

Abstract
Deficiency of glycogen branching enzyme (GBE) activity causes glycogen storage disease type IV (GSD IV), an autosomal recessive error of metabolism. Abnormal glycogen accumulates in myocytes, hepatocytes, and neurons, causing variably progressive, benign to lethal organ dysfunctions. A naturally occurring orthologue of human GSD IV was described previously in Norwegian forest cats (NFC). Here we report that while most affected kittens die at or soon after birth, presumably due to hypoglycemia, survivors of the perinatal period appear clinically normal until onset of progressive neuromuscular degeneration at 5 months of age. Molecular investigation of affected cats revealed abnormally spliced GBE1 mRNA products and lack of GBE cross-reactive material in liver and muscle. Affected cats are homozygous for a complex rearrangement of genomic DNA in GBE1, constituted by a 334 bp insertion at the site of a 6.2 kb deletion that extends from intron 11 to intron 12 (g. IVS11+1552_IVS12-1339 del6.2 kb ins334 bp), removing exon 12. An allele-specific, PCR-based test demonstrates that the rearrangement segregates with the disease in the GSD IV kindred and is not found in unrelated normal cats. Screening of 402 privately owned NFC revealed 58 carriers and 4 affected cats. The molecular characterization of feline GSD IV will enhance further studies of GSD IV pathophysiology and development of novel therapies in this unique animal model.
Keywords: glycogenosis, glycogen storage, branching enzyme, enzymopathy, neuromuscular degeneration, hypoglycemia, genomic rearrangement
Introduction
The glycogen storage diseases (GSDs), are a group of autosomal recessive disorders of glycogen synthesis and degradation that result in disturbed glucose homeostasis and glycogen accumulation in various tissues [1]. The GSDs are categorized according to the deficient enzyme activity and have been numbered in the chronological order of first documentation. Clinical signs of GSDs vary, depending on which enzyme is deficient, the severity of the enzyme defect, and the type and site of glycogen accumulation. Phenotypic variations between species have also been observed and may be due to metabolic adaptations leading to the relative dependence of species on dietary carbohydrate for glucose homeostasis.
Glycogen storage disease type IV (GSD IV; OMIM 232500) is a deficiency of the glycogen branching enzyme (GBE; α-1,4glucan: α-1,4glucan 6-glucosyl transferase; EC 2.4.1.18; gene symbol GBE1; OMIM 607839). GBE is an enzyme of glycogen synthesis, deficiency of which results in tissue deposition of an abnormal and poorly soluble poly-glucan resembling amylopectin [2, 3]. The clinical presentation in humans is quite heterogeneous [4]. Many patients exhibit failure to thrive before 1 year of age, develop early hepatic cirrhosis, and die or receive a liver transplant before 4 years of age. A few reported patients exhibit nonprogressive liver disease, and some children have exhibited later onset cardiac failure or skeletal muscular disease with or without liver involvement. More recently, some cases of fetal akinesis, severe hypotonia, or cardiopulmonary collapse at birth have been attributed to GSD IV. Although few GBE mutations have been reported, it has been postulated that the phenotypic variability is at least partly due to variable residual enzyme activity in patients with different GBE mutations [5–7].
GSD IV has also been reported as an autosomal recessive trait in horses and cats. In quarter horses, the disorder is caused by a GBE1 nonsense mutation (p.Y34X) presents variably as midgestational abortion, stillbirth, or foals surviving from one to18 weeks with hypoglycemic seizures, persistent weakness, cardiac abnormalities, and/or sudden death [8–10]. In a family of Norwegian forest cats (NFC), deficiency of GBE activity was observed in 4 kittens; one died 6 hours postnatally, one died suddenly at 5 months of age due to an intercurrent pulmonary abscess, and two had onset of progressive neuromuscular and cardiac disease at 5 months of age [11]. Both of the latter had severe skeletal muscle disease by 8 months; one was euthanized at 9 months, and the other died of sudden cardiac failure at 13 months of age. Another affected NFC developed neuromuscular and cardiac disease, first observed at 5 months of age, that progressed within a month to tetraparesis with severe muscle atrophy [12]. That cat also had cardiac conduction defects with cardiomyocyte degeneration and hypoglycemic seizures that led to the decision for euthanasia.
We established an outbred GSD IV breeding colony derived from a purebred GSD IV carrier related to the originally reported [11] affected NFCs. Here we report clinical and pathologic observations from breeding experiments within this colony as well as the molecular characterization of the GBE1 mutation and GBE expression in affected cats.
Materials and Methods
Animals
We established a feline GSD IV breeding colony by mating a carrier male NFC (cat A in figure 1) to an unrelated female domestic shorthair cat (cat B in figure 1). Matings were performed by placing females housed under 16 hours of light/day in a room occupied by a specific male until pregnancy could be detected by abdominal palpation. Deliveries generally occurred unobserved and unaided during nighttime hours. We performed all matings, specimen collection, determination of clinical phenotype, and euthanasia according to protocols approved by Institutional Animal Care and Use Committees and conforming to National Institutes of Health guidelines.
Figure 1.

Feline GSD IV pedigree. Offspring of each mating are arranged on a horizontal line connecting vertical lines that descend from symbols indicating the parents. Squares indicate males, circles indicate females, filled symbols indicate GSD IV affected offspring, half-filled symbols indicate obligate carriers determined by breeding, and half-filled and open symbols indicate clinically normal cats. Numerals inside of symbols are the number of offspring, when greater than one, of the gender and clinical status indicated by the symbol, produced by that mating. All offspring were examined histologically or were observed clinically to adulthood. The colony founder cats, marked A and B, were a purebred NFC and an unrelated domestic shorthair cat, respectively. The cats labeled A, B, and C are the same cats so labeled in figure 7.
Phenotype determination
Newborn kittens were regularly examined and weighed daily until euthanasia or adoption. Routine electromyographic (EMG) studies were performed under general anesthesia induced by intramuscular administration of ketamine/valium and maintained by halothane inhalation. In vivo phosphorous nuclear magnetic resonance ([31P] NMR) spectra were acquired via a 2 cm diameter circular surface coil (81 MHz, sweep width 5000 Hz, 2K complex data, TR 3 s, 45 degree pulse at coil center) placed over the belly of the triceps surae muscle group of a GSD IV affected cat and a normal littermate. Anesthesia for this procedure was achieved by preanesthetic acetyl promazine/atropine administration and intravenous infusion of propofol (Abbott Laboratories, North Chicago, IL). Tissues of offspring that died in the perinatal period and all other sacrificed cats were preserved in 10 % neutral buffered formalin. Formalin-fixed biopsy specimens were embedded in paraffin and stained for light microscopy with hematoxylin & eosin (H&E), periodic acid-Schiff (PAS), and iodine. GBE enzyme activity was determined in homogenates of leukocytes, liver and/or muscle as previously described [11].
Feline GBE cloning and sequencing
An 0.85 kb fragment of feline GBE cDNA for use as a hybridization probe was amplified by RT-PCR as described [13] from normal cat fibroblast RNA using degenerate PCR primers deduced from areas of high homology between the yeast and human GBE sequences (GenBank accession nos. M76739 and L07956, respectively). The cDNA fragment was [32P] labeled in a random-primed DNA polymerase reaction (Roche Diagnostics, Inc., Indianapolis, IN) and used to screen 5 x 105 plaques of a normal feline liver cDNA bacteriophage λ library [14]. Plasmids containing hybridizing inserts were rescued into the phagemid pBluescript® (Stratagene, La Jolla, CA) and purified with a Qiagen® plasmid preparation kit (Qiagen, Inc., Chatsworth, CA). Of 3 hybridizing clones that were purified, 2 had apparently full-length inserts and were sequenced on both strands by dideoxy-chain-termination, cycle-sequencing methods on an ABI 373A automated sequencer (Applied Biosystems, Inc., Foster City, CA). DNA sequences were assembled and analyzed (DNASTAR, Madison, WI) and submitted to GenBank (accession no. AY439007). A feline genomic clone was similarly isolated by screening ~ 750,000 clones of a bacteriophage λFIX® II library (Stratagene, La Jolla, CA). The hybrization probe was a 475 bp Bam HI/Stu I restriction fragment of the feline GBE cDNA that included exons 11–13 and a portion of exon 14. Phage DNA of 3 purified clones was prepared, digested with Eco RI, and transferred to a nylon membrane. A single 4 kb Eco RI fragment common to 2 of the phage clones hybridized to the library-screening probe and was subcloned into pUC18 for sequencing (GenBank accession no. DQ979412). Authenticity of the clone was verified by finding GBE exons 13 and 14 within its sequence, as well as by mapping a dinucleotide repeat marker found in the sequence to feline chromosome C2 in a context homologous to human chromosome 3p11–13 [15].
Long range PCR
Long range PCR of genomic DNA templates to obtain sequence for introns 11 and 12 was performed in 50 μL reactions using the Takara LA PCR kit (PanVera, Inc., Madison, WI) and 30 cycles of 98°C for 20 s, 63°C for 1 min, and 68°C for 20 min. Primers for intron 11 were 5’-GATGAAGATTGGAATATGGGCAATA-3’ (1431 F) in exon 11 and 5’-CACCGAGAGCATGAGTAATGAGTC-3’ (1671R) in exon 12, and for intron 12 were 5’-GCTCTCGGTGGAGAAGGCTATC-3’ (1662F) in exon 12 and 5’-CAGATGCCGTAGGAACCCAAGTC-3’ (CAR) in intron 12. PCR across the rearrangement breakpoints in DNA of affected cats used primers 1431F and CAR. In some reactions we used the Taq plus Long™ reagents (Stratagene, La Jolla, CA). PCR products were gel purified and sequenced directly as described above.
Northern and Southern blotting
PolyA RNA was selected by oligo dT chromatography from 100 μg of total liver RNA of a normal and a GSD IV affected cat as previously described [16]. RNA samples were electrophoresed on 1% formaldehyde gels and transferred to a nitrocellulose membrane by standard blotting methods [17]. The membrane was sequentially hybridized to labeled probes of full-length feline GBE cDNA and partial feline β-actin cDNA. Genomic DNA was isolated from samples of cat spleen by phenol/chloroform extraction; 10 μg aliquots were digested with various restriction enzymes and electrophoresed on 0.8 % agarose gels. Southern blots [17] were hybridized to random-prime labeled probes of partial (exons 11–14, as indicated above, or exon12 only) or full-length feline GBE cDNA.
Immunoblotting
Frozen liver samples were thawed, homogenized in 2.5 volumes of cold pH 7 buffer (containing 15 mM 2-mercaptoethanol, 10 mM ethylenediaminetetraacetic acid, and 1 mM phenylmethylsulfonyl fluoride [PMSF]), and centrifuged at 150,000 x g for 60 min. proteins were immunoprecipitated from aliquots of supernatant (100 μl) containing 3 mg of total protein by sequential incubations on ice with goat serum-agarose beads (Sigma, St. Louis, MO) for preclearing, goat anti-rabbit liver GBE [7–9] or nonspecific goat serum, and anti-goat IgG-agarose beads. Bead-associated immune complexes were washed in cold 50 mM Tris-HCl, pH 7.5, containing 200 mM NaCl, 2% Na cholate, and 1 mM PMSF, and boiled in 2X Laemli electrophoresis buffer. Proteins were separated by SDS-polyacrylamide (7.5 %) gel electrophoresis and transferred to nitrocellulose membranes. Blots were blocked in 2% gelatin and incubated with goat anti-rabbit liver GBE (1:6000). After washing, blots were incubated anti-goat IgG-alkaline phosphatase conjugate (1:15,000), and immune complexes were detected by incubation in p-nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate.
Mutation detection and carrier testing
PCR for GBE amplification of genomic DNA samples isolated from blood [18] was done in separate reactions using a common forward primer and either of 2 allele-specific (normal or deleted) reverse primers flanking the site of the 5’ breakpoint. PCR primers were 5’-TGTATCTTCCACATCAGAGCAGTG-3’ (GSD F), 5’-ATAGTGTAGGTGTTGCATCCCTAAT-3’ (normal allele R), and 5’-GACACACCTGTAATCAAGTACCTCT-3’ (deleted allele R). Approximately 500 ng of genomic DNA template was amplified in 50 μL reactions using the 2.5 U of AmpliTaq® (Perkin Elmer, Culver City, CA), 1.5 mM MgCl2, 0.25 μM each primer, 0.25 μM each dNTP. Thermocycling included a manual “hotstart” and an automated “touchdown annealing” procedure in 35 cycles of 94°C for 30 s, 70 → 60°C (dropping 1°C /cycle for 10 cycles and 60°C thereafter) for 30 s, and 72°C for 5 min.
Results
Breeding experiments
We identified a purebred NFC male as a carrier of GSD IV by measurement of approximately half-normal GBE activity in peripheral blood leukocytes and liver (cat A in figure 1). A breeding colony was established by mating this cat with an unrelated healthy domestic shorthair cat of known lineage (cat B of figure 1). GBE activity was measured in leukocytes of offspring from 4 litters produced by this mating, and females considered to have half-normal activity were raised for matings with the colony-founding male. In a subsequent litter, an addition male and female were identified as carriers by enzyme assay and used in further matings. All offspring, including those that died in the perinatal period, were examined histologically or were raised to at least 6 months of age.
Two of the females, cats C and D in figure 1, produced 32 and 19 clinically normal kittens, respectively, in matings with obligate carrier males and none that were GSD IV affected. Therefore, in contradiction to their GBE enzyme activity determinations, the probability that each was actually a GSD IV carrier was < 0.0001 and < 0.0042, respectively. The incorrect designation of these cats as carriers was confirmed when the GBE1 mutation was determined (below), but results of these carrier X normal matings comprised an informative control group. The carrier X carrier matings produced 16 affected cats, 9 male and 7 female, among 94 offspring. Previously reported [11] biochemical investigation demonstrated that GBE deficiency sufficient to cause clinical disease is an autosomal recessive trait in these cats. While 17 % affected offspring with even gender distribution is close to the expected proportion, it is statistically different (χ2=3.19, df=1, p=0.074) from to what is expected for a fully penetrant, simple autosomal recessive trait. It is likely, therefore, that some affected kittens were resorbed during gestation or were not accounted for postnatally. Excluding offspring of a carrier female that routinely cannibalized her newborn kittens, litter sizes (average 5.3 kittens/litter; n=12; range 4–8) produced in carrier X carrier matings did not differ significantly from those of carrier X normal matings (5.1 kittens/litter; n=10; range 4–6) or from historical norms in this feline research facility, but the proportion of affected kittens was still only 11 of 63 offspring (17.5%).
In litters produced in carrier X carrier matings, again excluding those of the cannibalizing female, there were 14 kittens among 63 offspring that were stillborn or died in the first few hours postpartum (22 % perinatal deaths). Carrier X normal matings produced litters in which there were only 7 kittens among 54 offspring that died perinatally (13 %). The difference was even more pronounced (21% vs. 2.5 % perinatal mortality) when each cat’s first litter was not considered, in order to correct for variable mothering skills in first-litter queens. Upon histochemical analysis of tissues from offspring of the carrier X carrier matings, 10 of the 14 kittens that died perinatally had deep blue inclusions in all iodine-stained tissues examined, the chromatic reaction being indicative of abnormal glycogen synthesized in the absence of GBE activity [8, 11]. The same distribution of abnormal glycogen was found whether the affected kitten had breathed (as evidenced by aeration of the lungs) or not. In contrast, among offspring produced in the carrier X normal matings, none of 7 kittens that died in the perinatal period had the deep blue staining inclusions that characterized the affected cats in skeletal or cardiac myocytes or neurons. Instead, if the kitten had not breathed, the iodine stain revealed reddish or light mahogany-colored glycogen deposits, found mostly in the center of skeletal muscle fibers. In kittens of this group that had breathed, however, there were non-staining spaces in the center of skeletal myocytes.
Thus, in contrast to previous clinical reports [11, 12] of privately owned NFC, among the 16 affected kittens produced in this outbred colony, only 2 survived the perinatal period. Having recognized that perinatal mortality was the primary phenotype of feline GSD IV, we gave greater attention to newborn litters in later stages of the investigation. We observed 2 neonates, a male in one litter and a female in another, that were failing to thrive in the first hours postpartum. The kittens stopped nursing and exhibited reduced voluntary movement. Both were given oral and subcutaneous glucose and fluid support for 2 days and returned to normal activity within an hour of initiating therapy. After these first days of life, both kittens were clinically unremarkable until, at 23 weeks of age, each became mildly hyperthermic (rectal temperatures of 39.4–41°C; normal range 38.5–39°C). Eight and 24 days later, respectively, the male and then the female developed fine generalized muscle tremors with occasional twitches. In both cats, the hyperthermia persisted, and the neuromuscular signs increased in amplitude and frequency, leading to progressive and generalized skeletal muscle atrophy. Generalized EMG abnormalities were observed as described previously [11, 12] in the affected female at 23 weeks but not at 18 weeks of age. Similarly, elevations of muscle phosphodi- and mono-ester peaks in the [31P] NMR spectrum of the affected male (figure 2) were evident at 23 weeks but not at 18 weeks of age, findings suggestive of later-onset membrane damage and altered glycogenolysis, respectively, and consistent with a progressive myopathy of metabolic origin [19]. Necropsy of the affected male (28 weeks of age) and of the affected female (43 weeks) revealed neuromuscular pathologic changes as previously described [11, 12] though neither cat had extensive myocardial degeneration. Additional findings in the affected female, not previously reported, were mild to moderate storage of abnormal glycogen in retinal ganglion cells with some minimal degeneration of the optic nerve, characterized by dilated myelin sheaths and occasional deposits of debris in the older cat. Accompanying these changes was severe glycogen storage with degeneration, necrosis, and myocyte regeneration in the extra-ocular muscles. Immunohistochemical staining for glial fibrillary acidic protein in the affected male’s spinal cord revealed exuberant astrocytosis diffusely throughout both dorsal and ventral horn gray matter.
Figure 2.

Muscle [31P] NMR spectra of a GSD IV affected cat and a normal littermate. Panel A shows the spectrum collected from the triceps surae of a normal male cat and panel B shows that collected from his GSD IV affected male littermate, each at 18 weeks of age. Panel C shows the spectrum collected from the affected cat at 23 weeks of age. Spectral peaks representative of inorganic phosphate, phosphocreatine, and ATP labeled in the γ, α, and β phosphate positions, are indicated by Pi, PCr, γ, α, and β, respectively. PME and PDE indicate peaks of phosphomono- and phosphodiesters, respectively.
Cloning and sequence of feline GBE1
The full-length feline GBE1 cDNA was cloned and sequenced (GenBank Accession no. AY439007). It is composed of an 86 bp 5’ untranslated region (UTR), a 2097 bp open reading frame (ORF), and a 716 bp 3’ UTR. The deduced amino acid sequence is 699 residues with 94%, 92%, 90%, 89%, 77%, 69%, 66%, 61%, 59% and 57% identity to GBE of dog, horse, human and mouse, rat, frog, chicken, fish, fruitfly, Saccharomyces cerevisiae, and Arabidopsis thaliana sequences, respectively, found by BLASTP search [20] in GenBank. A BLAT search [21] in the dog and human genomes for homology to the feline cDNA sequence indicates that feline GBE1 is composed of 16 exons and 15 introns. If the genomic DNA spanned by the cDNA is similar in length to that in dog, the gene is ~265 kb.
Analysis of GBE1 expression in GSD IV affected cats
Northern blots of normal feline liver RNA demonstrated an ~ 3 kb mRNA species in polyA selected RNA, but no signal was observed in GSD IV affected cat liver (figure 3A). Despite this result, we obtained and sequenced overlapping RT-PCR products from affected cat fibroblast RNA after random hexamer priming of the RT reaction. However, when we used primers in exons 8 and 14 for the PCR reaction, we obtained multiple smaller products, instead of the predicted single product of 848 bp (figure 3B). The two largest of the abnormal products from GSD IV affected cat were sequenced, and they corresponded to mRNA transcripts missing precisely the 172 bp of exon 12 or the 283 bp of exons 11 and 12 together, respectively. RT-PCR products of affected cats that did not cross exon 12, cumulatively covering the full extent of the GBE1 cDNA, were of expected size and normal sequence. Both incorrectly spliced products predicted shifts of the reading-frame and a premature translation stop 10 codons into exon 13. Western blots of proteins immunoprecipitated from normal cat liver, but not from that of a GSD IV affected cat, demonstrated a single protein band of ~ 80 kDa that reacted to anti-rabbit liver GBE antiserum (figure 3C). Thus it appears that in liver and muscle (the latter tissue not shown) of GSD IV affected cats, whatever aberrant GBE1 mRNAs may exist are not stable or do not encode a stable, immunodetectable protein.
Figure 3.
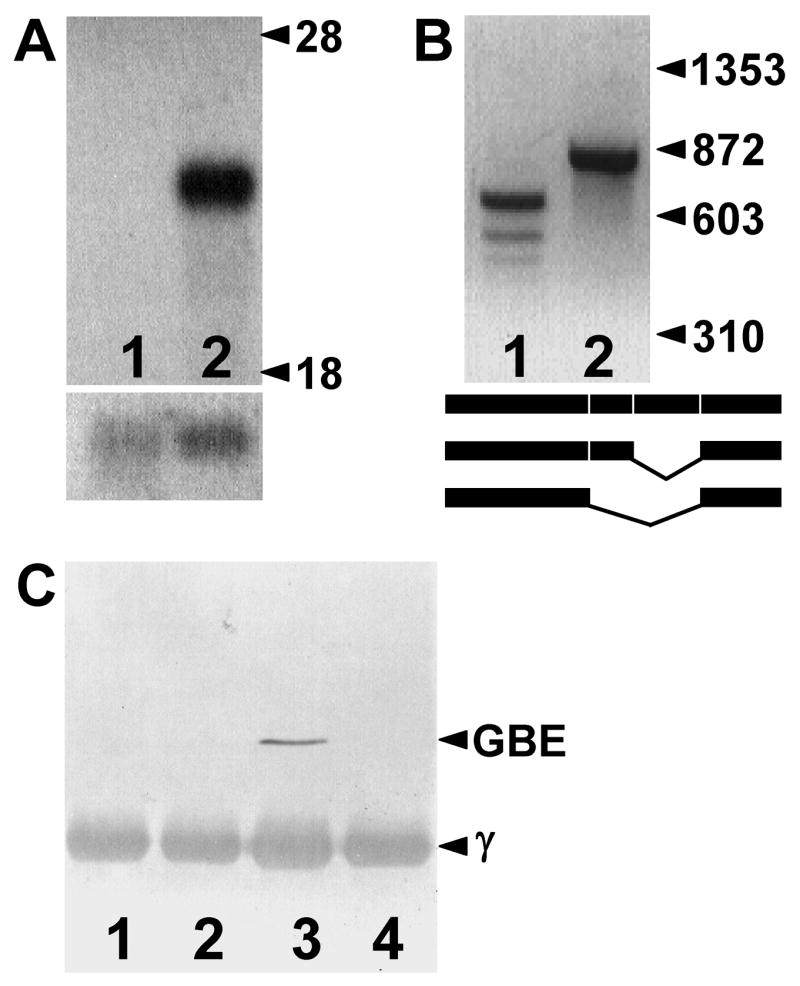
GBE expression. Panel A shows a northern blot of polyA-selected liver RNA of a GSD IV affected (lane 1) and a normal (lane 2) cat that was sequentially hybridized to feline GBE1 (upper blot) and ACTB (β-actin; lower blot) cDNA probes. Migration of the 28S and 18S ribosomal RNAs are indicated. Panel B shows RT-PCR products amplified from affected (lane 1) and normal (lane 2) cat liver RNA using PCR primers in GBE1 exons 8 and 14. Migration of φX174 Hae III fragments is indicated on the right. Lane 2 shows the expected product of 848 bp. Sequence of the two larger products from the affected cat demonstrated they were aberrantly spliced and missing exon 12 and exons 11 and 12, respectively, as shown in the schematic. Panel C shows a western blot of GSD IV affected (lanes 1 and 2) and normal (lanes 3 and 4) cat liver proteins immunoprecipitated using goat anti-rabbit liver GBE (lanes 1 and 3) or non-immune (lanes 2 and 4) serum. Proteins on the blot were detected by reaction with the anti-rabbit liver GBE serum. The IgG heavy chain (γ) migrating at ~ 50 kDa in all lanes was detected by reaction with the secondary antibody- (anti-goat IgG) alkaline phosphatase conjugate and provided an internal loading control.
Genomic analysis of the mutant allele
The data presented above suggested that part or all of exon 12 was missing and/or that one or more of the flanking consensus splice site recognition sequences was altered in GSD IV affected cats. Southern blot analysis comparing hybridization of the full-length feline GBE1 cDNA to restriction endonuclease digests of genomic DNA from normal and affected cats demonstrated multiple restriction fragment length differences, suggesting a sizable deletion in the affected cat GBE1 gene (not shown). To determine the extent of the deletion, an additional Southern blot was hybridized sequentially first to a cDNA probe of exons 11–14 and then to a probe of exon 12 only (figure 4A). These data demonstrated that the putative deletion excluded only exon 12 of the cDNA and that exon 12 is encoded within ~ 3.6 kb Hind III and ~ 5.1 kb Eco RI fragments of genomic DNA in normal cats.
Figure 4.

Genomic analysis of the GBE1 locus and affected cat mutation. Panel A shows a Southern blot of normal (lanes 1 and 3) and affected cat DNA (lanes 2 and 4) digested with Hind III (lanes 1 and 2) or Eco RI (lanes 3 and 4) hybridized to a GBE1 cDNA probe of exons 11–14 (upper) and thereafter to a probe of only exon 12 (lower). Migration of λ Hind III fragments is indicated on the right. Exons 11–14 of feline GBE1 cDNA have no Eco RI or Hind III recognition sites. Panel B shows PCR products amplified from genomic templates of a normal (lanes 1 and 2) and a GSD IV affected cat (lane 3). The products were variously amplified with primers in exons 11 and 12 (lane 1), in exon 12 and at a site in intron 12 that is 613 bp 5’ of exon 13 (lane 2), and in exon 11 and the same intron 12 site as in lane 2 (lane 3). Migration of λ Hind III/Eco RI double-digest fragments is indicated on the right. Panel C is a schematic indicating the normal cat exon-intron arrangement deduced from the size of products in panel B, sequencing the products in lanes 1 and 3 of panel B, and sequencing a genomic clone containing part of intron 12 through part of intron 14. The dashed portion of intron 12 was not sequenced. Exons (e11–e13) and Eco RI (R) and Hind III (H) sites are indicated. Sequence of the affected cat product in lane 3 of panel B revealed a deletion (dotted line) of ~ 6.2 kb, as indicated at the bottom.
To obtain the genomic sequence surrounding exon 12, intron 11 was amplified by PCR from normal cat DNA using primers in exons 11 and 12 (figure 4B), and 1591 bp of the most 3’portion of intron 12 was obtained as part of a 4 kb Eco RI fragment isolated from a feline genomic library. Both were sequenced (GenBank accession no. DQ979412), and PCR primers were chosen to amplify across the uncloned portion of normal cat intron 12, thus establishing the normal genomic organization of the region (figure 4C). Finding that intron 12 is ~ 5.3 kb in the cat suggests that feline GBE1 may be more compact overall than in the dog and human genomes, where the same intron is 38 kb and 40 kb, respectively.
An ~ 2.6 kb PCR product (figure 4B) obtained using primers chosen to amplify across the putative deletion in affected cat DNA, from exon 11 to the 3’ portion of intron 12, was sequenced to determine the rearrangement breakpoints. The 5’ breakpoint was in an AT-rich region 1552 bp 3’ of exon 11, where the identity of the affected cat sequence diverged from the normal sequence by deletion of ~ 6.2 kb, extending into intron 12, and insertion of 334 bp (figure 5). The 101 bp at the 3’ end of the inserted sequence was a near-perfect inverted duplication of the 97 bp of normal sequence immediately 3’ of the 3’ breakpoint. The long palindrome thus formed predicted formation of a highly stable stem loop (ΔG ~ 105 kcal/mol; equivalent melting temperature of 93°C) [22, 23] in the DNA of the rearranged allele.
Figure 5.

Sequence of the GBE1 rearrangement breakpoints. The PCR product shown in lane 3, panel B of figure 4 was sequenced. Sequence identical to that of the normal cat is in upper case, and the insertion is in lower case. The 2 portions of an inverted, near-perfect duplication of a partial feline SINE are underlined by solid and dotted arrows. The sequence underlined by dashes and the contiguous SINE sequence underlined by the solid arrow is identical to the sequence of a Felis catus genome sequence trace that is orthologous to sequence of the assembled dog genome sequence ~ 17 Mb 3’ of GBE1 exon 12.
The ~100 bp of inverted repeat sequence at the 3’ rearrangement breakpoint of the deleted allele is the split-block RNA polymerase III promoter sequence of a short interspersed element (SINE) found in many copies throughout the domestic and related cat genomes [24], as well as in dog, ferret, and spotted hyena genomes, as determined by BLASTn search [25] of the nonredundant nucleotide (nr) database in GenBank. While the full-length (~230 bp) SINE was present in the normal cat sequence of intron 12 at the 3’ breakpoint, the only SINE in intron 11 of the normal cat sequence was 1.5 kb 3’ of the 5’ rearrangement breakpoint. In investigating the origin of the inserted sequence, the 5’-most 31 bp had no identity to any sequence in the Felis catus WGS trace database queried by discontiguous MegaBLAST [26], nor was there significant homology to any other sequence found in GenBank by BLASTn search. Using the next 5’-most 200 bp of inserted sequence, not extending into the SINE sequence portion, in a discontiguous MegaBLAST search of the nr sequence database, we found ~50 bp of homology to intergenic sequences on several human chromosomes that appeared to be diverged remnants of the endogenous retrovirus element ERV1-B1 (GenBank accession no. AC013829). We also found identity of this 200 bp portion to a sequence trace in the Felis catus WGS database that we subsequently used to nucleate an iterative search of the same database, each time eliminating SINE sequences from the query of the subsequent search. The resulting 5.56 kb contig contained 6 SINEs, including 2 oriented oppositely to the SINE in the sequence inserted in the rearranged allele of GBE1. We used the entire contig in a BLAT search of the dog genome and found homology along its full length, except in the SINEs, to an intergenic site (CFA31: 25,712,697–25,717,744) on the chromosome harboring GBE1 (CFA31: 8,747,146–9,013,108), but at a distance 3’ of exon 12 of ~17 Mb.
Allele-specific carrier testing in NFC
An allele-specific mutation detection assay was devised using a common forward PCR primer in the normal intron 12 sequence just 5’ of the rearrangement breakpoint and allele-specific reverse primers in the normal or affected cat (inserted) sequence 3’ of the 5’ rearrangement breakpoint. The rearranged allele detected in this manner segregated perfectly with the disease allele in the originally reported family of NFC [11, 12] as well as among founders of the breeding colony (figure 6). Altogether we tested 19 affected cats, 14 obligate carriers, including both parents of the cat reported by Coates et al. [12] and the 3 normal cats that were shown highly unlikely to be carriers in breeding experiments. We also tested 60 unrelated normal cats of different breeds and did not observe the rearranged allele, thus indicating that the inserted sequence was not a polymorphism of cats generally that would be found in NFC without the disease-causing deletion. Among 402 privately owned NFC, we found 4 affected, 58 carrier, and 341 genetically normal cats, suggesting a mutant allele frequency of 0.082, albeit in a biased sample. Two of the carriers had been imported to the USA independently from different catteries in Europe.
Figure 6.

GSD IV carrier detection in NFC. The result of GBE1 allele-specific PCR amplification for each cat on the gel below is aligned with the cat’s symbol in the pedigree above (same conventions as in figure 1). PCR products of the normal (n) and disease (a) alleles are indicated. The cats labeled A, B, and C are the same cats so labeled in figure 1.
Discussion
In humans, the age of onset, organ distribution, and clinical course of GSD IV is highly variable, ranging from mild nonprogressive hepatopathy to fetal neuromuscular dysfunction and immediate-postnatal cardiopulmonary collapse [4]. This clinical heterogeneity has been attributed to differing GBE1 mutations that, in turn, are presumed to provide varied amounts of residual enzyme activity, albeit the available assays for GBE activity measurement are imprecise at lower activities and vary between tissues screened. Compared to other enzymes, assay of GBE activity is particularly troublesome because it is an indirect assay, and the substrate and product are both heterogeneous polymers. Although alternatively spliced mRNAs were observed in affected cat liver by RT-PCR, no GBE1 mRNA or protein of any size was seen on northern and western blots. This is consistent with nonsense-mediated decay of the abnormally spliced RNAs because they were each predicted to harbor premature stop codons in exon 13 [27]. Hence, the GBE1 mutation in NFC creates a null allele, and the so-called residual GBE activity (0.8–3.8 % of normal) observed in assays of affected cat tissues [11] was likely due to inaccuracies inherent in the enzyme activity assay, including non-enzymatic in vitro activities. We believe this to be true also of residual activities reported in a number of human patients. A more routine difficulty with enzyme assays was also illustrated in this study when 2 cats were misidentified as GSD IV carriers. As is true for many inborn errors of metabolism [28], the range of enzyme activities observed in GSD IV heterozygous animals overlaps the normal range, especially when assays are conducted in leukocyte or fibroblast preparations rather than in the most affected tissues. The very low probability, determined from breeding experiments, that either of these 2 cats was truly a carrier correlated with DNA tests demonstrating that neither had the rearranged disease allele.
In humans, 22 GSD IV-causing mutations have been identified [29] (Human Gene Mutation Database [http://www.hgmd.cf.ac.uk/ac/index.php] accessed Dec., 2006) and many patients are heteroallelic, which may well explain some of the clinical heterogeneity, but the association between mutation and enzyme function is not yet clear. In contrast, affected animals of the same breed are generally homozygous for the same mutation, so the phenotypic variation should be less pronounced. Indeed the phenotypic spectrum of GSD IV in quarter horses ranges only slightly, from fetal to neonatal illness and death by a few weeks of age due to severe hypoglycemia [8–10]. Hence, it was surprising to observe two very different GSD IV presentations, perinatal death and juvenile-onset progressive neuromuscular disease, in NFC despite their having the same GBE1 genotype, thus illustrating that other genes or nongenetic factors can modify the “typical” phenotype of single gene disorders such as GSD IV.
In this study, stillbirth or death in the immediate postnatal period was recognized as the most common presentation of cats with GSD IV. As in humans, the perinatal period is very critical with respect to intermediary metabolism and the ability to maintain glucose homeostasis in vital organs. During the birth process and until a newborn kitten nurses, substrates for gluconeogenesis are limited, and the kitten is dependent on efficient degradation of tissue glycogen stores. Finding of abundant dark blue staining glycogen deposits in muscle of affected kittens that had breathed and, therefore, had lived for some period suggests that the abnormal (amylopectin-like) glycogen stored in tissue in the absence of GBE activity is not efficiently degraded when needed. Thus, perinatal death in the affected kittens was most likely due to profound hypoglycemia with ensuing cardiac failure, failure of the CNS respiratory center, and/or neurogenic coma rather than intracellular accumulation of the insoluble glucan polymer, similar to what is suspected in equine GSD IV and the human neonatal form.
Affected cats that survived the critical immediate postnatal period, 2 in the colony described here with short-term glucose supplementation, and 4 previously reported cats [11, 12] that were not treated, showed no obvious clinical signs until ~ 5 months of age, a time in a cat’s life past the most rapid growth phase and near the onset of puberty. The first clinical sign in affected cats examined prospectively was persistent hyperthermia, beginning well before signs of progressive neuromuscular disease. The pathogenesis of the hyperthermia is not known, but its onset before chronic involuntary muscle activity suggests that proinflammatory insults due to accumulation of abnormal glycogen and/or ensuing cell dysfunction or necrosis may have contributed. The rapid progression of neuromuscular pathologies observed here and previously in juvenile GSD IV cats occurred while they were normoglycemic, undoubtedly due to the high capacity for gluconeogenesis from amino acid and fatty acid precursors of more mature cats [30], and therefore, were most likely due to the intracellular accumulation of aberrant insoluble glucans.
These clinicopathologic studies in kittens with GSD IV may have implications for humans with this disorder. First, an GSD IV infant who could otherwise succumb to dysregulation of glycogen utilization and hypoglycemia in the perinatal period will likely survive to an age when other treatments, such as enzyme replacement, liver transplantation or gene therapy, could be instituted if hypoglycemia is ascertained quickly and treated aggressively. We do not, however, believe that the perinatal hypoglycemia phenotype in the kittens is strictly homologous to the reported fetal or neonatal onset forms of GSD IV of humans in which there may be arthrogryposis due to fetal akinesis or cardiopulmonary collapse and severe muscle hypotonia at birth because when present in these patients, hypoglycemia has been recognized and corrected but did not produce the dramatic clinical benefit observed in the kittens.
Secondly, if the progressive hepatic and later-onset neuromuscular forms of GSD IV are indeed due to cytotoxic or proinflammatory effects of accumulating aberrant and insoluble glucans, some of the phenotypic variation in human GSD IV may be due to environmental and/or genetic factors that modulate other components of glucose/glycogen homeostasis, the activity of glycogen synthase (GS; EC 2.4.1.11) for instance. Control of glycogen structure is a balance between the activities of GS and GBE [31], as was more recently illustrated in vivo when GS overexpression in a mouse line created GSD IV-like lesions [32]. Since GS of brain, muscle, and liver are different gene products, and all have highly regulated and rate-limiting activities in glycogen synthesis [1], the same GBE1 mutation on different GS genetic backgrounds may well create differing tissue-specific pathology and clinical signs. By whatever means, effective definitive therapy for GSD IV will most likely need to be delivered directly to the majority of cells in all vital organs and must abrogate abnormal glycogen storage.
The GBE1mutation causing GSD IV in NFC is a unique complex genomic rearrangement, a class of mutation not previously described in human patients. It was created by a moderately sized insertion, including sequences from at least 2 distant origins, and a large deletion that excised exon 12 and portions of the flanking introns. Complex rearrangements are an uncommon group of disease-causing mutations, constituting only 0.7 % of all mutations in the Human Gene Mutation Database, and the mechanisms by which they occur are seldom obvious. Insertions and deletions mediated by SINE retrotransposition (e.g. Alu sequences in humans) occur in many species, and a number of human and canine genetic disorders have been so caused [33–37]. Additionally, several genetic disease-causing deletions caused by unequal homologous recombination between Alu sequences have been reported [36]. Although 10% of L1 endonuclease-mediated retrotransposition events in humans also cause deletions [34], some of which are of the magnitude observed in the feline GSD IV mutation, the additional DNA inserted immediately 5’ of the inserted SINE sequence at the mutation site argues against recent SINE retrotransposition as the mechanism generating this mutation. Transduction of a long stretch of DNA 5’ of a SINE has not been observed during Alu insertion. What may have happened, however, is a segmental duplication, as are associated with flanking Alu sequences in humans [38], caused by replication slippage [39] that newly placed a SINE with some 5’ DNA in intron 11, followed by a deletion mediated by unequal homologous recombination between SINEs. An additional mechanism such as the postulated capture of preformed DNA oligonucleotides during double stranded DNA breaks [39] might have to be invoked to explain the most-5’ 31bp of the insertion. Alternatively, a model of intrachromosomal serial replication slippage in trans has been put forth recently [40]. It is consistent with many of the disease-causing complex rearrangements catalogued in HGMD and is less cumbersome than other proposed mutational mechanisms for examples of this class of lesion. The feline mutation described here may be compatible with that model, but we must look forward to the scheduled increased sequence coverage and assembly of the feline genome for help to determine the origin(s) of the insertion sequences.
For breeders of NFC, clinicopathologic and molecular characterization of GSD IV and development of a DNA-base screening test for the mutant allele has been beneficial. For some time, GSD IV remained an occult disease among the population of purebred NFC, even when several carriers were active in breeding programs, assuredly due to the loss of most affected kittens as stillborn or neonatal deaths that were not further examined. While few affected NFC were ascertained by DNA testing, the frequency of carrier animals, and hence the mutant allele frequency, was high in the studied, albeit biased, NFC population. Genetic diversity of a small genetically isolated population, as is the NFC breed, can be a concern when selecting against a specific disease, but the available DNA test allows owners to select solely against the disease allele, while maintaining desirable attributes of their animals. We expect that GSD IV will be relegated to the archives of NFC history.
As a large animal (larger than mouse) model of human genetic disease and yet easily maintained in research colonies, various studies of pathophysiology that are directly relevant to human medicine can be applied to GSD IV cats, as was done in this study and in previous investigations of canine GSD Ia and VII [41, 42]. GSD IV cats can also serve uniquely as subjects in preclinical studies to evaluate innovative treatment modalities of practical utility in human patients, particularly those that address the need for global tissue delivery of GBE activity.
Acknowledgments
This work was supported by the NIH Referral Center – Animal Models of Human Genetic Disease (RR02512) at the University of Pennsylvania School of Veterinary Medicine and the Genetics Research Fund of the Michigan State University College of Veterinary Medicine. We thank Adam Seng and Polly Foureman, DVM, for DNA testing of privately owned cats, and Barry Prior, currently of the Department of Veterinary Biomedical Sciences, University of Missouri, Columbia, for the muscle nuclear magnetic resonance spectroscopy studies. We especially thank Allen M. Gold, Ph.D., Columbia University Health Sciences, New York, for the generous gift of anti-GBE serum. New sequences reported here have been deposited in GenBank (accession nos. AY439007 and DQ979412)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen Y-T. Glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. 8. McGraw Hill; New York: 2001. pp. 1521–1551. [Google Scholar]
- 2.Andersen DH. Familial cirrhosis of the liver with storage of abnormal glycogen. Lab Invest. 1956;5:11–20. [PubMed] [Google Scholar]
- 3.Brown BI, Brown DH. Lack of an alpha-1,4-glucan: alpha-1,4-glucan 6-glycosyl transferase in a case of type IV glycogenosis. Proc Nat Acad Sci, USA. 1966;56:725–729. doi: 10.1073/pnas.56.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2:177–188. doi: 10.2174/1566524024605815. [DOI] [PubMed] [Google Scholar]
- 5.Bao Y, Kishnani P, Wu JY, Chen YT. Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest. 1996;97:941–948. doi: 10.1172/JCI118517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruno C, van Diggelen OP, Cassandrini D, Gimpelev M, Giuffre B, Donati MA, Introvini P, Alegria A, Assereto S, Morandi L, Mora M, Tonoli E, Mascell S, Traverso M, Pasquini E, Bado M, Vilarinho L, van Noort G, Mosca F, DiMauro S, Zara F, Minetti C. Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV) Neurology. 2004;63:1053–1058. doi: 10.1212/01.wnl.0000138429.11433.0d. [DOI] [PubMed] [Google Scholar]
- 7.Tay SKH, Akman HO, Chung WK, Pike MG, Muntoni F, Hays AP, Shanske S, Valberg SJ, Mickelson JR, Tanji K, DiMauro S. Fatal infantile neuromuscular presentation of glycogen storage disease type IV. Neuromusc Disord. 2004;14:253–260. doi: 10.1016/j.nmd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Render JA, Common RS, Kennedy FA, Jones MZ, Fyfe JC. Amylopectinosis in fetal and neonatal quarter horses. Vet Pathol. 1999;36:157–160. doi: 10.1354/vp.36-2-157. [DOI] [PubMed] [Google Scholar]
- 9.Valberg SJ, Ward TL, Rush B, Kinde H, Hiraragi H, Nahey D, Fyfe J, Mickelson JR. Glycogen branching enzyme deficiency in quarter horse foals. J Vet Intern Med. 2001;15:572–580. doi: 10.1892/0891-6640(2001)015<0572:gbediq>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Ward TL, Valberg SJ, Adelson DL, Abbey CA, Binns MM, Mickelson JR. Glycogen branching enzyme (GBE1) mutation causing equine glycogen storage disease IV. Mamm Genome. 2004;15:570–577. doi: 10.1007/s00335-004-2369-1. [DOI] [PubMed] [Google Scholar]
- 11.Fyfe JC, Giger U, Van Winkle TJ, Haskins ME, Steinberg SA, Wang P, Patterson DF. Glycogen storage disease type IV: inherited deficiency of branching enzyme activity in cats. Pediatr Res. 1992;32:719–725. doi: 10.1203/00006450-199212000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Coates JR, Paxton R, Cox NR, Braund KG, Steiss JE, Baker HJ, Simpson ST. A case presentation and discussion of type IV glycogen storage disease in a Norwegian forest cat. Prog Vet Neurol. 1996;7:5–11. [Google Scholar]
- 13.Henthorn PS, Somberg RL, Fimiani VM, Puck JM, Patterson DF, Felsburg PJ. IL-2Rγ gene microdeletion demonstrates that canine X-linked severe combined immunodeficiency is a homologue of the human disease. Genomics. 1994;23:69–74. doi: 10.1006/geno.1994.1460. [DOI] [PubMed] [Google Scholar]
- 14.Jackson CE, Yuhki N, Desnick RJ, Haskins ME, O’Brien SJ, Schuchman EH. Feline arylsulfatase B (ARSB): Isolation and expression of the cDNA, comparison to human ARSB, and gene localization to feline chromosome A1. Genomics. 1992;14:403–411. doi: 10.1016/s0888-7543(05)80233-2. [DOI] [PubMed] [Google Scholar]
- 15.Menotti-Raymond M, David VA, Roelke ME, Chen ZQ, Menotti KA, Sun S, Schäffer AA, Tomlin JF, Agarwala R, O'Brien SJ, Murphy WJ. Second-generation integrated genetic linkage/radiation hybrid maps of the domestic cat (Felis catus) J Hered. 2003;94:95–106. doi: 10.1093/jhered/esg008. [DOI] [PubMed] [Google Scholar]
- 16.Fyfe JC, Kurzhals RL, Lassaline ME, Henthorn PS, Alur PRK, Wang P, Wolfe JH, Giger U, Haskins ME, Patterson DF, Sun H, Jain S, Yuhki N. Molecular basis of feline β-glucuronidase deficiency: An animal model of mucopolysaccharidosis VII. Genomics. 1999;58:121–128. doi: 10.1006/geno.1999.5825. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. pp. 7.31–7.44. [Google Scholar]
- 18.Kawasaki ES. Sample preparation from blood, cells, and other fluids. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, editors. PCR Protocols: a Guide to Methods and Applications. Academic Press; San Diego, CA: 1990. pp. 146–152. [Google Scholar]
- 19.Argov Z, Lofberg M, Arnold DL. Insights into muscle diseases gained by phosphorus magnetic resonance spectroscopy. Muscle & Nerve. 2000;23:1316–1334. doi: 10.1002/1097-4598(200009)23:9<1316::aid-mus2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 20.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent WJ. BLAT - The BLAST-Like Alignment Tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Markham NR, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slattery JP, Murphy WJ, O'Brien SJ. Patterns of diversity among SINE elements isolated from three Y-chromosome genes in carnivores. Mol Biol Evol. 2000;17:825–829. doi: 10.1093/oxfordjournals.molbev.a026361. [DOI] [PubMed] [Google Scholar]
- 25.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Schwartz S, Wagner L, Miller W. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- 27.Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- 28.Harris H. The Principles of Human Biochemical Genetics. 3. Elsevier/North-Holland Biomedical Press; New York: 1980. pp. 247–256. [Google Scholar]
- 29.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 30.Kettlehut IC, Foss MC, Migliorini RH. Glucose homeostasis in a carnivore (cat) and in rats feds a high-protein diet. Am J Physiol. 1980;239:R437–R444. doi: 10.1152/ajpregu.1980.239.5.R437. [DOI] [PubMed] [Google Scholar]
- 31.Smith EE. Enzymic control of glycogen structure. In: Whelan WJ, editor. Control of Glycogen Metabolism. Academic Press; New York: 1968. pp. 203–213. [Google Scholar]
- 32.Raben N, Danon M, Lu N, Lee E, Shliselfeld L, Skurat AV, Roach PJ, Lawrence JC, Jr, Musumeci O, Shanske S, DiMauro S, Plotz P. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739–1745. doi: 10.1212/wnl.56.12.1739. [DOI] [PubMed] [Google Scholar]
- 33.Callinan PA, Wang J, Herke SW, Garber RK, Liang P, Batzer MA. Alu retrotransposition-mediated deletion. J Mol Biol. 2005;348:791–800. doi: 10.1016/j.jmb.2005.02.043. [DOI] [PubMed] [Google Scholar]
- 34.Chen JM, Stenson PD, Cooper DN, Férec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- 35.Clark LA, Wahl JM, Rees CA, Murphy KE. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci USA. 2006;103:1376–1381. doi: 10.1073/pnas.0506940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 37.Pele M, Tiret L, Kessler JL, Blot S, Panthier JJ. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum Mol Genet. 2005;14:1417–1427. doi: 10.1093/hmg/ddi151. [DOI] [PubMed] [Google Scholar]
- 38.Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Meta-analysis of gross insertions causing human genetic disease: Novel mutation mechanisms and the role of replication slippage. Hum Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- 40.Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Intrachromosomal serial replication slippage in trans give rise to diverse genomic rearrangements involving inversions. Hum Mutat. 2005;25:362–373. doi: 10.1002/humu.20230. [DOI] [PubMed] [Google Scholar]
- 41.Beaty RM, Jackson M, Peterson D, Bird A, Brown T, Benjamin DK, Jr, Juopperi T, Kishnani P, Boney A, Chen Y-T, Koeberl DD. Delivery of glucose-6-phosphatase in a canine model for glycogen storage disease, type Ia, with adeno-associated virus (AAV) vectors. Gene Ther. 2002;9:1015–1022. doi: 10.1038/sj.gt.3301728. [DOI] [PubMed] [Google Scholar]
- 42.Giger U, Argov Z, Schnall M, Bank W, Chance B. Metabolic myopathy in canine muscle-type phosphofructokinase deficiency studied by P-NMR. Muscle & Nerve. 1988;11:1260–1265. doi: 10.1002/mus.880111210. [DOI] [PubMed] [Google Scholar]