

Abstract
The regulation of molecular motors is an important cellular problem, as motility in the absence of cargo results in futile adenosine triphosphate hydrolysis. When not transporting cargo, the microtubule (MT)-based motor Kinesin-1 is kept inactive as a result of a folded conformation that allows autoinhibition of the N-terminal motor by the C-terminal tail. The simplest model of Kinesin-1 activation posits that cargo binding to nonmotor regions relieves autoinhibition. In this study, we show that binding of the c-Jun N-terminal kinase–interacting protein 1 (JIP1) cargo protein is not sufficient to activate Kinesin-1. Because two regions of the Kinesin-1 tail are required for autoinhibition, we searched for a second molecule that contributes to activation of the motor. We identified fasciculation and elongation protein ζ1 (FEZ1) as a binding partner of kinesin heavy chain. We show that binding of JIP1 and FEZ1 to Kinesin-1 is sufficient to activate the motor for MT binding and motility. These results provide the first demonstration of the activation of a MT-based motor by cellular binding partners.
Introduction
Long-distance intracellular trafficking is driven by kinesin and dynein motors that carry cargoes along microtubule (MT) tracks. Steady advances have been made in our understanding of the structure and mechanics of motor proteins (Schliwa and Woehlke, 2003; Mallik and Gross, 2004). However, less is known about how motor proteins bind to the proper cargo, become activated for transport, and deliver that cargo to the correct cellular locale.
Several aspects of motor protein function are likely to be regulated in cells, most notably regulation of the motor–cargo and motor–MT interactions (Guzik and Goldstein, 2004; Mallik and Gross, 2004). Recent work has begun to elucidate how motors attach to the appropriate cargoes (for review see Verhey and Rapoport, 2001; Hirokawa and Takemura, 2005). A general picture is emerging whereby kinesin family members use adaptor/scaffolding proteins to link to their cargoes, although examples of direct interactions with transmembrane proteins exist (for review see Hirokawa and Takemura, 2005). In the case of Kinesin-1 (formerly conventional kinesin or Kif5), the kinesin light chain (KLC) subunit binds directly to JNK-interacting protein 1 (JIP1), JIP2, and JIP3/Syd. As scaffolding proteins, the JIP proteins function to organize JNK signaling as well as to link Kinesin-1 to vesicular cargoes (for review see Verhey and Rapoport, 2001; Hirokawa and Takemura, 2005).
For regulation of the motor–MT interaction, most of our understanding comes from work on Kinesin-1 (Verhey and Rapoport, 2001). In the absence of cargo, Kinesin-1 is thought to be inactive as a result of a folded conformation that enables autoinhibition of the N-terminal motor domain by C-terminal tail domains. Autoinhibition leads to a simple prediction for how Kinesin-1 is activated: cargo binding to the Kinesin-1 tail frees the motor domains for ATP-driven motility. Although recombinant kinesin heavy chain (KHC) constructs can be activated in vitro by binding artificial cargoes such as glass slides or beads (Jiang and Sheetz, 1995; Coy et al., 1999), activation by cellular binding partners remains to be demonstrated. Alternatively, cargo binding may not be sufficient to activate Kinesin-1, and subsequent events may be required (Verhey and Rapoport, 2001).
We set out to test these models for Kinesin-1 activation. We show that binding of the JIP1 cargo protein to Kinesin-1 is not sufficient for activation. Thus, secondary mechanisms must contribute to regulation of the motor–MT interaction. We identify fasciculation and elongation protein ζ (FEZ) as a binding partner for the KHC tail. We then show that JIP1 and FEZ1 cooperate to activate Kinesin-1 for MT binding and motility.
Results and discussion
Binding of the JIP1 cargo protein is not sufficient to activate Kinesin-1
To test the model that cargo binding activates the motor, we investigated whether the binding of JIP1 activates Kinesin-1. Coexpression of myc-KHC + HA-KLC in mammalian cells (Fig. 1 A) results in a complete Kinesin-1 holoenzyme that can be immunoprecipitated with antibodies to the myc (Fig. 1 B, lane 5) or HA tags (Verhey et al., 1998). When coexpressed, Flag-JIP1 bound to the inactive Kinesin-1 molecule (myc-KHC + HA-KLC), as shown by coimmunoprecipitation with antibodies to the myc or Flag tags (Fig. 1 B, lanes 11 and 12). No proteins were precipitated in the absence of specific antibodies (Fig. 1 B, lanes 1, 4, 7, and 10).
Figure 1.
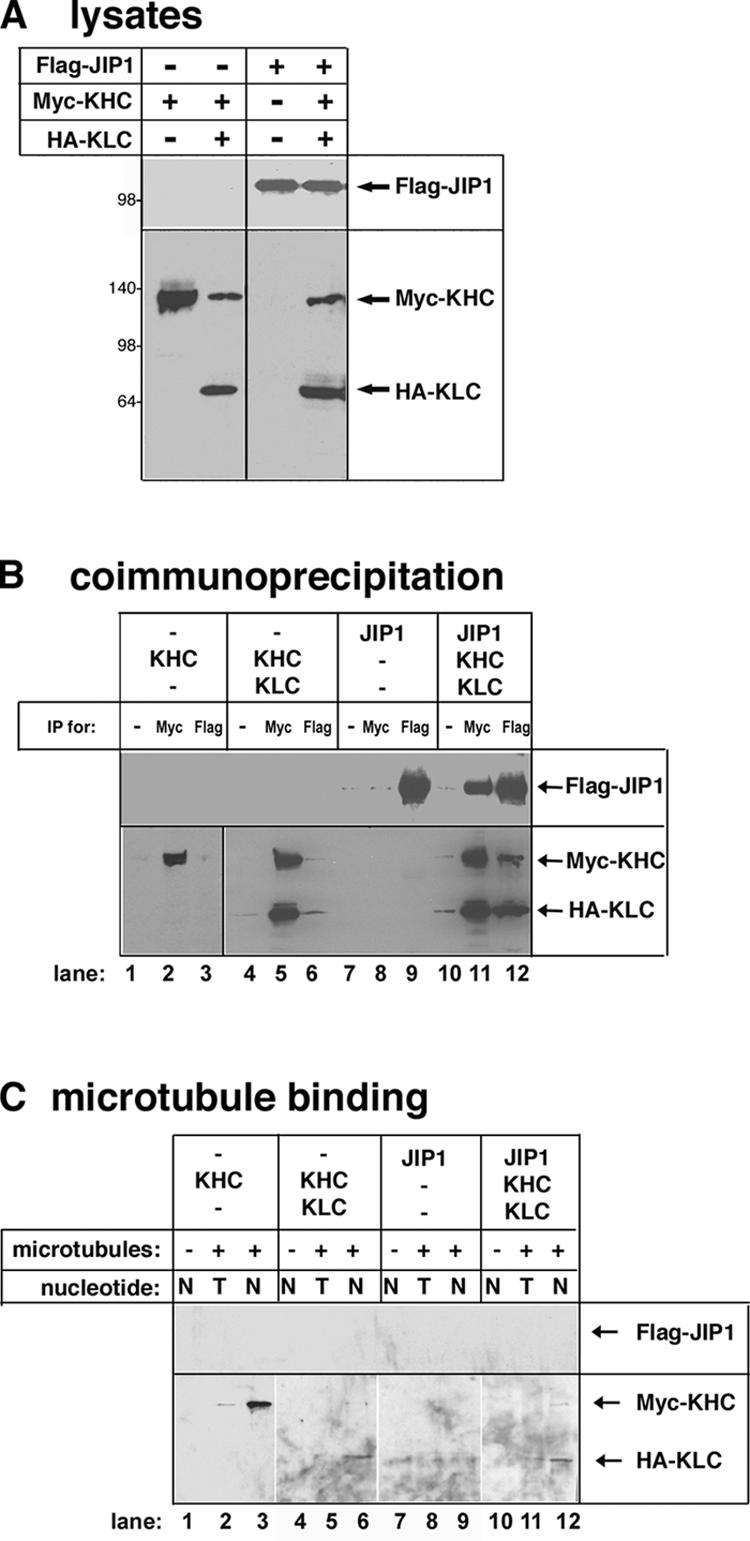
Binding of JIP1 to Kinesin-1 is not sufficient for activation. (A) COS cell lysates expressing the indicated proteins were immunoblotted with antibodies to the Flag (top) or myc and HA tags (bottom). (B) The indicated lysates were immunoprecipitated (IP) with antibodies to the myc or Flag tags or no antibody as a control (−). Precipitates were immunoblotted with antibodies to the Flag tag (top) or myc and HA tags (bottom). (C) Prepolymerized taxol-stabilized MTs were added (+) or not added (−) to the indicated lysates with either ATP (T) or AMPPNP (N). After sedimentation, the MT pellets were immunoblotted with antibodies to the Flag (top) or myc and HA tags (bottom). Panels are from different parts of the same gel.
We then used a MT-binding assay to test whether the binding of Flag-JIP1 to Kinesin-1 activates the motor. Myc-KHC expressed alone is not autoinhibited and can be cosedimented with MTs in the presence of 5′-adenylylimidodiphosphate (AMPPNP), a nonhydrolyzable analogue of ATP (Fig. 1 C, lane 3). Coexpression of myc-KHC + HA-KLC recreates the autoinhibited Kinesin-1 holoenzyme (Fig. 1 C, lane 6; Verhey et al., 1998). Interaction with Flag-JIP1 was not sufficient for activation, as the myc-KHC–HA-KLC–Flag-JIP1 complex did not cosediment with MTs (Fig. 1 C, lane 12). Control experiments demonstrated that the sedimentation of KHC is dependent on the presence of MTs (Fig. 1 C, lanes 1, 4, 7, and 10) and that the hydrolysis of ATP allows the motor to release from the MT (Fig. 1 C, lanes 2, 5, 8, and 11).
These results suggest that an additional event is required to activate Kinesin-1. One possibility is that phosphorylation or some other posttranslational modification is involved (Verhey and Rapoport, 2001). However, we have been unable to detect any role for phosphorylation in the activation of Kinesin-1 (unpublished data). As the complete autoinhibition of Kinesin-1 requires both the KHC inhibitory tail and the KLC subunit (Verhey et al., 1998; Adio et al., 2006), we hypothesized that the autoinhibitory effects of both of these regions must be relieved for activation.
FEZ1/UNC-76 binds to the inhibitory tail of KHC
To identify potential cargoes and/or regulators of the KHC tail, we performed a two-hybrid screen of a human brain library using the stalk/tail regions of rat KHC (KHC(750–955); Fig. 2) as a bait. 27 of the positive clones contained sequences encoding either FEZ1 and FEZ2 (Fig. 2 A), which is consistent with an interaction between the Drosophila melanogaster FEZ1 homologue, UNC-76, and Drosophila KHC (Gindhart et al., 2003). The interaction between mammalian FEZ1 and KHC was verified in vitro (Fig. S1 B, available at http://www.jcb.org/cgi/content/full/jcb.200605099/DC1). FEZ/UNC-76 likely plays an important if ill-defined role in axonal transport. First, the loss of Unc-76 function results in defects in locomotion and axon outgrowth that are similar to Kinesin-1 mutants (Bloom and Horvitz, 1997; Gindhart et al., 2003). Second, genetic interactions have been demonstrated between Unc-76 and both Khc and Klc (Gindhart et al., 2003). Third, FEZ1/2 can facilitate neurite outgrowth in cultured cells (Kuroda et al., 1999; Miyoshi et al., 2003; Okumura et al., 2004; Suzuki et al., 2005). Finally, FEZ1 can be cosedimented with MTs (Suzuki et al., 2005).
Figure 2.

FEZ1 binds to the inhibitory tail of KHC. (A) Schematic illustration of the domain structures of FEZ1, KHC, KLC, and JIP1. Interacting regions are indicated with a double arrow. TPR, tetritricopeptide repeat; PTB, phosphotyrosine binding. (B) Sequence alignment of KHC tail domains from Homo sapiens (Hs; GenBank/EMBL/DDBJ accession no. BAA25457), Drosophila (Dm; P17210), Caenorhabditis elegans (Ce; AAA28155), and N. crassa (Nc; P48467). Sequences were aligned using T-Coffee (Notredame et al., 2000), converted to Clustal-W, and modified by ESPript 2.2. Green overline, conserved coiled tail implicated in cargo binding; black overline, globular tail implicated in autoinhibition; dashed underline, folding site; asterisks, IAK sites; white text highlighted in red, identical residues; red text on white background, conserved residues. (C) Ability of truncated or mutant versions of KHC(750–955) to interact with FEZ1.
Two important domains have been identified within the KHC tail: the highly conserved coiled tail and the inhibitory globular tail (Fig. 2 B). Within the KHC inhibitory tail, the folding site interacts with the KHC motor/neck regions to generate the folded conformation, thus positioning the IAK site for inhibition of MT-stimulated ADP release (Fig. 2 B; Cross and Scholey, 1999; Stock et al., 1999; Hackney and Stock, 2000; Seiler et al., 2000; Bathe et al., 2005; Yonekura et al., 2006). To determine which of the KHC tail regions are required for interaction with FEZ1, we used a directed two- hybrid assay. Truncation of the KHC inhibitory tail or mutation of the folding site in the inhibitory tail abolished the interaction with FEZ1 (Fig. 2 C). Interestingly, mutation of the C-terminal residues of KHC, which are critical for the proper functioning of Neurospora crassa KHC (Seiler et al., 2000), also abolished the interaction with FEZ1 (Fig. 2 C). Importantly, FEZ is the first protein identified that binds to the inhibitory globular tail of KHC. These data indicate that FEZ1 is not likely to be strictly a cargo of Kinesin-1. This is supported by the diffuse localization of FEZ1 in neuronal cells (Fig. S1 E; Miyoshi et al., 2003) and the lack of UNC-76 accumulation in Khc axonal jams (Gindhart et al., 2003). Collectively, these results suggest that FEZ1 binding to the KHC folding site could play a critical role in Kinesin-1 activation, perhaps by relieving the folded conformation.
FEZ1 and JIP1 cooperate to activate Kinesin-1 in vitro
To test whether FEZ1 binding is sufficient to activate Kinesin-1, COS cell lysates expressing tagged versions of KHC, KLC, and FEZ1 (Fig. 3 A) were used in coimmunoprecipitation and MT-binding experiments. Immunoprecipitation of KHC brought down a complex of myc-KHC, HA-KLC, and FEZ1-hsv (Fig. 3 B), demonstrating that FEZ1 interacts with inactive Kinesin-1. This interaction is not sufficient for the activation of Kinesin-1, as the myc-KHC–HA-KLC–FEZ1-hsv complex did not cosediment with MTs in the presence of AMPPNP (Fig. 3 C).
Figure 3.

FEZ1 and JIP1 cooperate to activate Kinesin-1 in vitro. (A) COS cell lysates expressing the indicated proteins were immunoblotted with antibodies to the Flag tag (top) or to FEZ1 and the myc and HA tags (bottom). (B) The indicated lysates were mixed and immunoprecipitated (IP) with antibodies to KHC or the Flag tag or to no primary antibody as a control (−). Precipitates were immunoblotted with antibodies to the Flag tag (top), FEZ1 (middle), or myc and HA tags (bottom). (C) MT-binding assay as in Fig. 1 C. MT pellets were immunoblotted with antibodies to the Flag tag (top), FEZ1 (middle), or myc and HA tags (bottom). T, ATP; N, AMPPNP. (B and C) Panels are from different parts of the same gel. (D–F) Single-molecule motility assay. (D) Myc-KHC + 3xmCit-KLC lysates were mixed with lysates of mock-transfected cells (left) or cells expressing Flag-JIP1 and FEZ1-hsv (right). Representative motile events along Cy5-labeled MTs are shown in the kymographs (13 frames; 100-ms intervals). Bar, 1.0 μm. (E) Speed histogram. Myc-KHC + 3xmCit-KLC motors activated by JIP1 and FEZ1 move at a mean speed of 0.71 ± 0.31 μm/s (n = 47). Data are pooled from three separate experiments. (F) Single exponential decay fitting (red line) of the run length histogram shows that the same motors move processively for 0.46 ± 0.05 μm/run.
We next tested whether the binding of both FEZ1 and JIP1 to their respective autoinhibitory regions, the KHC inhibitory tail and the KLC subunit, is required to relieve autoinhibition. Inactive Kinesin-1 motors (myc-KHC + HA-KLC) were mixed with tagged versions of both FEZ1 and JIP1 (Fig. 3 A). An interaction between Kinesin-1 and both Flag-JIP1 and FEZ1-hsv was then demonstrated by coimmunoprecipitation (Fig. 3 B) and a protease protection assay (Fig. S1 F). Control experiments demonstrated that no interaction between Flag-JIP1 and FEZ1-hsv could be detected in the absence of Kinesin-1 (unpublished data). Importantly, the interaction of JIP1 and FEZ1 with Kinesin-1 was sufficient to activate Kinesin-1 for MT binding (Fig. 3 C).
To test whether the activation of Kinesin-1 by JIP1 and FEZ1 resulted in MT-based motility, we generated fluorescent protein (FP)–tagged versions of Kinesin-1. Monomeric versions of the FPs enhanced CFP (mCFP) and citrine (mCit; a brighter variant of enhanced YFP; Griesbeck et al., 2001) were used to minimize dimerization artifacts. To increase the FP–Kinesin-1 signal over the cellular autofluorescence and to ensure that observed motility was caused by assembled Kinesin-1 holoenzymes rather than active KHC subunits, the nonmotor KLC subunit was labeled with three tandem copies of mCit (3xmCit-KLC). Extraction of FP–Kinesin-1 from COS cells resulted in the movement of fluorescent spots along MTs in an in vitro motility assay (Fig. 3 D; Cai et al., 2007). When lysates expressing myc-KHC + 3xmCit-KLC were mixed with lysates of mock-transfected cells, very few motile events were observed (Fig. 3 D, left). However, when the same lysates were mixed with lysates expressing Flag-JIP1 and FEZ1-hsv, a dramatic increase in the number of motile events was observed (Fig. 3 D, right). These activated motors moved with a speed (0.71 ± 0.31 μm/s; Fig. 3 E) similar to recombinant KHC motors in the same assay, although the processivity of our mammalian-expressed Kinesin-1 was lower (0.46 ± 0.5 μm/run; Fig. 3 F). These results demonstrate that JIP1 and FEZ1 cooperate to activate Kinesin-1 for MT-based motility.
FEZ1 and JIP1 cooperate to activate Kinesin-1 in live cells
To test whether JIP1 and FEZ1 cooperate to activate Kinesin-1 in vivo, bicistronic plasmids were generated for the coexpression of KHC-mCit, KLC-mECFP, FEZ1, and JIP1 (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200605099/DC1). To reveal the amount of Kinesin-1 that is in an active state in vivo, we developed an assay to trap active FP–Kinesin-1 molecules on MTs by the addition of AMPPNP to live cells transiently permeabilized with streptolysin O (Fig. S3 A). Exposure to AMPPNP resulted in the rapid accumulation of KHC-mCit on MTs (Fig. 4 A and Video 1), whereas KHC-mCit + KLC-mCFP remained cytosolic (Fig. 4 B and Video 2), indicating that the Kinesin-1 holoenzyme is inactive in vivo. Coexpression with either JIP1 or FEZ1 alone was not sufficient to activate Kinesin-1 (Fig. S3 B and Videos 3 and 4). In contrast, coexpression with both JIP1 and FEZ1 resulted in the rapid accumulation of Kinesin-1 on MTs (Fig. 4, C and D; and Video 5). A truncated version of FEZ1 (FEZ1(1–308)) could not activate Kinesin-1 even in the presence of JIP1 (Fig. 4 E). Importantly, both JIP1 and FEZ1 accumulated on MTs with Kinesin-1 upon AMPPNP addition (Fig. 4, C and D). For the first time, these results demonstrate the activation of a MT-based motor by cellular binding partners. Binding of both JIP1 to the KLC subunit and FEZ1 to the KHC tail likely relieves autoinhibition, freeing the motor domains for MT-based motility (Fig. S3 C).
Figure 4.

FEZ1 and JIP1 cooperate to activate Kinesin-1 in live cells. (A–E) COS cells expressing the indicated proteins were exposed to AMPPNP for 10 min and were fixed and stained. Bar, 25 μm. (F) Fluorescence relocation index of Kinesin-1 over time was determined from videos (available at http://www.jcb.org/cgi/content/full/jcb.200605099/DC1) taken during permeabilization and AMPPNP treatment. n values in parentheses indicate the number of videos. Error bars represent SD. #, P > 0.5; **, P < 0.001 compared with KHC + KLC.
Dominant-negative constructs of FEZ1 disrupt Kinesin-1 transport
A role for FEZ1 in the activation of Kinesin-1 suggests that in the absence of FEZ1, Kinesin-1 should remain inactive. Indeed, the loss of UNC-76 function results in defects in axonal transport (Bloom and Horvitz, 1997; Gindhart et al., 2003). To test whether the loss of FEZ1 function specifically disrupts Kinesin-1 transport, we analyzed the targeted delivery of JIP1 to the tips of neurites in differentiated CAD cells. As we have been unable to knock down FEZ1 protein expression by RNAi (not depicted), we used the truncated FEZ1 protein (FEZ1(1–308)) that cannot activate Kinesin-1 (Fig. 4 E). The overexpression of myc-FEZ(1–308) caused a significant decrease (P < 0.001) in JIP1 localization to the tips of neurites (Fig. 5 C) that was similar to the loss of JIP1 transport upon the expression of a Kinesin-1 dominant-negative protein (Verhey et al., 2001). The overexpression of full-length myc-FEZ1 or truncated myc-FEZ1(1–230) had less of an effect (Fig. 5, A and B).
Figure 5.

Dominant-negative FEZ1 disrupts Kinesin-1 transport of JIP1. (A–D) Differentiated CAD cells expressing myc-FEZ1 (A), myc-FEZ1(1–230; B), or myc-FEZ1(1–308; C) or untransfected cells (D) were double labeled for the myc tag and endogenous JIP1 protein. Arrows indicate tips of neurites in transfected cells. Bar, 10 μm. (E) Quantification of JIP1 tip staining. n values in parentheses indicate the number of cells. Error bars represent SD. *, P ≈ 0.01; **, P < 0.001 compared with untransfected cells.
Our results show that FEZ plays an important role in the activation of Kinesin-1. Other FEZ partner proteins such as PKCζ, Disrupted-in-Schizophrenia-1, necdin, and E4B (Kuroda et al., 1999; Miyoshi et al., 2003; Okumura et al., 2004; Lee et al., 2005) may contribute to the regulation of Kinesin-1. Alternatively, FEZ may function as both a regulator of Kinesin-1 and a cargo-linker molecule for the Kinesin-1 transport of FEZ-binding partners to the growth cone (Miyoshi et al., 2003), which is reminiscent of the multiple roles dynactin plays in cytoplasmic dynein transport processes (Schroer, 2004).
Autoinhibition of Kinesin-1 in the absence of cargo prevents futile ATP hydrolysis and allows the rapid and specific control of motor activity both temporally and spatially. Two intramolecular interactions contribute to the autoinhibition of Kinesin-1. First, the KHC inhibitory tail blocks the KHC motor–MT interaction, and, second, the KLC tetritricopeptide repeat motifs push the two KHC motor domains apart (Cai et al., 2007). Here, we demonstrate that binding partners of both the KHC tail and KLC subunit (FEZ1 and JIP1, respectively) are required for the full activation of Kinesin-1 for MT binding and motility (Fig. S4 C, available at http://www.jcb.org/cgi/content/full/jcb.200605099/DC1). Cargo binding may be a general mechanism for activating molecular motors. For example, autoinhibition of the kinesin-3 family member KIF1A is likely relieved by the cargo-induced localized dimerization of weak monomers (Klopfenstein et al., 2002; Lee et al., 2004). Autoinhibition and cargo-dependent activation are also likely to play a role in the regulation of actin-based myosin motors (Li et al., 2005).
Materials and methods
Plasmids and antibodies
Myc-tagged rat KHC, HA-tagged rat KLC, and Flag-tagged human JIP1 have been described previously (Verhey et al., 1998, 2001). Mutation of loop 12 (Δloop12 = H275, R279, and K282 to Ala) and all other deletions or mutations were made by QuikChange (Stratagene) or overlapping PCR and were verified by sequencing. A full-length clone of FEZ1 was isolated from a Marathon-Ready human brain cDNA library (CLONTECH Laboratories, Inc.) and inserted into the pLP-CMV-myc vector (CLONTECH Laboratories, Inc.) or pCDNA3 with an hsv-HIS tag. FP-tagged KHC and KLC were created in the vectors mCit/mECFP-N1 and mCit/mECFP-C1 (CLONTECH Laboratories, Inc.). A four-aa linker (SGAG) was inserted between KHC and the FP, whereas a five-aa linker (GPVAT) was inserted between KLC and the FP. For bicistronic vectors, a synthetic intron and the internal ribosome entry site (IRES) from encephalomyocarditus virus (pIRES-puro3; CLONTECH Laboratories, Inc.) were inserted immediately upstream of the KHC-mCit or HA-KLC-mECFP start codons, which were shifted to a more optimal position for translation (i.e., to the 11th AUG from the oligopyrimidine tract; Ohlmann and Jackson, 1999). Full-length FEZ1 or JIP1 sequences were then inserted immediately upstream of the synthetic intron/IRES cassette.
Antibodies used were obtained as follows: myc and HA (Santa Cruz Biotechnology, Inc.), Flag (Sigma-Aldrich), KHC (1614 [Chemicon] and 13 [Verhey et al., 1998]), and JIP1 (152 [Verhey et al., 2001]). Polyclonal antibodies to FEZ1 were generated against peptides comprising aa 1–19 (MEAPLVSLDEEFEDLRPSC; 429) or 342–362 (CLNTVIPYEKKASPPSVEDLQ; 432) as described previously (Verhey et al., 1998). Fluorescein- and Rhodamine red-X–labeled secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Two-hybrid screen
Plasmid pGBKT7-KHC(750–955) was expressed in yeast strain AH109. A Matchmaker pretransformed human fetal brain library (CLONTECH Laboratories, Inc.) in strain Y187 was screened by yeast mating. 46 of the positive clones contained sequences encoding KLC as expected. 22 clones containing fragments of FEZ1 and five clones containing fragments of FEZ2 were isolated from 7.5 × 106 transformants. For directed two-hybrid analysis, cDNAs were cloned into plasmids pGBKT7 and pACT2 and expressed in yeast strains AH109 and Y187, respectively. A positive interaction was noted by growth on plates lacking histidine and plates lacking adenine.
Cell culture, immunoprecipitation, MT binding, and fluorescence microscopy
COS and CAD cells were cultured and transfected as described previously except that TransIT-LT1 (Mirus) was used for transfection (Verhey et al., 2001). Cell lysis, immunoprecipitation, and MT binding were performed as described previously (Verhey et al., 1998). Cells were processed for immunofluorescence (Verhey et al., 2001) and mounted in 50% glycerol and 0.5% n-propyl gallate in PBS. Images of fixed cells were taken on a microscope (BX51; Olympus) with a UplanFl 60× NA 1.25 oil immersion objective (Olympus) and CCD camera (DP70; Olympus) or on a microscope (Axioplan; Carl Zeiss MicroImaging, Inc.) with a plan-Apochromat 63× NA 1.4 oil immersion objective (Carl Zeiss MicroImaging, Inc.) and color camera (Axiocam; Carl Zeiss MicroImaging, Inc.). ImageJ software (National Institutes of Health [NIH]) was used to quantify JIP1 fluorescence using the Free Hand option to select neurite tips and cell bodies. The mean cell body fluorescence was subtracted from the mean neurite tip fluorescence to provide a measure of JIP1 transport in each cell. Higher fluorescence in tips relative to the cell body represents positive JIP1 transport. All cells in each experimental condition were pooled for statistical analysis (t test; Microsoft Excel). Images were prepared with Photoshop (Adobe).
Live cell permeabilization and imaging
A coverglass was assembled in a Leiden's chamber and maintained at 37°C in Ringer's buffer (10 mM Hepes, 155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, and 10 mM glucose, pH 7.2). 0.1 μg/ml streptolysin O with 10 mg/ml BSA was added for 30 s, and the cells were rapidly washed three times with buffer I (25 mM Hepes, 5 mM MgCl2, 115 mM KOAc, 5 mM NaOAc, 0.5 mM EGTA, pH 7.2, and 10 mg/ml BSA) and placed in 1 mM AMPPNP in buffer I. Images were acquired using an inverted microscope (Eclipse TE-300; Nikon) with a planApo 60× NA 1.4 oil-immersion objective (Nikon) and a cooled CCD camera (Photometrics Quantix; Roper Scientific) controlled by MetaMorph 6.2r6 software (Universal Imaging Corp.). Excitation and emission wavelengths were selected using a filter set (model 86006; Chroma Technology Corp.) and filter wheel controller (Lambda 10-2; Sutter Instrument Co.).
Relocation index was calculated on a frame by frame basis. In ImageJ, the MT pattern in the last frame was masked as MT Mask. The rest of the cell, excluding the nucleus and any aggregated motor in the extreme cell periphery, was masked as Other Region Mask. These masks were applied to each frame to measure the total fluorescence pixel intensities (Sumi, MT and Sumi, Other Region). The ratio (Ri) of Sumi, MT over Sumi, Other Region was calculated for each frame. The Relocation indexi was defined as the percent change of Ri as compared with before the addition of AMPPNP. A two-tailed t test was used for statistical analysis.
In vitro single-molecule total internal reflection fluorescence microscopy
A 25 × 25-mm #1.5 coverglass and a microscope slide were assembled into a flow chamber with double-sided tape (chamber volume of ∼30 μl). Cy5-labeled MTs in P12 buffer (12 mM Pipes/KOH, 1 mM EGTA, and 2 mM MgCl2, pH 6.8) with 10 μM taxol were flowed into the chamber and incubated at room temperature for 2 min. 15 mg/ml BSA (in P12 buffer with 10 μM taxol) was then flowed in and incubated for 10 min. 50 μl of oxygen scavenger buffer (1 mM DTT, 1 mM MgCl2, 2 mM ATP, 10 mM glucose, 0.1 mg/ml glucose oxidase, 0.08 mg/ml catalase, 10 mg/ml BSA, and 10 μM taxol in P12) containing 10 μl total of COS lysate was flowed in, and the chamber was sealed with wax. Objective-type total internal reflection fluorescence microscopy was performed on a custom-modified microscope (Axiovert 135TV; Carl Zeiss MicroImaging, Inc.) equipped with an α-plan Fluar NA 1.45 objective, 2.5× optovar, 505DCXR dichroic and HQ540/70M emission filters (Chroma Technology Corp.), and a back-illuminated EMCCD camera (Cascade 512B; Roper Scientific). The 488-nm line of a tunable, single-mode, fiber-coupled argon ion laser with Littrow prism (Schäfter und Kirchhoff; Melles Griot) at an incident power of 0.55 mW was used for capturing video sequences at 10 Hz. Videos and images were prepared with ImageJ, Photoshop, and Illustrator (Adobe).
Online supplemental material
Fig. S1 shows the characterization of FEZ1 and its interaction with KHC. Fig. S2 shows the coexpression of FP–Kinesin-1 with FEZ1 and JIP1 via bicistronic plasmids. Fig. S3 shows a live cell MT-binding assay. Videos 1 and 2 show the MT-binding activity of KHC-mCit (Video 1) and KHC-mCit + KLC-mECFP (Video 2) in live COS cells. Video 3 shows the MT-binding activity of KHC-mCit + KLC-mECFP expressed with JIP1 in live COS cells. Video 4 shows the MT-binding activity of KHC-mCit + KLC-mECFP expressed with FEZ1 in live COS cells. Video 5 shows the MT-binding activity of KHC-mCit + KLC-mECFP expressed with JIP1 and FEZ1 in live COS cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200605099/DC1.
Supplementary Material
Acknowledgments
We gratefully acknowledge T. Rapoport for ongoing support and J. Swanson and E. Meyhofer for the use of microscopes and helpful discussions.
This work was supported, in part, by a grant to K.J. Verhey (NIH grant GM070862).
Abbreviations used in this paper: AMPPNP, 5′-adenylylimidodiphosphate; FEZ, fasciculation and elongation protein ζ; FP, fluorescent protein; IRES, internal ribosome entry site; JIP, JNK-interacting protein; KHC, kinesin heavy chain; KLC, kinesin light chain; MT, microtubule.
References
- Adio, S., J. Reth, F. Bathe, and G. Woehlke. 2006. Review: regulation mechanisms of Kinesin-1. J. Muscle Res. Cell Motil. 27:153–160. [DOI] [PubMed] [Google Scholar]
- Bathe, F., K. Hahlen, R. Dombi, L. Driller, M. Schliwa, and G. Woehlke. 2005. The complex interplay between the neck and hinge domains in kinesin-1 dimerization and motor activity. Mol. Biol. Cell. 16:3529–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, L., and H.R. Horvitz. 1997. The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc. Natl. Acad. Sci. USA. 94:3414–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, D., A.D. Hoppe, J.A. Swanson, and K.J. Verhey. 2007. Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 176:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coy, D.L., W.O. Hancock, M. Wagenbach, and J. Howard. 1999. Kinesin's tail domain is an inhibitory regulator of the motor domain. Nat. Cell Biol. 1:288–292. [DOI] [PubMed] [Google Scholar]
- Cross, R., and J. Scholey. 1999. Kinesin: the tail unfolds. Nat. Cell Biol. 1:E119–E121. [DOI] [PubMed] [Google Scholar]
- Gindhart, J.G., J. Chen, M. Faulkner, R. Gandhi, K. Doerner, T. Wisniewski, and A. Nandlestadt. 2003. The kinesin-associated protein UNC-76 is required for axonal transport in the Drosophila nervous system. Mol. Biol. Cell. 14:3356–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck, O., G.S. Baird, R.E. Campbell, D.A. Zacharias, and R.Y. Tsien. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. J. Biol. Chem. 276:29188–29194. [DOI] [PubMed] [Google Scholar]
- Guzik, B.W., and L.S. Goldstein. 2004. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr. Opin. Cell Biol. 16:443–450. [DOI] [PubMed] [Google Scholar]
- Hackney, D.D., and M.F. Stock. 2000. Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2:257–260. [DOI] [PubMed] [Google Scholar]
- Hirokawa, N., and R. Takemura. 2005. Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6:201–214. [DOI] [PubMed] [Google Scholar]
- Jiang, M.Y., and M.P. Sheetz. 1995. Cargo-activated ATPase activity of kinesin. Biophys. J. 68:283S–284S. [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein, D.R., M. Tomishige, N. Stuurman, and R.D. Vale. 2002. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 109:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda, S., N. Nakagawa, C. Tokunaga, K. Tatematsu, and K. Tanizawa. 1999. Mammalian homologue of the Caenorhabditis elegans UNC-76 protein involved in axonal outgrowth is a protein kinase C ζ-interacting protein. J. Cell Biol. 144:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.R., H. Shin, J. Choi, J. Ko, S. Kim, H.W. Lee, K. Kim, S.H. Rho, J.H. Lee, H.E. Song, et al. 2004. An intramolecular interaction between the FHA domain and a coiled coil negatively regulates the kinesin motor KIF1A. EMBO J. 23:1506–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S., C.L. Walker, B. Karten, S.L. Kuny, A.A. Tennese, M.A. O'Neill, and R. Wevrick. 2005. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum. Mol. Genet. 14:627–637. [DOI] [PubMed] [Google Scholar]
- Li, X.D., R. Ikebe, and M. Ikebe. 2005. Activation of myosin Va function by melanophilin, a specific docking partner of myosin Va. J. Biol. Chem. 280:17815–17822. [DOI] [PubMed] [Google Scholar]
- Mallik, R., and S.P. Gross. 2004. Molecular motors: strategies to get along. Curr. Biol. 14:R971–R982. [DOI] [PubMed] [Google Scholar]
- Miyoshi, K., A. Honda, K. Baba, M. Taniguchi, K. Oono, T. Fujita, S. Kuroda, T. Katayama, and M. Tohyama. 2003. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol. Psychiatry. 8:685–694. [DOI] [PubMed] [Google Scholar]
- Notredame, C., D.G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. [DOI] [PubMed] [Google Scholar]
- Ohlmann, T., and R.J. Jackson. 1999. The properties of chimeric picornavirus IRESes show that discrimination between internal translation initiation sites is influenced by the identity of the IRES and not just the context of the AUG codon. RNA. 5:764–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura, F., S. Hatakeyama, M. Matsumoto, T. Kamura, and K.I. Nakayama. 2004. Functional regulation of FEZ1 by the U-box-type ubiquitin ligase E4B contributes to neuritogenesis. J. Biol. Chem. 279:53533–53543. [DOI] [PubMed] [Google Scholar]
- Schliwa, M., and G. Woehlke. 2003. Molecular motors. Nature. 422:759–765. [DOI] [PubMed] [Google Scholar]
- Schroer, T.A. 2004. Dynactin. Annu. Rev. Cell Dev. Biol. 20:759–779. [DOI] [PubMed] [Google Scholar]
- Seiler, S., J. Kirchner, C. Horn, A. Kallipolitou, G. Woehlke, and M. Schliwa. 2000. Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat. Cell Biol. 2:333–338. [DOI] [PubMed] [Google Scholar]
- Stock, M.F., J. Guerrero, B. Cobb, C.T. Eggers, T.G. Huang, X. Li, and D.D. Hackney. 1999. Formation of the compact conformer of kinesin requires a C-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J. Biol. Chem. 274:14617–14623. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Y. Okada, S. Semba, Y. Orba, S. Yamanouchi, S. Endo, S. Tanaka, T. Fujita, S. Kuroda, K. Nagashima, and H. Sawa. 2005. Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules. J. Biol. Chem. 280:24948–24956. [DOI] [PubMed] [Google Scholar]
- Verhey, K.J., and T.A. Rapoport. 2001. Kinesin carries the signal. Trends Biochem. Sci. 26:545–550. [DOI] [PubMed] [Google Scholar]
- Verhey, K.J., D.L. Lizotte, T. Abramson, L. Barenboim, B.J. Schnapp, and T.A. Rapoport. 1998. Light chain-dependent regulation of Kinesin's interaction with microtubules. J. Cell Biol. 143:1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey, K.J., D. Meyer, R. Deehan, J. Blenis, B.J. Schnapp, T.A. Rapoport, and B. Margolis. 2001. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekura, H., A. Nomura, H. Ozawa, Y. Tatsu, N. Yumoto, and T.Q. Uyeda. 2006. Mechanism of tail-mediated inhibition of kinesin activities studied using synthetic peptides. Biochem. Biophys. Res. Commun. 343:420–427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}