Ci-155 might be reduced by direct PKA phosphorylation.
This idea was tested previously using a Ci derivative lacking amino acids 611−760 (Ci-U; Figure 1A) that was thought to be resistant to PKA-dependent proteolysis because it could not be processed to the Ci-75 repressor (Methot and Basler, 1999). Expression of Ci-U throughout the wing disc induced ectopic ptc-lacZ only in posterior (P) cells where Hh is present, whereas a Ci-U derivative lacking PKA sites 1−3 induced ptc-lacZ also in anterior (A) cells that are not exposed to Hh (Wang et al., 1999). It was concluded that phosphorylation of PKA sites 1−3 reduces the specific activity of Ci-U in anterior wing disc cells. However, it was recently proposed that exactly those sequences absent from Ci-U collaborate with the zinc finger region of wild-type Ci to arrest digestion of ubiquitinated Ci-155 by the proteasome in order to spare the N-terminal Ci-75 repressor derivative (Tian et al., 2005). Thus, Ci-U may be defective for processing to Ci-75 but not for PKA-dependent ubiquitination and consequent complete proteolysis by the proteasome. We therefore re-examined the properties of Ci-U carrying epitope tags at each terminus.
A UAS-Flag/HA-Ci-U-Myc transgene was expressed evenly throughout the wing disc using C765-GAL4 and, as previously reported, induced ectopic ptc-lacZ only in P cells (Figure 5B) and largely failed to repress hh-lacZ in posterior smo mutant clones, indicating deficient processing to Ci-75 (Figure S3B). However, staining for a C-terminal Myc epitope showed that full-length Ci-U protein levels were higher in P cells, where Hh is present, than in A cells that are not exposed to Hh (Figure S7B). Furthermore, full-length Ci-U levels were reduced in posterior smo mutant clones, where Hh signaling is blocked (Figure S7H) and were elevated in anterior smo PKA double mutant clones, where PKA phosphorylation of Ci-U is blocked (Figure S7K). Thus, Ci-U appears to be proteolyzed in a PKA-dependent (and Hh-regulated) manner, just as seen for wild-type Ci (Figures S7A, S7G and S7J). Hence, the activation of Hh target genes in A cells of wing discs that was previously observed when Ala residues replaced PKA sites 1−3 of Ci-U is likely attributable, at least in part, to stabilization of Ci-U and certainly cannot be taken as evidence of increased Ci-U specific activity.
Figure 5. Su(fu) and Fu kinase regulate the activity of proteolysis-resistant Ci-155.
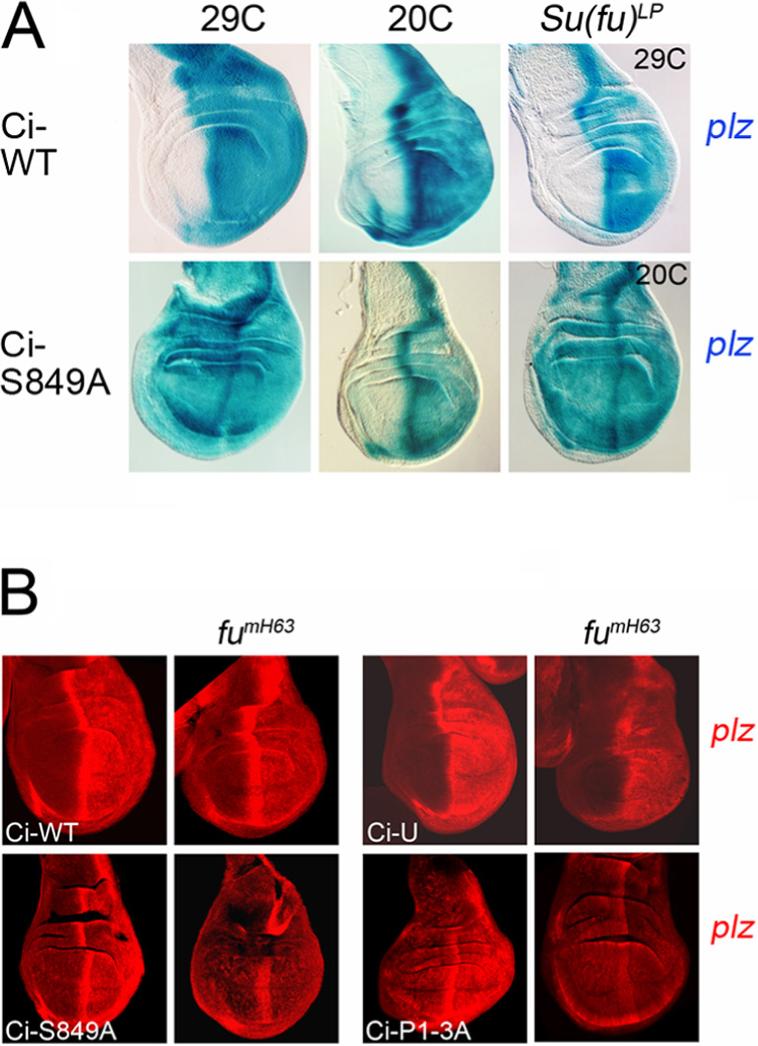
(A) Ectopic induction of ptc-lacZ (blue; assayed by β-galactosidase activity) by ci transgenes driven by C765-GAL4 was seen only in posterior wing disc cells (right) for Ci-WT at 29C and 20C but was also seen in anterior cells for Ci-S849A at 29C. At 20C, Ci-S849A induced anterior ptc-lacZ only when Su(fu) was absent. (B) Selective induction of ptc-lacZ (red; detected by antibody to β-galactosidase) in posterior cells (right) by Ci-S849A and Ci-P1−3A expressed at 20C using C765-GAL4 was eliminated in fumH63 wing discs lacking Fu kinase activity but was only reduced in discs expressing Ci-WT (here at 20C) or Ci-U (here at 29C).
PKA regulates Ci-155 activity by affecting Ci-155 levels but not Ci-155 specific activity
From our studies of GST-Ci phosphorylation and Slimb binding in vitro we expected Ci-S849A to be completely refractory to PKA-dependent proteolysis but susceptible to phosphorylation at all residues other than S849. Uniform transcription of a Ci-S849A transgene in wing discs resulted in full-length Ci-S849A protein levels, detected by a C-terminal Myc epitope, that were slightly higher in A cells than in Hh-expressing P cells (Figure S7C), as expected for a Ci variant that is subject only to Hh-stimulated, Cul3-dependent proteolysis (Jiang, 2006). When Cul3-dependent proteolysis was eliminated by using wing discs that lack Fu kinase activity, Ci-S849-Myc levels were equal in A and P cells, whereas Ci-WT-Myc and Ci-U-Myc levels were clearly higher in P cells (Figure S7D-F). Ci-S849A Myc epitope levels were neither decreased in posterior smo mutant clones (Figure S7I) nor increased in anterior smo PKA double mutant clones (Figure S7L), confirming that Ci-S849A is not subject to PKA-dependent proteolysis or regulation of this proteolysis by Hh. Ci-P1−3A has Ala substitutions at PKA sites 1−3 (Price and Kalderon, 2002) and is also completely refractory to PKA-dependent proteolysis, exhibiting the same C-terminal Myc epitope staining patterns as described above for Ci-S849A (data not shown). Comparison of Ci-S849A and Ci-P1−3A can therefore reveal whether any effects of PKA on Ci-155 specific activity involve PKA sites 1−3.
Ci-S849A and Ci-P1−3A transgenes consistently induced ectopic ptc-lacZ in both A and P cells whereas Ci-WT induced ectopic ptc-lacZ only in P cells when the transgenes were expressed using C765-GAL4 and assayed at 25C or 29C (Figure 5A and data not shown). Thus, in contrast to conclusions drawn previously from the properties of Ci-U (Methot and Basler, 1999), resistance to proteolysis can suffice for Ci-155 to activate the prototypical Hh target gene, ptc, presumably simply by increasing the levels of Ci-155.
If assayed at 20C, at which temperature GAL4 activity is roughly four-fold lower than at 29C (Morimura et al., 1996), Ci-S849A and Ci-P1−3A transgenes induced ectopic ptc-lacZ almost exclusively in P cells (Figures 5A and 5B) and only when those cells retained smo function (Figures 6D and 6G), indicating Hh-dependent activity. Thus, at 20C, Ci-S849A and Ci-P1−3A do not quite accumulate sufficiently to escape inhibition by stoichiometric binding partners in the absence of Hh signaling, but should be poised to induce ptc-lacZ in response to any increase in Ci-155 specific activity. However, we saw no increase in ptc-lacZ expression in posterior smo pka clones compared to posterior smo mutant clones in wing discs expressing either Ci-S849A or Ci-P1−3A at 20C (Figures 6D-E and 6G-H). We conclude that the activity of proteolysis-resistant Ci-155 is not significantly constrained by direct PKA phosphorylation in the absence of Hh signaling. By contrast, Ci-WT and Ci-U transgenes induced higher levels of ptc-lacZ in posterior smo pka double mutant clones than in posterior smo mutant clones at both 20C and 29C (Figures 6A and 6B; data not shown). This induction presumably results entirely from the stabilization of Ci-155.
Figure 6. Specific activity of proteolysis-resistant Ci-155 is not significantly reduced by PKA or by Cos2.

(A-I) Activation of the ptc-lacZ reporter (red) by Ci-WT (A-C), Ci-S849A (D-F) and Ci-P1−3A (G-I) expressed using C765-GAL4 at 20C was compared in posterior clones (arrowheads), marked by loss of GFP (green), that lacked activity of Smo alone (A, D, G) or Smo together with either PKA (B, E, H) or Cos2 (C, F, I). ptc-lacZ induction was increased by the additional loss of PKA and Cos2 for Ci-WT (B, C vs A) but not for Ci-S849A (D-F) or Ci-P1−3A (G-I).
Factors that regulate the activity of stabilized Ci-155
In the absence of Hh, Cos2 is thought to facilitate PKA-dependent Ci-155 proteolysis and to limit Ci-155 activity also by reducing nuclear import of Ci-155 (Wang et al., 2000; Wang and Holmgren, 2000). These two proposed activities have not, however, been uncoupled to assess each individual contribution. We found that Ci-S849A and Ci-P1−3A did not induce significantly higher levels of ptc-lacZ in posterior smo cos2 double mutant clones than in smo mutant clones at 20C (Figures 6F and 6I). Thus, for the levels of proteolysis-resistant Ci-155 in this assay, Cos2 does not significantly limit Ci-155 activity by cytoplasmic tethering or any other mechanism. We tested the validity of this conclusion under normal conditions of ci expression by comparing the phenotypes of pka cos2 double mutant clones with single pka and cos2 mutant clones in otherwise normal wing discs. Loss of Cos2 function in anterior clones induces high levels of Ci-155 and ptc-lacZ but almost no ectopic expression of Engrailed (En), which responds only to high levels of Hh pathway activity (Ingham and McMahon, 2001) (Figure S8B). PKA mutant clones induce strong ectopic ptc-lacZ and clear ectopic En expression, albeit weaker than Hh-independent En expression in P cells and Hh-induced En expression in AP border cells (Figure S8A). These different phenotypes are somewhat surprising in view of the fact that nuclear accumulation of Ci-155 is more rapid in cos2 than in PKA mutant cells of wing discs treated with Leptomycin B to inhibit nuclear export (Wang et al., 2000; Wang and Holmgren, 2000). However, since inhibition of Ci-155 proteolysis might be less complete in cos2 than in PKA mutant clones, we also examined PKA cos2 double mutant clones, in which Ci-155 levels should be equivalent to those in PKA mutant clones. Ectopic En expression was induced in PKA cos2 double mutant clones to the same level as observed in PKA mutant clones, but no higher (Figure S8C). Thus, in the absence of PKA phosphorylation and consequent proteolysis, endogenous Ci-155 activity is not significantly restrained by Cos2.
Cos2 also has a role in transducing a Hh signal but evidence for this activity in wing discs has been limited to assaying high-level Hh target genes (Wang et al., 2000; Wang and Holmgren, 2000) because other target genes, like ptc, are induced simply by eliminating the inhibitory action of Cos2 on Ci. Since Cos2 does not significantly limit the activity of Ci-S849A at 20C in the absence of Hh signaling we were able to test the positive role of Cos2 in cells responding to Hh in isolation. Loss of Cos2 activity in posterior wing disc clones reduced Hh-dependent induction of ptc-lacZ by Ci-S849A to a similar extent as seen in smo mutant clones (Figure S9). This is consistent with the possibility that Cos2 may be necessary to transduce all aspects of the Hh response (Lum et al., 2003).
In wing discs expressing Ci-S849A at 20C, loss of Su(fu) activity resulted in the induction of ptc-lacZ in A cells to almost the same level as in Hh-responding P and AP border cells (Figure 5A). By contrast, Ci-WT did not induce ptc-lacZ expression in A cells in the absence of Su(fu) even at 29C (Figure 5A). These observations indicate that Su(fu) is the key factor that limits the activity of Ci-155 protected from PKA-dependent proteolysis.
Fu kinase activity is required for Hh to counter the inhibitory action of Su(fu) on Ci-155 (Ingham and McMahon, 2001). Accordingly, in wing discs lacking Fu kinase activity, Ci-S849A and Ci-P1−3A induced almost no ptc-lacZ expression in posterior cells at 20C (Figure 5B). Induction of ptc-lacZ in P cells by Ci-WT and Ci-U was also reduced by loss of Fu kinase activity but remained higher than in A cells, especially at higher temperatures, presumably because of selective stabilization of full-length Ci in P cells (Figure 5B). Altogether, these observations indicate that PKA-dependent proteolysis normally suffices to silence Ci-155 in anterior wing disc cells without any requirement for Su(fu), whereas complete inhibition of PKA-dependent proteolysis raises endogenous Ci-155 levels to a level just beyond the silencing capacity of Su(fu).
In summary, we have used proteolysis-resistant Ci variants to establish that PKA and Cos2 limit Ci activity principally by regulating its proteolysis, and that only Su(fu) significantly restrains full-length Ci activity independent of proteolysis.
The efficiency of Ci-155 proteolysis influences Hh target gene induction
To investigate the consequences of reducing, rather than eliminating phosphorylation-stimulated binding of Ci to SCFSlimb we assayed the properties of a number of variant ci transgenes expressed at 29C using C765-GAL4 in wing discs. We chose those conditions to approximate, or slightly exceed the expression level of endogenous ci in anterior wing disc cells (based on induction of ptc-lacZ by endogenous Ci in PKA but not slimb mutant clones and by Ci-S849A at 29C but not 20C).
Ci-G3A (S888A), Ci-G2,3E (S852E/S888E) and Ci-SRRAS (S855A/S891A), which exhibited two- to five-fold reductions in Slimb binding in vitro, substantially repressed hh-lacZ in posterior smo mutant clones (Smelkinson and Kalderon, 2006) (Figure 3, Figure S1 and Figure S4), but exhibited higher C-terminal Myc epitope staining in A cells than P cells, indicating inefficient proteolysis in vivo (Figure 7). Aberrantly high anterior levels of Ci-155 coupled to strong hh-lacZ repression in posterior smo clones has also been seen for Ci-SL, which bound Slimb at least as strongly as wild-type Ci in vitro (but may not be phosphorylated as efficiently in vivo), and for endogenous Ci in cells either lacking GSK3 activity or with compromised SCF function due to Nedd8 inactivation (Ou et al., 2002; Smelkinson and Kalderon, 2006) (Figure S3). Thus, the hh-lacZ assay detects even low levels of processing whereas staining for full-length Ci easily detects reductions in proteolytic efficiency. More importantly, two- to five-fold reductions in our in vitro Slimb binding assay reflect changes in affinity in vivo that substantially inhibit Ci-155 proteolysis.
Under the conditions of our assay, where endogenous Ci is supplemented by a Ci variant expressed at a slightly higher level, Ci-G3A, Ci-G2,3E and Ci-SRRAS induced ectopic anterior ptc-lacZ in all lines (Figure 7). Ci-Us3 and Ci-Ds2 (Figure 1B), which had only minimal Slimb-binding deficits in vitro (Figure 3 and Figure S4) and fully repressed hh-lacZ in posterior smo mutant clones (Figure S1), exhibited abnormally high C-terminal Myc epitope staining in some lines but not others (Figure 7), presumably because marginally compromised proteolysis was overwhelmed only in lines where transgene expression was higher. The resulting increase in anterior Ci-155 levels was sufficient to induce anterior ptc-lacZ in some Ci-Ds2 lines but not in any Ci-Us3 lines (Figure 7). These results show that small differences in Ci-155 levels that are not readily detected by direct staining can determine whether ptc is induced or not.
We cannot predict from these experiments whether the impairment of proteolysis in variants such as Ci-G3A would suffice to induce ectopic ptc in anterior wing disc cells if expressed alone and at exactly the same level as endogenous ci. However, we do surmise that the resulting Ci-155 levels would be close to the threshold for ptc induction and might readily cross that threshold in response to a small increase in Ci-155 specific activity. Similarly, in PKA mutant clones, complete disruption of PKA-dependent Ci-155 proteolysis suffices for activation of ptc, but an increase in Ci-155 specific activity is also required to induce en strongly (Figure S6A).
DISCUSSION
Regulation of Ci-155 specific activity
The central task of Hedgehog signal transduction in Drosophila is to regulate the activity of the transcription factor, Ci. This is accomplished by regulating the levels of Ci-155 activator and Ci-75 repressor and the specific activity of Ci-155. To examine the regulation of Ci-155 specific activity in isolation we developed a Ci variant (Ci-S849A), which escapes PKA-dependent proteolysis but has a minimally altered pattern of PKA-initiated phosphorylation. Ci-S849A was expressed in wing discs (via C765-GAL4) at roughly the same level as endogenous Ci at 29C but at distinctly lower levels at 20C. At 29C, Ci-S849A induced the Hh target gene reporter, ptc-lacZ in anterior cells that are not stimulated by Hh. Since ptc induction requires Ci-155 activator and is not accomplished simply by loss of Ci-75 repressor (Methot and Basler, 1999), we conclude that elevated levels of Ci-155 can suffice to confer some activator function. At 20C, Ci-S849A induced ptc-lacZ in anterior cells only when Su(fu) was removed. By contrast, Ci-S849A activity was not detectably increased by loss of either PKA or Cos2 activity in posterior smo mutant clones, where Hh signaling is blocked. Thus, Su(fu) is the principal component that limits Ci-155 specific activity when Ci-155 is protected from PKA-dependent proteolysis, whereas PKA and Cos2 normally limit Ci-155 activity simply by promoting its proteolysis.
The prior assertion that PKA limits Ci-155 specific activity by direct phosphorylation was based on the use of an inappropriate reagent (Ci-U), which was mistakenly thought to be inert to PKA-dependent proteolysis (Wang et al., 1999). A similar role for Cos2 was previously inferred principally from the observation that Ci-155 accumulated more rapidly in the nuclei of anterior Leptomycin B-treated wing disc cells when those cells lack Cos2 activity (Wang et al., 2000; Wang and Holmgren, 2000). It appears that the inferred cytoplasmic retention of Ci-155 by Cos2 contributes very little quantitatively to limiting the activity of stabilized Ci-155. That conclusion is supported by our observation that loss of Cos2 function did not enhance the weak induction of En seen in PKA mutant clones of otherwise wild-type wing discs. The remaining, long-standing observations suggesting roles for PKA and Cos2 in limiting Ci-155 specific activity are the different degrees of Hh target gene induction in PKA (strongest), cos2 (intermediate) and slimb (weakest) mutant wing disc clones. We suggest that this might result from different degrees of disruption of PKA-dependent proteolysis in these clones. This suggestion is consistent with the proposed role of Cos2 in facilitating Ci-155 phosphorylation (Zhang et al., 2005) and with our observation that ptc-lacZ can be induced in slimb mutant clones when PKA activity is halved (Figure S6).
Regulation of Ci-155 activity through inhibition of PKA-dependent proteolysis
We have found that both GST-Ci association with SCFSlimb and Ci-155 proteolysis depend on two phosphorylated regions of Ci. The first region provides an essential Slimb binding site that can be created by five successive CK1 phosphorylations primed initially by PKA site 1. Of the four phosphorylated residues within the motif (844SpTpYYGSpMQSp852) that interacts directly with Slimb, at least one (S849) is essential for binding and two others (S844, S852) enhance binding (T845 is essential, but priming and binding functions have not been separated). The second critical phosphorylated region of Ci includes two GSK3 sites (S884, S888) that are primed by PKA site 3. This region enhances, but does not suffice for, binding to SCFSlimb.
The requirement for multiple successive phosphorylations by PKA, CK1 and GSK3 to create a high affinity SCFSlimb binding domain on Ci-155 has two important consequences. First, it demands a special mechanism for facilitating Ci phosphorylation that is met by Cos2. Second, it provides a mechanism through which a small change in Ci-155 phosphorylation, induced for example by limited dissociation of protein kinases from Cos2, can be translated into a substantial inhibition of Ci-155 proteolysis. The sharp increase in Ci-155 levels at the anterior limit of Hh signaling territory shows that a low dose of Hh does indeed severely curtail PKA-dependent Ci-155 proteolysis (Hooper and Scott, 2005). We found that Hh could inhibit proteolytic processing of Ci variants driven by either PKA-primed GSK3 sites (Ci-SL) or PKA-primed CK1 sites (Ci-G2,3E and Ci-Y846G), but complete inhibition was observed only for the latter pair. This suggests that the sensitive response of Ci proteolysis to Hh depends principally on CK1.
How does inhibition of Ci-155 proteolysis affect Ci-155 activity? Previously, the properties of Ci-U and slimb mutant clones were taken as evidence that inhibition of proteolysis does not suffice for Ci activation. Since we have now shown that Ci-U is subject to PKA-dependent proteolysis and that PKA does not affect the specific activity of Ci-155, we rely instead on the properties of Ci-S849A and PKA mutant clones to conclude that complete inhibition of PKA-dependent proteolysis does suffice to induce the Hh target gene, ptc. This, in turn, suggests that the high Ci-155 levels anterior to the stripe of elevated ptc expression at the AP border of wing discs result from substantial, but incomplete inhibition of Ci-155 proteolysis.
It has generally been assumed that inhibition of Ci-155 proteolysis is uniformly strong throughout the AP border and that the activation of ptc and en in nested domains is due solely to changes in Ci-155 specific activity elicited by increasing levels of Hh. However, several factors suggest to us that there may also be a significant gradient of residual PKA-dependent proteolysis at the AP border that contributes to morphogen action.
First, the precise degree of substantially inhibited Ci-155 proteolysis can determine whether Hh target genes are induced or not. This is evident from differences in ptc-lacZ induction among proteolytically impaired Ci variants (Fig. 7) and between PKA and slimb mutant clones.
Second, we have found that Su(fu) is the principal regulator of Ci-155 activity when Ci-155 levels are elevated, yet Hh instructs an almost unchanged morphogenetic response in the absence of Su(fu). It is likely that a proteolytic gradient is critical under these conditions, although it is also possible that Cos2 assumes a more significant role in regulating Ci-155 specific activity when Su(fu) is absent.
Third, we found that the loss of any one of four phosphoserines that contribute to Ci-Slimb binding (S844, 852, 884, 888) diminishes but does not abolish Slimb binding. For S888A (G3A) this results in elevated Ci-155 levels and an increased activity but residual proteolysis is clearly evident from the ability to generate sufficient Ci-75 to repress hh-lacZ (Figure S1D and Figure 7). Thus, dispersion of direct Slimb binding determinants among several phosphorylatable residues provides a mechanism for Hh to elicit graded inhibition of Ci-155 proteolysis. We speculate that at the highest levels of Hh, most Ci-155 molecules will not bind to SCFSlimb at all because they lack at least two of the six key phosphorylated residues, whereas a large proportion of Ci-155 molecules may bind SCFSlimb with intermediate affinity at low or intermediate Hh levels because they lack only one critical phosphoserine.
Fourth, regulation of Ci-155 specific activity depends on Ci-155 levels. Thus, Ci-155 is only activated by loss of Su(fu) when Ci-155 levels are elevated by Hh or appropriate mutations, presumably because other stoichiometric binding partners such as Cos2 act redundantly with Su(fu) when their Ci-155 binding capacity is not saturated. We would therefore expect the release of Ci-155 from repressive partners to be progressively facilitated as the relative levels of Ci-155 increase. This would allow increasing Hh levels to enhance Ci-155 activity through synergistic effects on Ci-155 levels and Ci-155 specific activity.
Properties of a variant Slimb/β-TRCP binding motif
The archetypal β-TRCP/Slimb substrates, β-catenin and IKB, contain a single, dually phosphorylated, high affinity binding site (DSpGxxSp) that triggers rapid substrate proteolysis (Willems et al., 2004). The primary direct Slimb binding motif that we defined (SpTpYYGSpMQSp) in Ci differs notably by the presence of Tyr instead of Gly at the third position, by the inclusion of a fourth electronegative residue at its C-terminus and by binding with lower affinity, permitting additional influences on Ci-SCFSlimb association. The fourth electronegative residue (pS852) likely interacts with at least one of two positively charged Slimb surface residues (R333, R353) based on their potential proximity and reduced GST-Ci binding to the R333A/R353A Slimb variant.
Most known β-TRCP substrates include phosphorylated or acidic residues 2−4 residues C-terminal to the standard six amino acid binding motif (DSpGXXSp) but their contribution to binding has not generally been assessed. Even β-catenin includes such phosphorylated residues that are known to have an essential priming role but have not been tested rigorously for direct interactions with β-TRCP. The variant β-TRCP binding motif (EEGFGSpSSp) of mammalian Wee1A presents a notable exception, in which a β-TRCP Arg residue equivalent to R353 of Slimb interacts with the phosphoserines at position 6 and 8 of this motif (Watanabe et al., 2005). This suggests that positive surface residues of β-TRCP/Slimb may commonly stabilize association with extended binding motifs. We speculate that extended β-TRCP/Slimb binding motifs are likely to be especially important and prevalent in substrates lacking Gly at the third position because we found that R333 and/or R353 of Slimb promotes binding to Ci-WT but not to Ci-SL, and pS852 contributes significantly to Slimb binding in Ci-WT but not in Ci-Y846G.
Vertebrate Gli homologs of Ci also have a residue other than Gly at the third position (generally Ala) and a potentially phosphorylated Ser at the C-terminus of a putative extended β-TRCP binding motif of 9−11 residues (SSAYx(x)SRRSS). Both a second Wee1A binding motif (DSAFQEPDS) and a β-TRCP binding motif of the p100 precursor of NFKB p52 (DSAYGSQSVE) also lack a Gly residue at the third position and include residues beyond the 6 amino acid core motif that might, by analogy to Ci, potentiate binding (Amir et al., 2004; Liang et al., 2006; Watanabe et al., 2005).
Our studies of Ci provide a clear precedent for the use of an extended β-TRCP/Slimb binding motif to translate regulated substrate phosphorylation into regulated proteolysis. However, we also found that Slimb binding cannot be predicted by focusing only on the interactions of charged residues. Thus, fully phosphorylated Ci includes two sequences with a similar distribution of charged residues to that of the primary SCFSlimb binding site (837DSpQNSpTpASpTp and 858SpSpQVSpSpIPTp compared to 844SpTpYYGSpMQSp) but those sites neither suffice for Slimb binding (in Ci-S849A) nor enhance binding significantly in vitro (as revealed by D837A, T842A, S858A, S859A and Ds2 variants). This probably reflects significant binding contributions of non-polar residues in positions 3−5 of an extended Slimb/β-TRCP binding motif, as suggested by the presence of Tyr or Phe at position 4 of the functional motifs of Ci, Gli, p100 and Wee1A.
Co-operative SCFSlimb binding via two phosphorylated regions of Ci and SCF oligomerization
Several F-box proteins that use WD40 repeats to bind substrate (“FBW” proteins) also include a dimerization domain that directs assembly of higher order SCF complexes (Tang et al., 2007; Willems et al., 2004). Some substrates of these SCF complexes (for example, Cyclin E for Fbw7 and Wee1A for β-TRCP) contain more than one phosphorylated region capable of interacting with the same WD40 binding surface of the FBW protein (Hao et al., 2007; Watanabe et al., 2005). This raises the question of whether the co-operative function of two or more such regions depends on SCF dimerization and simultaneous interaction with two FBW subunits of a dimeric complex.
We found that Ci also contains two phosphorylated regions that contribute to SCF association and that Slimb molecules can bind to each other within higher order functional SCF complexes. We also found that Slimb self-association enhanced binding to GST-Ci relative to GST-Ci-SL, which contains a single DSGxxS motif; however, it was not required for both phosphorylated regions of GST-Ci to stimulate binding. Hence, simultaneous binding to separate Slimb monomers within a larger complex can be excluded as a requisite mechanism for co-operativity between the two phosphorylated regions of Ci. This is consistent with recent structural studies that predict a wider separation of WD40 binding surfaces within an SCF dimer than can be spanned readily by the two critical phosphorylated regions of a single Ci molecule (Tang et al., 2007).
Does the region preceding PKA site 3 of Ci bind directly to the FBW component (Slimb) of an SCF complex as for Cyclin E and Wee1A? That model was proposed for Gli-2/3 proteins, which include a recognizable, potentially extended, variant Slimb-binding motif (DSYDPISTDAS) (Pan et al., 2006; Tempe et al., 2006). The analogously positioned sequence in Drosophila (SFYDPISPGCS) retains the YDPIS sequence and the two GSK3 sites at position 7 and 11 (underlined) but lacks a Ser at position 2 (bold), which is required in Gli-2/3 for normal β-TRCP association and proteolysis. Also, Ala substitution of the first Ser in the Drosophila motif (together with three other Ser residues) had only a minor effect on Slimb binding in vitro and Ci-155 proteolysis in vivo. Thus, this region of Ci does not have a clearly recognizable and demonstrably functional, conventional β-TRCP/Slimb binding site. It is, nevertheless, conceivable that the conserved elements of the putative β-TRCP binding motif of Gli-2/3 might provide a very weak direct interaction with the WD40 domain of Slimb that is sufficient to enhance SCFSlimb association.
However, we also found that the GSK3 enhancement of Ci-Slimb binding conferred by the GSK3 sites preceding PKA site 3 was lost if Slimb lacked an F-box domain and consequent direct association with SkpA and its SCF complex partners. We interpret this result with some caution because Slimb-ΔF also bound less well than wild-type Slimb to a canonical β-catenin substrate and to GST-Ci phosphorylated only at its primary Slimb binding site. Nevertheless, the result suggests that the region of Ci immediately preceding PKA site 3 might augment SCF association by binding directly to an SCF component other than Slimb.
Whether GSK3 stimulates Ci binding to SCFSlimb via a direct interaction with Slimb, an unprecedented interaction with another SCF component, or through a conformational effect on the primary Slimb binding site of Ci, the Ci-G3A transgene reveals that the stimulation conferred by GSK3 phosphorylation is critical for efficient Ci-155 proteolysis and for Hh pathway silencing (Figure 7). Since Slimb self-association enhanced GST-Ci but not GST-Ci-SL binding in vitro, we suspect that this may also be important for Ci-155 proteolysis in vivo. We do not know if SCFSlimb dimerization (or oligomerization) is regulated, but the different modes of association of SCFSlimb with Ci and with β-catenin certainly provide several opportunities for SCF regulatory mechanisms or mutations to affect the Hh pathway without altering the Wnt/β-catenin pathway.
EXPERIMENTAL PROCEDURES
Mutagenesis and cloning
The entry vectors and destination vectors for P-element germline transformation and tissue culture cell transfection were made using Gateway Technology (Invitrogen) as described previously (Smelkinson and Kalderon, 2006). Additionally, for tissue culture cell transfection we used destination vectors pAFHW and PAMW, which encode N-terminal Flag/HA and N-terminal Myc tags, respectively. pGEX2T-Ci685−920, described previously (Price and Kalderon, 2002), was used to make the GST-Ci fusion proteins. Mutations were made in pENTR-D/TOPO-Ci, pENTR-D/TOPO-Slimb, and pGEX2T-Ci using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and altered coding regions were sequenced in their entirety.
Kc cell extracts, in vitro phosphorylation and GST pull-down assay
Cells were cultured as described previously (Smelkinson and Kalderon, 2006) but extracts were made 60 hours after providing fresh media following DNA transfection. In vitro phosphorylation and GST pull-down assays were described previously (Smelkinson and Kalderon, 2006). Proteins were visualized by Western blotting using 12CA5 mouse anti-HA (Jeff Field, Univ. of Pennsylvania), 9E10 mouse anti-Myc (Developmental Studies Hybridoma Bank; “DSHB”), rabbit anti-Cul1 (Zymed) and rabbit anti-GST primary antibodies. “5923” rabbit anti-Phospho-S852 (G2) primary antibody was raised and purified by Quality Controlled Biochemicals as a customized project. Alexaflour-680 and 800 (Molecular Probes) were used as secondary antibodies and visualized using the LI-COR Odyssey Infared Imager which allows quanitification of band intensities after background subtraction, as described previously (Smelkinson and Kalderon, 2006).
Immunoprecipitation from Kc cell extracts
10μg of pAFHW-slimb* (Slimb variant with N-terminal Flag/HA tag) was transfected alone or together with 10μg of pAMW-slimb (Slimb with N-terminal Myc tag). Following lysis, extracts were incubated with 25μl bed volume of M2 mouse anti-Flag affinity agarose gel (Sigma) for approximately 2 hours. Agarose was washed 3 times with Kc lysis buffer (Smelkinson and Kalderon, 2006), eluted with SDS-PAGE loading buffer, and then boiled for 5 minutes prior to loading.
Immunohistochemistry
Larvae were heat-shocked and dissected as described previously (Smelkinson and Kalderon, 2006). ptc-lacZ and hh-lacZ expression was assayed using rabbit anti-β-galactosidase antibody (Promega) and Alexaflour-594 or 647 secondary antibodies (Molecular probes), or according to activity by incubating with X-gal (1X PBS, 0.3% Triton-X 100, 1mM MgCl2, 0.138% K4Fe(Cn)6, 0.1% K3Fe(Cn)6, 2.4 mg/ml X-gal) for 1 hour at 37°C. Full-length Ci transgenes were stained with Cy3 conjugated 9E10 mouse anti-Myc antibody (Amersham) or 9E10 mouse anti-Myc (DSHB) and Alexaflour-594 secondary. Engrailed was stained using 4D9 mouse anti-En (DSHB) and Alexaflour-594 secondary. CD2 was stained using mouse anti-rat CD2 (Serotec) and Alexaflour-647 secondary.
Fly crosses can be found in Supplementary materials.
ACKNOWLEDGMENTS
We thank Amanda Cavallero, Pui-Leng Ip and Steve Marks for contributing data, critical reagents and excellent technical assistance. We thank Konrad Basler, Cheng-Ting Chien, Jeff Field, Terence Murphy, Elizabeth Silva, Gary Struhl, the Bloomington Stock Center and the Developmental Studies Hybridoma Bank for reagents, and Steve Marks, Josie Steinhauer, Liang Tong and Cindy Vied for thoughtful discussions. This work was supported by NIH research grant GM41815 to D.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
REFERENCES
- Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23:2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- Bustos VH, Marin O, Meggio F, Cesaro L, Allende CC, Allende JE, Pinna LA. Generation of protein kinase Ck1alpha mutants which discriminate between canonical and non-canonical substrates. Biochem J. 2005;391:417–424. doi: 10.1042/BJ20050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, Vielhaber EL, Giovanni A, Virshup DM. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Molecular & Cellular Biology. 2005;25:2795–2807. doi: 10.1128/MCB.25.7.2795-2807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26:131–143. doi: 10.1016/j.molcel.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Hooper JE, Scott MP. Communicating with Hedgehogs. Nature Reviews Molecular Cell Biology. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates. Development. 2006;133:3–14. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- Ilouz R, Kowalsman N, Eisenstein M, Eldar-Finkelman H. Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J Biol Chem. 2006;281:30621–30630. doi: 10.1074/jbc.M604633200. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes & Development. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature. 2002;416:548–552. doi: 10.1038/nature733. [DOI] [PubMed] [Google Scholar]
- Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell. 2005;9:819–830. doi: 10.1016/j.devcel.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Jiang J. Regulation of Hh/Gli Signaling by Dual Ubiquitin Pathways. Cell Cycle. 2006;5 doi: 10.4161/cc.5.21.3406. [DOI] [PubMed] [Google Scholar]
- Kobe B, Kampmann T, Forwood JK, Listwan P, Brinkworth RI. Substrate specificity of protein kinases and computational prediction of substrates. Biochim Biophys Acta. 2005;1754:200–209. doi: 10.1016/j.bbapap.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Liang C, Zhang M, Sun SC. beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. 2006;18:1309–1317. doi: 10.1016/j.cellsig.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Lum L, Zhang C, Oh S, Mann RK, von Kessler DP, Taipale J, Weis-Garcia F, Gong R, Wang B, Beachy PA. Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Molecular Cell. 2003;12:1261–1274. doi: 10.1016/s1097-2765(03)00426-x. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Current Topics in Developmental Biology. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. Hedgehog controls limb development by regulating the activities of distinct transcriptional activator and repressor forms of Cubitus interruptus. Cell. 1999;96:819–831. doi: 10.1016/s0092-8674(00)80592-9. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development. 2000;127:4001–4010. doi: 10.1242/dev.127.18.4001. [DOI] [PubMed] [Google Scholar]
- Methot N, Basler K. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development. 2001;128:733–742. doi: 10.1242/dev.128.5.733. [DOI] [PubMed] [Google Scholar]
- Morimura S, Maves L, Chen Y, Hoffmann FM. decapentaplegic overexpression affects Drosophila wing and leg imaginal disc development and wingless expression. Dev Biol. 1996;177:136–151. doi: 10.1006/dbio.1996.0151. [DOI] [PubMed] [Google Scholar]
- Ou CY, Lin YF, Chen YJ, Chien CT. Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes & Development. 2002;16:2403–2414. doi: 10.1101/gad.1011402. [see comment] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nature Reviews Cancer. 2003;3:903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- Price MA, Kalderon D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell. 2002;108:823–835. doi: 10.1016/s0092-8674(02)00664-5. [DOI] [PubMed] [Google Scholar]
- Shirogane T, Jin J, Ang XL, Harper JW. SCFbeta-TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. Journal of Biological Chemistry. 2005;280:26863–26872. doi: 10.1074/jbc.M502862200. [DOI] [PubMed] [Google Scholar]
- Sisson BE, Ziegenhorn SL, Holmgren RA. Regulation of Ci and Su(fu) nuclear import in Drosophila. Dev Biol. 2006;294:258–270. doi: 10.1016/j.ydbio.2006.02.050. [DOI] [PubMed] [Google Scholar]
- Smelkinson MG, Kalderon D. Processing of the Drosophila hedgehog signaling effector Ci-155 to the repressor Ci-75 is mediated by direct binding to the SCF component Slimb. Current Biology. 2006;16:110–116. doi: 10.1016/j.cub.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Strigini M, Cohen SM. A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development. 1997;124:4697–4705. doi: 10.1242/dev.124.22.4697. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Chiba T, Suzuki T, Fujita T, Ikenoue T, Omata M, Furuichi K, Shikama H, Tanaka K. Homodimer of two F-box proteins betaTrCP1 or betaTrCP2 binds to IkappaBalpha for signal-dependent ubiquitination. J Biol Chem. 2000;275:2877–2884. doi: 10.1074/jbc.275.4.2877. [DOI] [PubMed] [Google Scholar]
- Tang X, Orlicky S, Lin Z, Willems A, Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F, Tyers M. Suprafacial orientation of the SCFCdc4 dimer accommodates multiple geometries for substrate ubiquitination. Cell. 2007;129:1165–1176. doi: 10.1016/j.cell.2007.04.042. [DOI] [PubMed] [Google Scholar]
- Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26:4316–4326. doi: 10.1128/MCB.02183-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Holmgren RA, Matouschek A. A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-kappaB. Nat Struct Mol Biol. 2005;12:1045–1053. doi: 10.1038/nsmb1018. [DOI] [PubMed] [Google Scholar]
- Wang G, Amanai K, Wang B, Jiang J. Interactions with Costal2 and suppressor of fused regulate nuclear translocation and activity of cubitus interruptus. Genes & Development. 2000;14:2893–2905. doi: 10.1101/gad.843900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang B, Jiang J. Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes & Development. 1999;13:2828–2837. doi: 10.1101/gad.13.21.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QT, Holmgren RA. Nuclear import of cubitus interruptus is regulated by hedgehog via a mechanism distinct from Ci stabilization and Ci activation. Development. 2000;127:3131–3139. doi: 10.1242/dev.127.14.3131. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, Osada H. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci U S A. 2005;102:11663–11668. doi: 10.1073/pnas.0500410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems AR, Schwab M, Tyers M. A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta. 2004;1695:133–170. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Molecular Cell. 2003;11:1445–1456. doi: 10.1016/s1097-2765(03)00234-x. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhao Y, Tong C, Wang G, Wang B, Jia J, Jiang J. Hedgehog-regulated Costal2-kinase complexes control phosphorylation and proteolytic processing of Cubitus interruptus. Developmental Cell. 2005;8:267–278. doi: 10.1016/j.devcel.2005.01.001. [see comment] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.