

Abstract
Aging is a highly complex biological process that is believed to involve multiple mechanisms. Mice that have small amounts of the mitotic checkpoint protein BubR1 age much faster than normal mice, but whether other mitotic checkpoint genes function to prevent the early onset of aging is unknown. In this study, we show that several aging-associated phenotypes appear early in mice that are double haploinsufficient for the mitotic checkpoint genes Bub3 and Rae1 but not in mice that are single haploinsufficient for these genes. Mouse embryonic fibroblasts (MEFs) from Bub3/Rae1 haploinsufficient mice undergo premature senescence and accumulate high levels of p19, p53, p21, and p16, whereas MEFs from single haploinsufficient mice do not. Furthermore, although BubR1 hypomorphic mice have less aneuploidy than Bub3/Rae1 haploinsufficient mice, they age much faster. Our findings suggest that early onset of aging-associated phenotypes in mice with mitotic checkpoint gene defects is linked to cellular senescence and activation of the p53 and p16 pathways rather than to aneuploidy.
Introduction
Maintenance of genetic stability requires faithful segregation of duplicated chromosomes during mitosis. Most human cancers consist of cells that have an abnormal chromosome content, which is also known as aneuploidy. However, how the aneuploidy develops remains poorly understood (Jallepalli and Lengauer, 2001; Draviam et al., 2004; Weaver and Cleveland, 2005). The spindle assembly checkpoint, commonly referred to as the mitotic checkpoint, is a surveillance mechanism that ensures accurate segregation of mitotic chromosomes by delaying metaphase-to-anaphase progression until each kinetochore has properly attached to the mitotic spindle (Kops et al., 2005). Kinetochores that are not yet attached to mitotic microtubules and chromosome pairs that lack tension across sister chromatids generated by the spindle poles activate the spindle assembly checkpoint (Yu, 2002; Rieder and Maiato, 2004). Various mitotic checkpoint proteins, including Bub3, Bub1, BubR1, Mad1, and Mad2, bind to kinetochores that lack attachment or tension and generate a “stop anaphase” signal that diffuses into the mitotic cytosol (Shah and Cleveland, 2000). This signal is believed to consist of Bub3, BubR1, and Mad2 protein complexes (Fang et al., 1998; Sudakin et al., 2001; Tang et al., 2001). These complexes bind to Cdc20 to prevent premature ubiquitination of cyclin B and securin by the anaphase-promoting complex (APC; Peters, 2002; Nasmyth and Haering, 2005). Checkpoint inactivation occurs after proper alignment of the mitotic chromosomes at the metaphase plate. This triggers release of Bub3, BubR1, and Mad2 protein complexes from Cdc20 and subsequent destruction of cyclin B and securin by the APC. Separase, which in mammalian cells is inhibited through association with securin and also by cyclin B/Cdk1-mediated phosphorylation, triggers sister chromatid disjunction by cleavage of the cohesin subunit Scc1 (Nasmyth and Haering, 2005). Various studies have addressed the physiological relevance of the spindle assembly checkpoint by deleting Mad or Bub genes in the mouse (for review see Baker et al., 2005). Mice that are homozygous null for Mad2, Bub3, or BubR1 die during the early stages of embryogenesis (Dobles et al., 2000; Kalitsis et al., 2000; Babu et al., 2003; Baker et al., 2004; Wang et al., 2004), with a small subset of Mad2-null embryos remaining viable until embryonic day 10.5 (Burds et al., 2005). On the other hand, mice that are heterozygous null for these genes are born alive and seem to exhibit no overt phenotypes. Thus, the challenge for studying the function of individual mitotic checkpoint genes in the mouse is to disrupt their function significantly but not so severely that the embryo dies.
Recently, we have reported the generation of a series of mice in which the expression of BubR1 is reduced in a graded fashion from normal levels to zero by the use of wild-type, knockout, and hypomorphic alleles (Baker et al., 2004). Of this series, mice with BubR1 levels as low as 10% of normal (BubR1H/H mice) are born alive and develop into adult mice. Remarkably, despite having severe aneuploidy, these animals do not have an escalated spontaneous tumor burden (Baker et al., 2004). Instead, these mice develop a variety of progeroid (resembling old age) features. In addition, mouse embryonic fibroblasts (MEFs) from BubR1H/H mice show profound premature cellular senescence. These observations, combined with the demonstration that BubR1 levels decline in ovary, testis, and spleen tissue as wild-type mice age, have suggested that this mitotic checkpoint protein is a key regulator of the normal aging process. However, whether aging is a general feature of spindle assembly checkpoint dysfunction remains unknown.
The mitotic checkpoint protein Bub3 is a seven-blade β propeller that has substantial sequence similarity with the mRNA export factor protein Rae1; the two proteins have 34% identity and 52% similarity in humans (Larsen and Harrison, 2004). Like Bub3 (Kalitsis et al., 2000), Rae1 is essential for early mouse embryogenesis, but mice that are haploinsufficient for Bub3 or Rae1 are viable and have a normal appearance (Babu et al., 2003). These heterozygous mice show remarkably similar mitotic defects, including spindle assembly checkpoint impairment and chromosome missegregation (Babu et al., 2003). Mice that are haploinsufficient for both Bub3 and Rae1 are also viable but develop much more severe mitotic phenotypes than single haploinsufficient mice. In this study, we have investigated the long-term phenotypes of mice in which the mitotic checkpoint proteins Bub3 and Rae1 are disrupted individually or in combination. We find that phenotypes associated with aging appear early in double haploinsufficient mice but not in single haploinsufficient mice. We also find that single and double haploinsufficient mice are not predisposed to spontaneous tumor development despite the accumulation of substantial numbers of aneuploid cells.
Results
Combined Bub3 and Rae1 haploinsufficiency reduces lifespan
To examine the long-term biological consequences of impaired spindle assembly checkpoint function, we generated cohorts of mice in which the mitotic checkpoint genes Bub3 and Rae1 were haploinsufficient individually or in combination (Babu et al., 2003). We monitored 70 wild-type, 56 Bub3+/−, 96 Rae1+/−, and 100 Bub3+/−/Rae1+/− mice daily for spontaneous tumor development or ill health for a period of >2 yr. During the first year of life, Bub3+/−/Rae1+/−, Bub3+/−, and Rae1+/− mice were indistinguishable from wild-type mice and exhibited no overt abnormalities (Fig. 1, A and B; and not depicted). However, in the second year, a substantial proportion of Bub3+/−/Rae1+/− mice began to develop an aged appearance (Fig. 1 D). Wild-type mice did not show this appearance (Fig. 1 C), nor did Bub3+/− or Rae1+/− mice. The median overall survival of Bub3+/− and Rae1+/− mice was slightly reduced in comparison with wild-type animals (24 vs. 25 mo, respectively), but these differences were not significant (Fig. 1 E). In contrast, the median survival of Bub3+/−/Rae1+/− mice was significantly reduced to 22 mo, which translates into a 12% decrease in lifespan. We note that the reduced lifespan of Bub3+/−/Rae1+/− mice may be the result of the sum of the lifespan reductions in Bub3+/− and Rae1+/− mice.
Figure 1.

Combined Bub3 and Rae1 haploinsufficiency reduces lifespan. (A–D) Representative photographs of wild-type and Bub3+/−/Rae1+/− mice at 5 (A and B) and 27 mo (C and D). These animals were not moribund at the time of the picture. Note the overt cataract, severe lordokyphosis, and cachetic appearance of the 27-mo-old Bub3+/−/Rae1+/− mouse pictured in D. (E) Overall survival curves for wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice. The curve marked with an asterisk is significantly different from that of wild-type mice using a log-rank test. We note that the lifespan of Bub3+/− and Rae1+/− mice is not statistically different from that of Bub3+/−/Rae1+/− mice.
Spontaneous tumorigenesis is not accelerated in Bub3+/−/Rae1+/− mice despite high aneuploidy
We performed biopsies on moribund mice from our cohorts to investigate whether chromosome number instability predisposes mice to spontaneous tumorigenesis. The tumor incidences of Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice were not significantly different from wild-type mice (Fig. 2 A). In addition, the tumor spectra exhibited in these groups were nearly identical. Comparison of the tumor-free survival curves of the various genotypes revealed a slight decrease in tumor latency in Bub3+/−/Rae1+/− mice compared with wild-type mice; however, this decrease is not significant (Fig. 2 B). The observed slight reduction in tumor latency most likely relates to the lifespan reduction of ∼3 mo in the Bub3+/−/Rae1+/− group. This difference is mirrored by the reduction in tumor latency by ∼4 mo. Thus, mice in which the mitotic checkpoint genes Bub3 and Rae1 are disrupted individually or in combination are not predisposed to spontaneous tumorigenesis.
Figure 2.
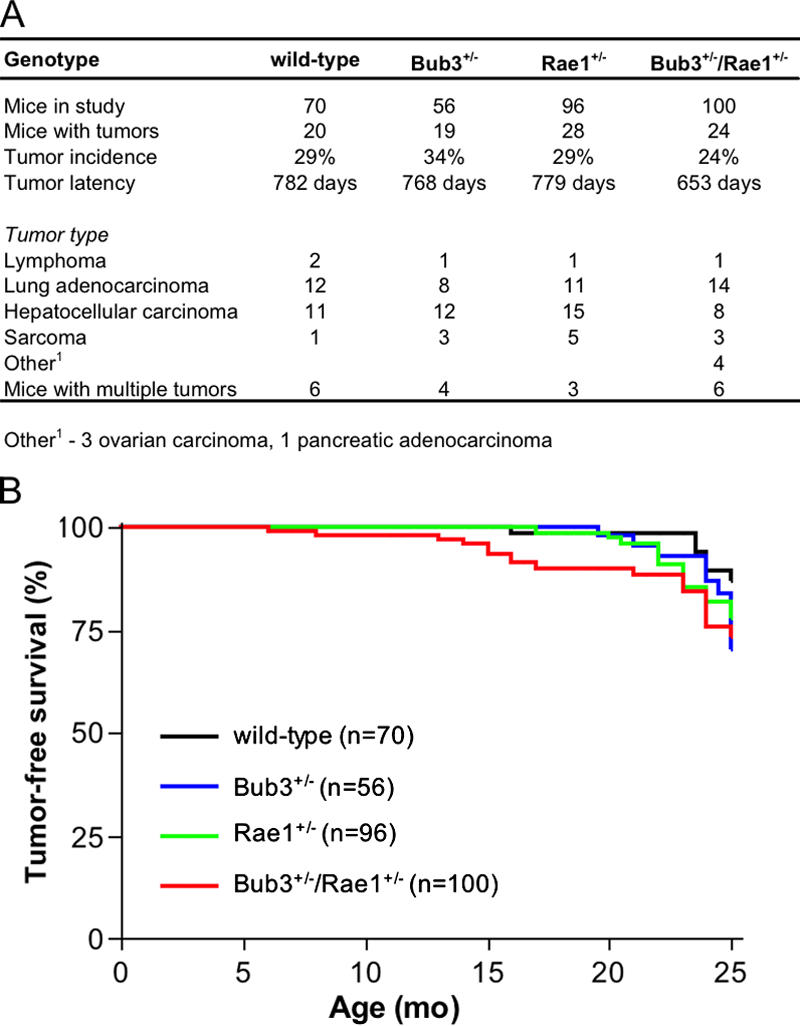
Bub3+/−/Rae1+/− mice develop spontaneous tumors at normal rates despite severe aneuploidy. (A) Spontaneous tumor incidence, latency, and spectrum of wild-type and Bub3+/−/Rae1+/− mice. Upon biopsy of moribund animals, all tissues were screened for tumors. Tumorous tissues were collected and processed for histological evaluation. (B) Tumor-free survival curves of wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice.
Aneuploidy progresses with age in mice with various mitotic checkpoint gene defects
Chromosome counts on splenocytes from 5-mo-old mice have shown that Bub3 and Rae1 single heterozygotes have significant numbers of aneuploid cells and that combined heterozygosity dramatically increases these numbers (Table I; Babu et al., 2003). We sought to determine whether aneuploidy progressed with age in Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice. Indeed, aneuploidy in single haploinsufficient Bub3 and Rae1 mice increased dramatically over time. Specifically, the aneuploidy in Bub3+/− splenocytes increased from 9% at 5 mo to 29% at 27 mo, whereas in Rae1+/− splenocytes, it increased from 9% at 5 mo to 33% at 24 mo (Table I). In splenocytes of Bub3+/−/Rae1+/− mice, aneuploidy was already 37% at 5 mo of age but further increased by 10% over the next 19 mo. Wild-type mice had no aneuploidy at 27 mo, but 3% aneuploidy was detected at 35 mo. Aside from aneuploidy, another feature of defective or weakened spindle assembly checkpoint activity in mice is premature sister chromatid separation (PMSCS; Michel et al., 2001; Baker et al., 2004). Mitotic figures from Bub3 and Rae1 single heterozygotes showed no PMSCS at 5 mo, but 10–11% of metaphases examined at 24–27 mo displayed this feature. As Bub3+/−/Rae1+/− mice aged, the percentage of cells exhibiting PMSCS also increased (Table I), going from 14% at 5 mo to 24% at 24 mo. The increases in PMSCS in Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice suggest that the activity of the spindle assembly checkpoint further declines as these animals age. Splenocytes from wild-type mice displayed no PMSCS at 27 mo, but 4% of splenocytes had PMSCS at 35 mo, suggesting that checkpoint activity declines as normal mice reach an extremely old age. We observed no chromosome breaks or fusions in metaphase spreads from 27-mo-old wild-type and 24-mo-old Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice (Table I). Thus, aneuploidy and PMSCS progress as mice in which Bub3 and Rae1 are disrupted individually or in combination age. Furthermore, mild aneuploidy and PMSCS are features of very old wild-type mice.
Table I.
Mice that are single or double haploinsufficient for Bub3 and Rae1 develop progressive aneuploidy
| Mouse genotype | Age (n) | Mitotic figures inspected |
Percent aneuploid figures (SD) |
Karyotypes with indicated chromosome number
|
Percent mitotic figures with PMSCS (SD) |
Percent mitotic figures with breaks |
Percent mitotic figures with fusions |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | |||||||
| Wild type | 5 mo (3) | 150 | 0 (0) | 150 | 0 (0) | ND | ND | ||||||||||
| 22 mo (3) | 150 | 0 (0) | 150 | 0 (0) | ND | ND | |||||||||||
| 27 mo (3) | 150 | 0 (0) | 150 | 0 (0) | 0 | 0 | |||||||||||
| 35 mo (3) | 150 | 3 (1) | 1 | 145 | 3 | 1 | 4 (1) | ND | ND | ||||||||
| Bub3+/− | 5 mo (3) | 150 | 9 (1) | 2 | 3 | 136 | 7 | 2 | 0 (0) | ND | ND | ||||||
| 27 mo (3) | 150 | 29 (3) | 2 | 3 | 5 | 106 | 20 | 9 | 5 | 11 (2) | 0 | 0 | |||||
| Rae1+/− | 5 mo (3) | 150 | 9 (1) | 3 | 2 | 136 | 6 | 3 | 0 (0) | ND | ND | ||||||
| 24 mo (3) | 150 | 33 (5) | 1 | 5 | 10 | 101 | 19 | 9 | 2 | 3 | 10 (2) | 0 | 0 | ||||
| Bub3+/−/Rae1+/− | 5 mo (4) | 200 | 36 (2) | 6 | 9 | 9 | 128 | 8 | 16 | 10 | 6 | 5 | 3 | 14 (1) | ND | ND | |
| 12 mo (3) | 150 | 39 (3) | 4 | 5 | 8 | 92 | 9 | 11 | 11 | 6 | 1 | 1 | 2 | 18 (2) | ND | ND | |
| 18 mo (3) | 150 | 45 (2) | 5 | 9 | 6 | 83 | 11 | 8 | 17 | 3 | 4 | 2 | 2 | 22 (2) | ND | ND | |
| 24 mo (3) | 150 | 47 (1) | 8 | 6 | 8 | 80 | 12 | 16 | 7 | 8 | 2 | 1 | 2 | 24 (5) | 0 | 0 | |
| BubR1H/H | 5 mo (4) | 200 | 15 (4) | 2 | 12 | 170 | 16 | 35 (4) | ND | ND | |||||||
| 12 mo (5) | 250 | 33 (4) | 9 | 22 | 168 | 44 | 7 | 24 (4) | ND | ND | |||||||
Empty spaces mean that there were no karyotypes with the indicated chromosome number.
Bub3+/−/Rae1+/− mice display early aging–associated phenotypes
The finding that several Bub3+/−/Rae1+/− mice displayed an aged appearance at a relatively young age prompted us to screen for aging-associated phenotypes in a systematic fashion. All mice of our cohort were screened every 2 wk for the development of cataracts and lordokyphosis (abnormal convexity in the curvature of the spine when viewed from the side). This monitoring strategy revealed that the incidence of cataract formation was significantly higher in Bub3+/−/Rae1+/− mice than in Bub3+/−, Rae1+/−, and wild-type mice (Fig. 3 A). Furthermore, the latency of cataract formation was shorter in Bub3+/−/Rae1+/− mice than in mice of the other genotypes. Histological examination confirmed that cataractous eyes from Bub3+/−/Rae1+/− mice had features reminiscent of human age–related cataracts (Fig. 3, B and C). Bub3+/−/Rae1+/− mice developed lordokyphosis at an earlier age and with greater severity than Bub3+/−, Rae1+/−, and wild-type mice (Fig. 4, A and B). Mutant mice with early lordokyphosis, but not age-matched control animals, showed clear signs of skeletal muscle atrophy and degeneration (Fig. 4 C).
Figure 3.
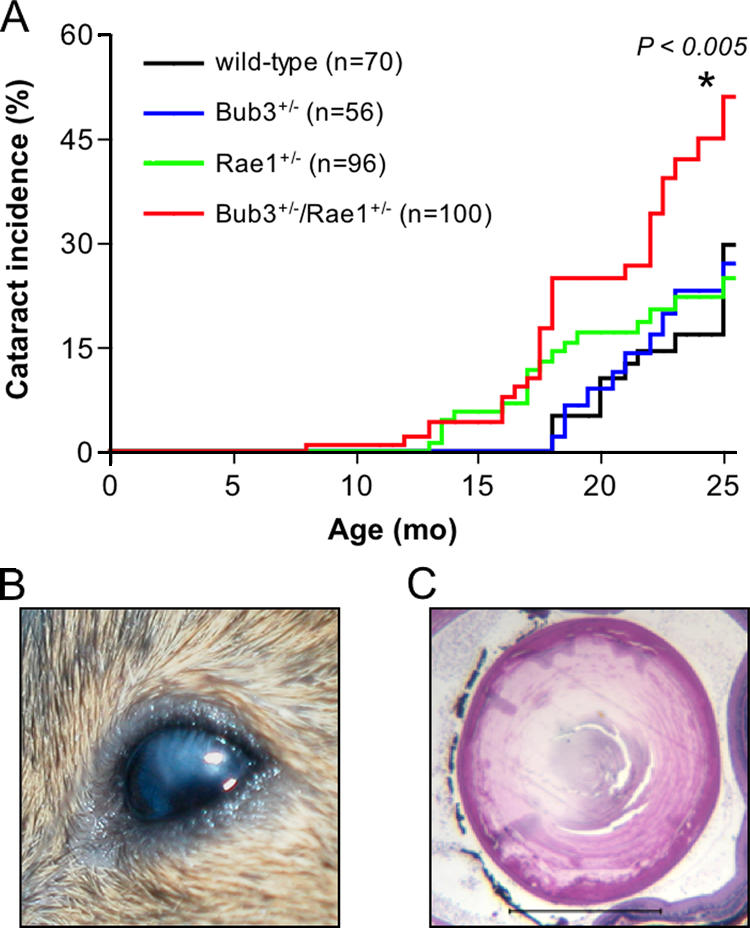
Combined Bub3 and Rae1 haploinsufficiency accelerates cataract formation. (A) Cataract incidence of wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice as detected by the use of slit light after dilating the eyes. The asterisk indicates that the curve is statistically different from that of wild-type mice using a log-rank test. (B) Overt cataract detected in a 15-mo-old Bub3+/−/Rae1+/− mouse. (C) Cross section of a cataractous lens from a 15-mo-old Bub3+/−/Rae1+/− mouse stained with hematoxylin and eosin. Note that the posterior portion of the eye has Morgagnian globules, whereas the inner portion has profound calcification. Bar, 1 mm.
Figure 4.

Lordokyphosis develops early in mice with combined Bub3 and Rae1 haploinsufficiency. (A) Incidence of lordokyphosis in wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice. The asterisk indicates that the curve is statistically different from that of wild-type mice using a log-rank test. (B) Skinned 19-mo-old Bub3+/−/Rae1+/−, 19-mo-old wild-type, and 35-mo-old wild-type mice. The Bub3+/−/Rae1+/− mouse and the 35-mo-old wild-type mouse exhibit lordokyphosis (arrowheads) and have little subcutaneous fat (arrows). (C) Cross section of gastrocnemius muscles of the animals shown in B. Arrowheads mark degenerated fibers, and arrows mark areas of fibroblast infiltration.
We next screened for additional aging-associated phenotypes by comparative analysis of mice from our Bub3+/−/Rae1+/− and wild-type cohorts. A well-established feature of aging is loss of body weight (MacIntosh et al., 2000). Although the weight of Bub3+/−/Rae1+/− mice at 5 mo was similar to that of age-matched wild-type mice, Bub3+/−/Rae1+/− mice had significantly lower body mass than wild-type mice at 24 mo (Fig. 5 A). Dual energy X-ray absorptiometry was used to determine whether this loss of weight was caused by a reduction in total body fat. At 5 mo of age, Bub3+/−/Rae1+/− and wild-type mice had similar percentages of total body adipose tissue; however, at 27 mo, Bub3+/−/Rae1+/− showed a dramatic reduction in body fat compared with wild-type mice (Fig. 5 B). Reduced dermal thickness and subdermal adipose are characteristics of aged mice (de Boer et al., 2002; Tyner et al., 2002). Histological examination of the skin revealed significant reductions in subcutaneous adipose thickness in 27-mo-old Bub3+/−/Rae1+/− mice but not in age-matched control animals (Fig. 5, C and E). The mean dermal thickness of 27-mo-old Bub3+/−/Rae1+/− mice was 21% lower than that of 5-mo-old Bub3+/−/Rae1+/− mice (Fig. 5 D), whereas in 27-mo-old wild-type mice, it was only 9% lower than in 5-mo-old wild-type mice (Fig. 5 D).
Figure 5.

Early aging–related phenotypes of combined Bub3/Rae1 haploinsufficient mice. (A) Body weight analysis of wild-type and Bub3+/−/Rae1+/− male mice (n ≥ 4 male mice per genotype). Bub3+/−/Rae1+/− mice have normal body weights at 5 mo but significantly reduced body weights at 24 mo. We note that neither Bub3+/− nor Rae1+/− mice show this reduction (not depicted). The decrease in body weight between the 24- and 35-mo-old wild-type mice is not significant but does show a trend of reduction in body weight. (B) Graph showing total body fat content in wild-type and Bub3+/−/Rae1+/− mice at 5, 27, and 35 mo (n = 4 male mice per genotype at each age). (C) Graph showing subcutaneous adipose layer thickness of wild-type and Bub3+/−/Rae1+/− mice at 5 and 27 mo along with 35-mo-old wild-type mice. Note the reduced thickness in 27-mo-old Bub3+/−/Rae1+/− mice and 35-mo-old wild-type mice (n = 4 males per genotype). (D) Graph showing dermal thickness of wild-type and Bub3+/−/Rae1+/− mice at 5 and 27 mo along with 35-mo-old wild-type mice. (A–D) Error bars represent SD. Asterisks mark values that are significantly different from wild-type values using a Mann-Whitney test. (E) Representative hematoxylin and eosin–stained dorsal skin sections of 5- and 27-mo-old wild-type and Bub3+/−/Rae1+/− from which the data in C and D were collected. Dermis (d) and adipose layer (a) are indicated. Bars, 100 μm.
We then investigated whether the age-related phenotypes present in Bub3+/−/Rae1+/− mice could also be observed in very old wild-type mice. We found that wild-type mice of 35 mo had lordokyphosis (Fig. 4 B), muscle atrophy (Fig. 4 C), and osteoporosis (not depicted). These mice also had significantly lower amounts of total body fat and subcutaneous adipose tissue than 5-mo-old wild-type mice (Fig. 5, B and C). As expected, the dermis of 35-mo-old wild-type mice was significantly thinner than that of 5-mo-old wild-type mice (Fig. 5 D). Together, the aforementioned data show that several aging-associated phenotypes appear early in mice that are double haploinsufficient for the mitotic checkpoint genes Bub3 and Rae1 but not in mice that are single haploinsufficient for these genes. The data also show that most of these phenotypes do occur in wild-type mice of extremely advanced age.
Combined haploinsufficiency of Bub3 and Rae1 results in premature senescence
We next investigated whether combined haploinsufficiency of Bub3 and Rae1 triggers cell death or cellular senescence, which are two processes that have been linked to aging (Campisi, 2005; Lombard et al., 2005). We intercrossed Bub3+/− and Rae1+/− mice to produce Bub3+/−/Rae1+/−, Bub3+/−, Rae1+/−, and wild-type MEFs from 13.5-d-old fetuses. At passage 5 (P5), we assayed Bub3+/−/Rae1+/− and wild-type MEFs for cell death by TUNEL staining. We observed no difference in amounts of TUNEL-positive cells in Bub3+/−/Rae1+/− and wild-type MEFs cultures (Fig. 6 A), indicating that combined haploinsufficiency of Bub3 and Rae1 did not increase the rate of cell death. Combined annexin V–FITC and propidium iodide (PI) staining of P5 Bub3+/−/Rae1+/− and wild-type MEFs showed that neither apoptotic nor nonapoptotic cell death was significantly increased in Bub3+/−/Rae1+/− MEFs (Fig. 6 B) despite high rates of chromosome missegregation in mitosis. This is remarkable because high rates of cell death have been observed in studies in which the mitotic checkpoint proteins Mad2 and BubR1 were depleted by short inhibitory RNA (Kops et al., 2004; Meraldi et al., 2004; Michel et al., 2004). Thus, it seems that cell death only occurs when mitotic checkpoint proteins drop below a critical threshold level and that aberrant chromosome segregation itself is insufficient to trigger cell death.
Figure 6.

Early onset of cellular senescence in Bub3/Rae1 double haploinsufficient MEFs. (A) Analysis of the percentages of cell death in Bub3+/−/Rae1+/− and wild-type MEFs cultures by TUNEL assay (n = 3 MEF lines per genotype). (B) Percentages of Bub3+/−/Rae1+/− and wild-type MEFs that are in the early stages of apoptosis (annexin V+/PI− cells), in the late stages of apoptosis, already dead (annexin V+/PI+ cells), that undergo nonapoptotic cell death (annexin V−/PI+ cells), or that are dead (PI+ cells). We used 3 MEF lines per genotype. We note that the differences between Bub3+/−/Rae1+/− and wild-type MEFs were not statistically significant using an unpaired t test. (C and D) Growth curves of wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− MEF cells at P3 (C) and P7 (D). 1.5 × 105 cells of indicated passages were seeded in duplicate on day 0 and counted for five consecutive days. Lines represent three independent MEF lines. Note that Bub3+/−/Rae1+/− MEF cells have reduced proliferation potential at P7, whereas single heterozygous Bub3+/− and Rae1+/− MEFs do not. (E) Percentages of wild-type, Bub3+/−, Rae1+/−, BubR1H/H, and Bub3+/−/Rae1+/− MEF cells that were positive for SA β-galactosidase activity at the indicated passages (n = 3 lines for each genotype at each passage). Asterisks in D and E mark values that are significantly different from those of wild-type mice using an unpaired t test. Error bars represent SD.
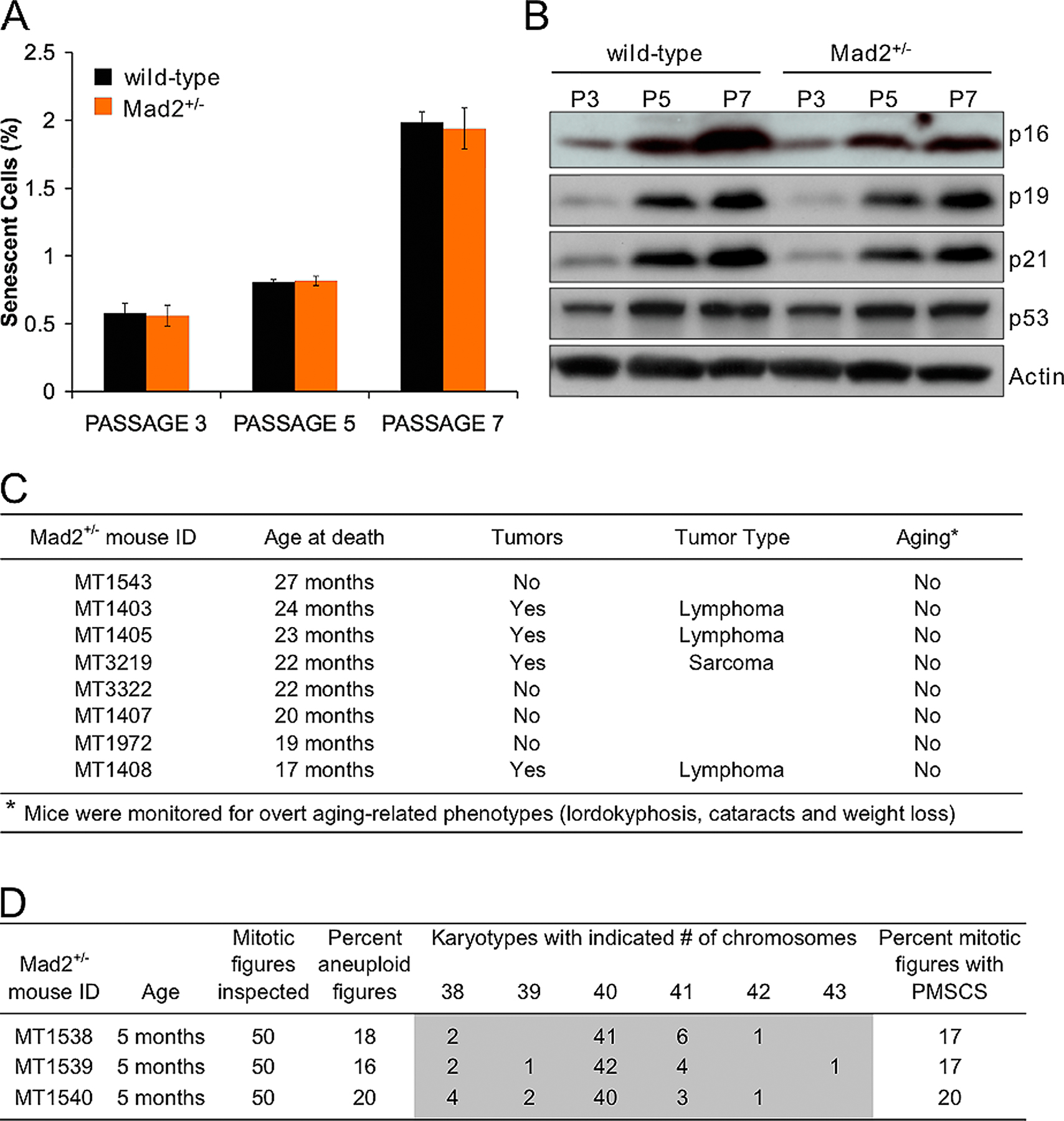
Cellular senescence is characterized in vitro by a decline in growth rate (Atadja et al., 1995). At P3, Bub3+/−/Rae1+/− growth rates were similar to those of Bub3+/−, Rae1+/−, and wild-type MEFs (Fig. 6 C). On the other hand, at P7, Bub3+/−/Rae1+/− MEFs showed significantly reduced growth rates compared with MEFs of the other three genotypes (Fig. 6 D). At P3, MEF cultures of all four genotypes had comparable numbers of cells that were positive for senescence-associated (SA) β-galactosidase activity (Fig. 6 E). However, at P5 and P7, Bub3+/−/Rae1+/− MEFs showed a profound increase in the number of cells staining positively for SA β-galactosidase in comparison with Bub3+/−, Rae1+/−, and wild-type MEFs. Consistent with these data, the senescence response genes p53, p21, p19, and p16 were induced earlier in Bub3+/−/Rae1+/− MEFs than in Bub3+/−, Rae1+/−, and wild-type MEFs (Fig. 7 A). Together, these findings indicate that compound heterozygosity of Bub3 and Rae1 causes early onset of cellular senescence.
Figure 7.

Bub3+/−/Rae1+/− MEFs have higher levels of SA molecular markers. (A) Western blot analysis of proteins in extracts from wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− MEFs at P3, P5, and P7. Blots were probed with antibodies against the indicated proteins. Note that the induction of senescence markers is delayed in Rae1+/− MEFs; this delay was consistently seen in three independent Rae1+/− MEF lines. (B) Western blots of protein extracts from P5 wild-type, Bub3+/−/Rae1+/−, and BubR1H/H MEFs probed with the indicated antibodies. The results shown in A and B are representative for three independently generated MEF lines of each genotype. (C–H) Analysis of the fidelity of distinct DNA damage repair pathways by measuring cell survival and colony formation ability after exposing early passage MEFs to various kinds of DNA-damaging agents. (C–E) Survival curves of wild-type and Bub3+/−/Rae1+/− MEFs after DNA damage by γ-irradiation (C), UV type B (D), or paraquat (E). (F–H) Colony-forming assay of wild-type and Bub3+/−/Rae1+/− MEFs after DNA damage by γ-irradiation (F), UV type B (G), or paraquat (H). Error bars represent SD.
The induction of p53, p21, and p19 indicates that the lesion triggering cellular senescence in Bub3/Rae1 double haploinsufficient MEFs activates the p53 DNA damage response pathway. To investigate whether activation of the p53 pathway might be caused by defective DNA repair, we analyzed the fidelity of distinct DNA damage repair pathways by measuring cell survival and colony formation ability after exposing Bub3+/−/Rae1+/− MEF cultures to various kinds of DNA-damaging agents. As shown in Fig. 7 (C–H), the DNA repair capacities of Bub3+/−/Rae1+/− and wild-type MEFs were very similar, suggesting that increased accumulation of DNA damage is an unlikely cause of increased senescence in Bub3+/−/Rae1+/− cultures.
The early aging–associated phenotypes that develop in Bub3+/−/Rae1+/− mice resemble those detected in mice with low levels of BubR1 (Baker et al., 2004). However, BubR1H/H mice had a much earlier onset of aging phenotypes than Bub3+/−/Rae1+/− mice. Consistent with this, BubR1 hypomorphic MEF cultures contained significantly higher numbers of SA β-galactosidase–positive cells than Bub3+/−/Rae1+/− MEF cultures (Fig. 6 E). In addition, BubR1H/H MEFs showed a more robust activation of the p53 and p16 pathways than Bub3+/−/Rae1+/− MEFs (Fig. 7 B). These data draw a clear correlation between the level of induction of senescence response genes and the rate of premature aging.
To further test this correlation, we prepared MEFs from Mad2 haploinsufficient mice and measured the degree of senescence at P3, P5, and P7. At each passage, the percentage of SA β-galactosidase–positive Mad2+/− MEFs was similar to that of wild-type MEFs (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200507081/DC1). Furthermore, at each passage, p53, p21, p19, and p16 levels of Mad2+/− MEFs were comparable with those of wild-type MEFs (Fig. S1 B). Consistent with these findings, we observed no early aging–related phenotypes in the small cohort of Mad2+/− mice that we followed for a period of 19–27 mo (Fig. S1 C). Chromosome counts showed that 18% of Mad2+/− splenocytes were aneuploid when mice were 5 mo of age (Fig. S1 D). Thus, Mad2 haploinsufficiency does not promote cellular senescence in MEFs and does not seem to trigger aging-related phenotypes in mice, although aneuploidy is increased.
Spindle assembly checkpoint activity measurements
Our chromosome counts suggested that there is a rather weak link between checkpoint dysfunction and the early onset of senescence and aging. To further investigate this issue, we obtained two additional measures for checkpoint activity by methods involving live cell imaging. The first assay is a nocodazole challenge assay. MEFs carrying various mitotic checkpoint gene defects were first transduced with a retrovirus carrying a YFP-tagged H2B gene to allow visualization of chromosomes by fluorescent microscopy. We then challenged the MEFs with nocodazole and measured the duration of the ensuing checkpoint-dependent mitotic arrest. This duration of arrest in mitosis was defined as the interval between nuclear envelope breakdown (NEBD; onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis). We used the time at which 50% of the cells had exited mitosis for comparison. Nocodazole-challenged wild-type MEFs typically remained arrested in prometaphase for 7.2 h (Fig. 8 A). However, single haploinsufficient MEFs of Bub3 and Rae1 showed a profound decrease in their ability to maintain this arrest, with 50% of the cells exiting at 3 h. Bub3+/−/Rae1+/− MEFs had an even further reduction in their ability to sustain arrest, with 50% of the cells exiting mitosis at 2.2 h. BubR1+/H and BubR1+/− MEFs, which express ∼60 and 30% of normal BubR1 protein, respectively (Baker et al., 2004), also had significant reductions in their ability to maintain a mitotic arrest, with 50% of the cells exiting at 5.3 and 3.8 h (Fig. 8 B). BubR1H/H MEFs (∼10% BubR1) had the most profound inability to maintain mitotic arrest, with 50% of the cells exiting at 1.1 h. It surprised us that the duration of arrest of BubR1H/H MEFs was substantially shorter than that of Bub3+/−/Rae1+/− MEFs, because BubR1H/H MEFs have less severe aneuploidy than Bub3+/−/Rae1+/− MEFs (Table II; also see Discussion). As an alternative measure for spindle checkpoint activity, we measured the accuracy of chromosome segregation during mitosis. In essence, we followed YFP-H2B–positive MEFs through an unchallenged mitosis by live cell imaging and determined the fraction of mitotic cells with chromosome segregation defects. We observed little or no misaligned chromosomes in wild-type, Bub3+/−, Rae1+/−, Bub3+/−/Rae1+/−, and BubR1H/H metaphases (Table III). However, a relatively large proportion of Bub3+/−/Rae1+/− and BubR1H/H anaphases had lagging chromosomes (Table III, Videos 1–3, and Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200507081/DC1). The incidence of lagging chromosomes was also increased in Bub3+/− and Rae1+/− anaphases, but to a lesser extent. It should be noted that lagging chromosomes are not necessarily a sign of failed mitotic checkpoint signaling because they could result from merotelically attached kinetochores that do not activate the mitotic checkpoint (Cimini et al., 2001). Bub3+/−/Rae1+/− MEFs had not only a somewhat higher percentage of anaphases with lagging chromosomes than BubR1H/H MEFs but also a higher percentage of abnormal anaphases with more than one lagging chromosome (Table III and Video 3). This may explain why the spectrum of chromosome losses and gains is wider for Bub3+/−/Rae1+/− MEFs than for BubR1H/H MEFs (Table II). Collectively, the aforementioned data confirm that spindle assembly checkpoint dysfunction and chromosome missegregation do not correlate well with cellular senescence and aging.
Figure 8.

Spindle assembly checkpoint activity analysis. (A) Analysis of mitotic checkpoint activity of wild-type, Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− MEFs. MEF cultures were challenged with nocodazole, and cells undergoing NEBD were marked. We continued to monitor these cells at 15-min intervals until their chromosomes decondensed. The duration of arrest in mitosis was defined as the interval between NEBD (onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis). The time at which 50% of the cells had exited mitosis has been indicated. This time was used as a measure for spindle assembly checkpoint activity. (B) Analysis of mitotic checkpoint activity of wild-type, BubR1+/H, BubR1+/−, and BubR1H/H MEFs. We note that BubR1+/H and BubR1+/− mice have a normal lifespan and lack early aging–associated phenotypes (Baker et al., 2004). Error bars represent SEM.
Table II.
Bub3/Rae1 double haploinsufficient MEFs develop more severe aneuploidy than BubR1 hypomorphic MEFs
| Mitotic MEF genotype (n) |
Mitotic figures inspected |
Percent aneuploid figures (SD) |
Karyotypes with indicated chromosome number
|
Percent mitotic figures with PMSCS (SD) |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 33 | 34 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | 44 | 45 | 46 | 47 | 49 | 51 | 53 | ||||
| Wild type (3) | 150 | 9 (1) | 7 | 136 | 7 | <1 | ||||||||||||||
| Bub3+/− (3) | 150 | 19 (2) | 1 | 4 | 9 | 121 | 8 | 2 | 2 | 3 | 0 | |||||||||
| Rae1+/− (3) | 150 | 19 (2) | 5 | 2 | 6 | 121 | 9 | 1 | 2 | 4 | 0 | |||||||||
| Bub3+/−/Rae1+/− (3) | 150 | 41 (2) | 1 | 6 | 5 | 3 | 3 | 15 | 88 | 6 | 2 | 3 | 4 | 4 | 5 | 1 | 2 | 1 | 1 | 19 (3) |
| BubR1H/H (3) | 125 | 36 (6) | 5 | 13 | 80 | 20 | 6 | 1 | 15 (3) | |||||||||||
Empty spaces mean that there were no karyotypes with the indicated chromosome number. Karyotyping was performed at passage 5.
Table III.
Chromosome missegregation rates in MEFs with various mitotic checkpoint gene mutations
| Genotype (n) | Mitotic cells inspected |
Metaphases with misaligned chromosomes |
Anaphases with lagging chromosomes |
Anaphases with indicated number of lagging chromosomes |
Percent abnormal anaphases with multiple lagging chromosomes |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 8 | |||||
| Wild type (6) | 114 | 0 (0%) | 3 (2.6%) | 111 | 3 | 0 | |||||
| Bub3+/− (5) | 152 | 1 (0.7%) | 9 (5.9%) | 143 | 8 | 1 | 11 | ||||
| Rae1+/− (5) | 100 | 0 (0%) | 7 (7.0%) | 93 | 7 | 0 | |||||
| Bub3+/−/Rae1+/− (6) | 130 | 0 (0%) | 19 (14.6%) | 114 | 10 | 3 | 3 | 1 | 1 | 1 | 47 |
| BubR1H/H (6) | 149 | 0 (0%) | 18 (12.1%) | 131 | 15 | 2 | 1 | 17 | |||
Empty spaces mean that there were no karyotypes with the indicated chromosome number.
Discussion
In this study, we used mice in which the mitotic checkpoint proteins Bub3 and Rae1 were disrupted individually or in combination to address whether premature aging is a common consequence of mitotic checkpoint gene defects. We found that Bub3 and Rae1 single haploinsufficient mice exhibit no signs of early aging despite their weakened checkpoint and progressive aneuploidy. Conversely, several aging-related phenotypes appear early in mice that are haploinsufficient for both Bub3 and Rae1. However, BubR1 hypomorphic mice age much faster but have less aneuploidy than Bub3/Rae1 haploinsufficient mice. This suggests that spindle assembly checkpoint dysfunction, which leads to the accumulation of aneuploidy, is unlikely to cause premature aging in BubR1 and Bub3/Rae1 mutant mice. We find that only mitotic checkpoint gene defects that can provoke cellular senescence in MEFs cause the early onset of aging-associated phenotypes in mice, with levels of induction of p19, p53, p21, and p16 highly correlating with the rate of cellular senescence and aging.
In the study of hypomorphic BubR1 mice (Baker et al., 2004), it became apparent that as these mice began to show signs of premature aging, they also started to accumulate aneuploid splenocytes. This led to the hypothesis that aneuploidy might be the primary lesion triggering cellular senescence and the process of aging. The use of Bub3 and Rae1 haploinsufficient mice allowed for rigorous testing of this hypothesis. At 5 mo, BubR1 hypomorphic mice have an aged appearance with detectable aneuploidy in 15% of their splenocytes (Baker et al., 2004). Age-matched single heterozygous Bub3 and Rae1 mice each have 9% aneuploid splenocytes but show no signs of early aging. One could argue that this may be the result of having aneuploidy that remains below a critical level. Splenocytes from 5-mo-old compound Bub3/Rae1 mice have 36% aneuploidy and a wide spectrum of chromosome losses and gains (Table I), but these animals show no overt features of aging at this age. If aneuploidy alone was a driving force of aging, these mice should have shown phenotypes of aging earlier than BubR1 hypomorphic mice. It is possible that the degree of aneuploidy in splenocytes is not representative of the degree present in tissues and cell types that develop early aging phenotypes. Although we cannot exclude this possibility with our current methods for measuring aneuploidy in vivo, the view that aneuploidy does not correlate with aging is further underscored by the finding that Bub3+/−/Rae1+/− MEFs are more aneuploid than BubR1 hypomorphic MEFs but exhibit lower levels of cellular senescence. In contrast, in mice with mitotic checkpoint gene defects, the rate of aging strongly correlates with the rate of cellular senescence. The induction of p53, p21, and p19 in both BubR1 hypomorphic and combined Bub3/Rae1 haploinsufficient cells but not Bub3 and Rae1 single haploinsufficient cells suggests that the lesion triggering cellular senescence and aging activates the p53 pathway. Like BubR1H/H MEFs (Baker et al., 2004), Bub3+/−/Rae1+/− MEFs have no detectable defects in DNA repair, suggesting that the lesion that triggers p53 activation is unlikely to be the accumulation of DNA damage or mutations. Various stress signals other than DNA damage are known to activate the p53 pathway (Ben-Porath and Weinberg, 2005; Harris and Levine, 2005), and future experiments will be necessary to determine whether any of these signals might be active in BubR1 hypomorphic and Bub3/Rae1 mutant mice.
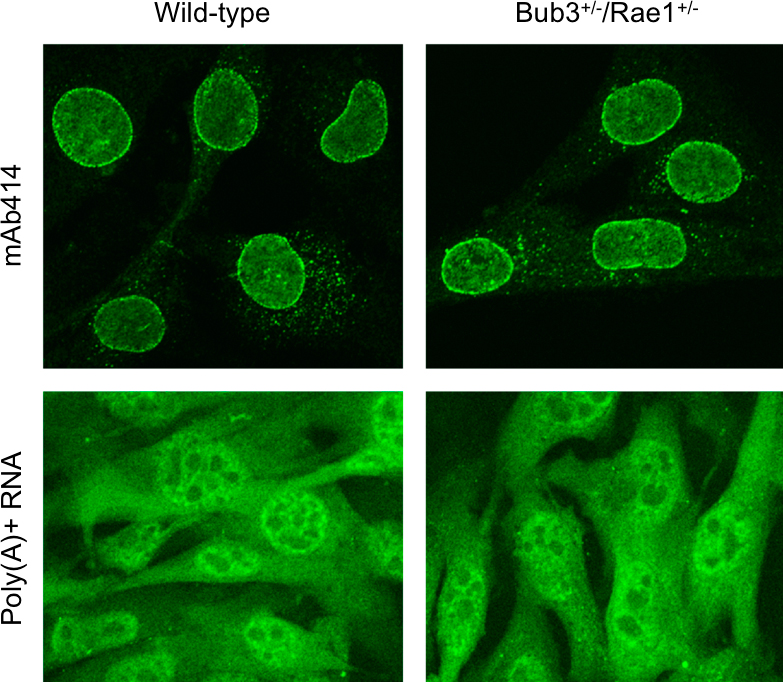
Rae1, Bub3, and BubR1 have all been implicated in functions outside of the spindle assembly checkpoint. For example, Rae1 was originally discovered as a nucleocytoplasmic transport factor that regulates the export of mRNA from the nucleus (Brown et al., 1995; Kraemer and Blobel, 1997; Pritchard et al., 1999). How Rae1 regulates nuclear transport and whether Bub3 functions also in the transport machinery has not yet been established. We found that combined Bub3/Rae1 haploinsufficiency does not cause overt defects in global mRNA export or nuclear pore complex biogenesis (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200507081/DC1). However, our experiments by no means represent a comprehensive analysis of the transport machinery, and it is certainly possible that Bub3/Rae1 haploinsufficient cells have subtle trafficking defects. A recent study has suggested that Bub3 can interact with histone deacetylases and that, in this context, Bub3 might act to repress gene transcription in interphase (Yoon et al., 2004). BubR1 can also repress gene transcription in a heterologous DNA-binding context, although not in conjunction with histone deacetylases (Yoon et al., 2004). BubR1 also seems to mediate apoptosis of cells that exit mitosis without chromosome segregation (Shin et al., 2003), and, in yeast, the BubR1 homologue Mad3 and Bub3 have both been linked to the accumulation of gross chromosomal rearrangements (Myung et al., 2004). Thus, it is possible that functions outside of the spindle assembly checkpoint are compromised in BubR1 hypomorphic and Bub3/Rae1 haploinsufficient mice and that early onset of aging-related phenotypes is linked to such defects.
Although BubR1 hypomorphic mice age much faster than Bub3/Rae1 mutant mice, both models have a common spectrum of age-related phenotypes that is distinctive from that of mouse strains in which DNA damage repair or telomere maintenance defects cause premature aging (Hasty et al., 2003; Lombard et al., 2005). For example, mice with mitotic checkpoint gene defects develop cataracts at high incidence, whereas the other models do not. Conversely, mice with defects in DNA damage repair or telomere maintenance show hematopoietic stem cell depletion and glucose intolerance, whereas mice with a weakened mitotic checkpoint do not (unpublished data). This distinction supports the idea that the lesion that triggers aging in BubR1 hypomorphic mice and Bub3/Rae1 mutant mice is distinct from that of other aging models. This suggestion is further supported by the demonstration that DNA damage repair and telomere maintenance functions are seemingly intact in Bub3/Rae1 haploinsufficient mice and BubR1 hypomorphic mice (Baker et al., 2004).
In this study, we have used a modified nocodazole challenge assay to measure the duration of sustained spindle checkpoint activity of MEFs with various mitotic checkpoint gene defects. In essence, we treated MEFs with nocodazole and determined how long individual cells remained in mitosis by live cell imaging and then used this time as a measure for spindle checkpoint activity. We validated our nocodazole challenge assay by using a series of MEFs with a graded reduction of BubR1 protein level. We found that the duration of arrest in mitosis declined as the amount of BubR1 decreased. We found further that this time of arrest correlated well with the rate of aneuploidy and chromosome missegregation. However, the finding that Bub3/Rae1 double-insufficient MEFs are more aneuploid than BubR1H/H MEFs (the percentage of aneuploid cells is higher and the spectrum of chromosome losses and gains is wider) appears inconsistent, as we find that Bub3/Rae1 haploinsufficient MEFs sustain their mitotic arrest in nocodazole for twice as long as BubR1H/H MEFs. A possible explanation for this discrepancy is that the nocodazole challenge assay may not provide a reliable measure for spindle assembly checkpoint activity; the reasoning is that the checkpoint functions to protect cells against chromosome missegregation rather than against microtubule poisoning. Alternatively, increased aneuploidy in Bub3/Rae1 haploinsufficient cells might be caused by the dysfunction of proteins involved in chromosome segregation but not in the spindle assembly checkpoint. Examples of such proteins are AdAPC (adenomatous polyposis coli; Kaplan et al., 2001), securin (Jallepalli and Lengauer, 2001), and separase (Waizenegger et al., 2002; Chestukhin et al., 2003).
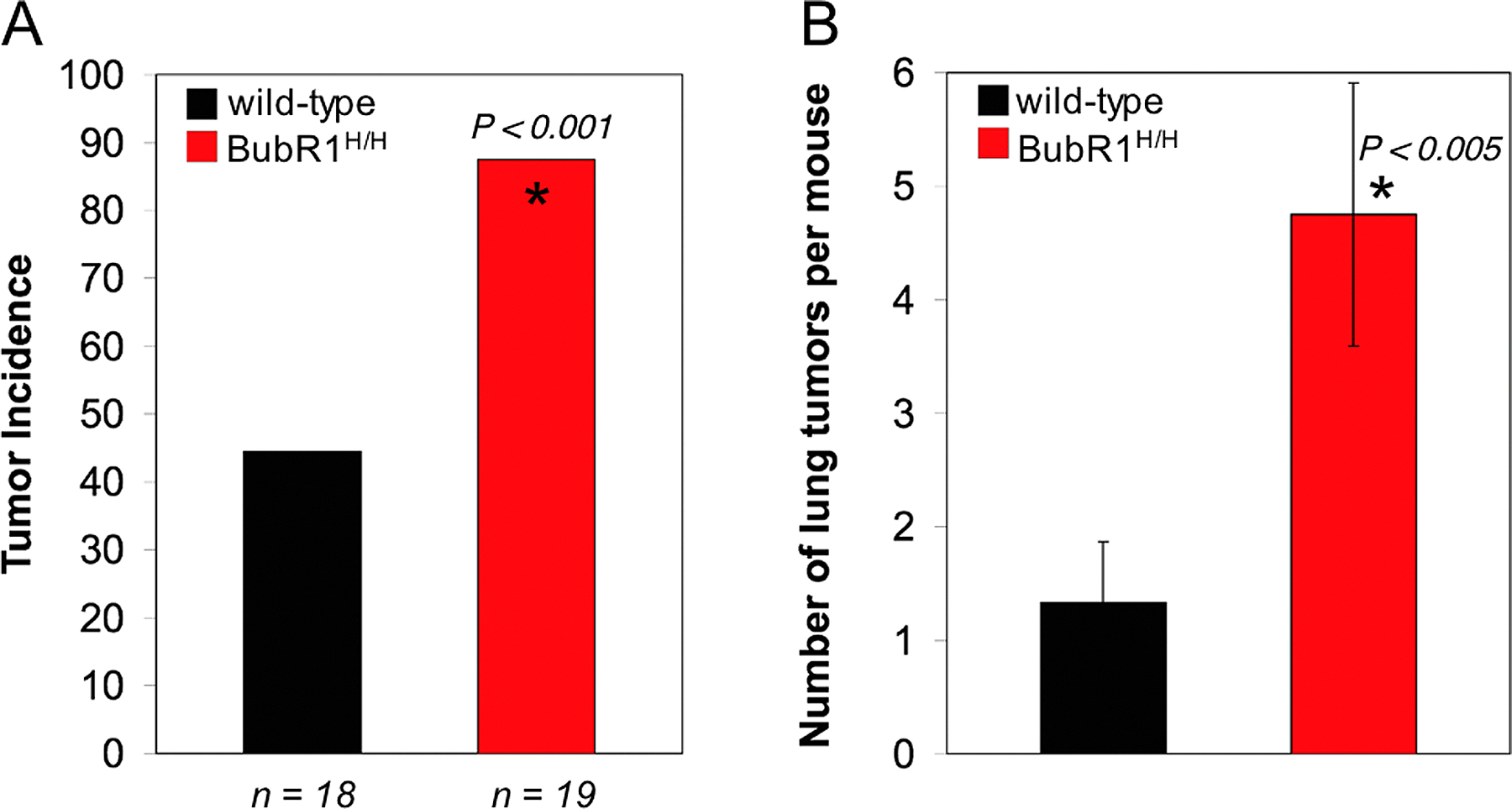
Abnormal numbers of chromosomes, termed aneuploidy, is one of the most common properties of cancer cells (Rajagopalan and Lengauer, 2004). Because aneuploidy occurs with such high frequency, it has been suggested that aneuploidy is an essential step in the process of tumorigenesis (Lengauer et al., 1998). In this study, we have demonstrated that Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− mice do not show any differences in both the rate of tumor formation and the type of tumor formed in comparison with wild-type animals. An earlier cancer susceptibility study on Bub3+/− mice yielded similar data (Kalitsis et al., 2005). That study further showed that even on a p53 or Rb1 heterozygous background, Bub3 haploinsufficiency had no significant effect on tumorigenesis. A suggested explanation for these findings was that the degree of impairment of the checkpoint machinery was too low to cause substantial aneuploidy (Kalitsis et al., 2005). However, although the aneuploidy in our double heterozygous Bub3/Rae1 mice is quite severe at 5 mo, we find no increased susceptibility to lifetime spontaneous tumor development. This result is consistent with our earlier observation that BubR1 hypomorphic mice are not tumor prone despite having severe aneuploidy (Baker et al., 2004). So far, only Mad2 haploinsufficiency has been shown to increase the risk for spontaneous tumors in mice (Michel et al., 2001). However, several studies have shown that impaired spindle checkpoint function increases the risk for carcinogen-induced tumors. For instance, we have shown previously that Rae1+/− and Bub3+/−/Rae1+/− mice are susceptible to lung adenoma formation after early postnatal treatment with dimethylbenzanthrene (DMBA; Babu et al., 2003). Others have shown that BubR1+/− mice have a susceptibility to azoxymethane-induced intestinal and lung tumor development (Dai et al., 2004), and, consistent with this finding, we observed that DMBA-treated BubR1H/H mice are prone to lung tumors (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200507081/DC1). All of these studies illustrate an important point that challenging these mice with carcinogenic agents introduces cooperating mutations that synergize with mitotic checkpoint failure to cause tumorigenesis and that these mutations may not occur under normal situations. It seems that the identification of gene mutations that cooperate with spindle assembly checkpoint gene alterations in tumorigenesis will be important to provide a better understanding of the role of aneuploidy in cancer. The link between spindle assembly checkpoint impairment and cancer predisposition is further supported by a recent report showing that a high proportion of individuals with mosaic variegated aneuploidy (MVA) syndrome carry bi-allelic mutations in the BubR1 gene (Hanks et al., 2004). MVA is a rare recessive disorder that is characterized by severe aneuploidy and a high risk for neoplasms such as rhabdomyosarcoma, Wilms tumor, and leukemia (Kajii et al., 2001; Plaja et al., 2001). Other characteristics of MVA syndrome include microcephaly, growth retardation, and eye abnormalities such as cataracts. Whether MVA patients with bi-allelic mutations in BubR1 develop progeroid features other than growth retardation and cataracts remains to be established: so far, only four patients with BubR1 mutations have been identified, three of which are <3 yr of age.
In a recent study, Kalitsis et al. (2005) confirmed that MEFs from Bub3 haploinsufficient mice have increased aneuploidy, but the increase was not as profound as the one we previously observed (Babu et al., 2003). The smaller increase most likely results from the fact that chromosome counts were performed at P3 instead of P5. Furthermore, BubR1+/− mice generated by Wang et al. (2004) typically exhibited splenomegaly and extramedullary megakaryopoiesis, but we did not detect these phenotypes in our BubR1+/− and BubR1H/H mice (Baker et al., 2004). It is unclear why that is, but it might be the result of the different methods used to create the models (enhancer trapping vs. homologous recombination), differences in the mouse housing facilities, and/or possible differences in genetic background.
Materials and methods
Generation of knockout mice and DMBA treatments
Bub3 and Rae1 heterozygous knockout mice and BubR1 hypomorphic mice were generated as described previously (Babu et al., 2003; Baker et al., 2004). These mice were derived from 129Sv/E embryonic stem cells and maintained on a mixed 129Sv/E × C57BL/6 genetic background. Mad2+/− mice were gifts from R. Benezra (Weill Medical College of Cornell University, New York, NY; Dobles et al., 2000). All mice were housed in a pathogen-free barrier facility for the duration of the study. Experimental procedures involving laboratory mice were reviewed and approved by the Institutional Animal Care and Use Committee of the Mayo Clinic. Prism software (GraphPad Software, Inc.) was used for the generation of survival curves and for statistical analysis. DMBA treatments were performed as described, except the duration of treatment was 4 mo instead of 5 (Babu et al., 2003).
Karyotype analysis
Metaphase spreads were prepared from splenocytes as described previously (Babu et al., 2003). The aneuploidy data for Bub3+/−, Rae1+/−, and Bub3+/−/Rae1+/− MEFs presented in Table II were first published in Babu et al. (2003); the data for BubR1H/H MEFs and mice were first published in Baker et al. (2004).
Collection and analysis of tumors
Moribund mice were killed, and major organs were screened for overt tumors using a dissection microscope. Collected tumors were processed for histopathology by standard methods. A chi square or Fisher's exact test was used to compare tumor incidence proportions across the genotypes for mice that developed tumors. Board-certified pathologists assisted in tumor analysis.
Cataract and body fat analysis
Biweekly, mice were screened for overt cataracts by examining dilated eyes with a slit light. At biopsy, cataractous eyes were collected and processed by standard methods for histological evaluation. Body fat content was measured using a small animal densitometer (Lunar PIXImus Corp.) as described previously (Nagy and Clair, 2000). A Mann-Whitney test was used for statistics.
Analysis of skin and skeletal muscle
Dorsal skin was dissected, fixed in 10% formalin, processed, and embedded in paraffin. 5-μm cross sections were prepared and stained with hematoxylin and eosin using standard procedures. Sections were stained, and the thickness of the dermal and adipose layers was calculated using a calibrated computer program (Spot Advanced; BioSpot). 40 random measurements were taken for each mouse of each genotype at the indicated age (n = 4 males). Histopathology on gastrocnemius muscle was performed as previously described (Kane et al., 2004; O'Cochlain et al., 2004). A Mann-Whitney test was used for statistics.
Generation and culture of MEFs
MEFs were generated from wild-type Bub3+/−, Rae1+/−, or Bub3+/−/Rae1+/− 13.5-d-old embryos as previously described (Babu et al., 2003). Growth curves of MEFs were generated as previously described (Baker et al., 2004). BubR1H/H, BubR1+/−, and BubR1+/H MEFs were previously generated (Baker et al., 2004).
Analysis of cell death, apoptosis, and DNA repair
TUNEL/Hoechst stainings were performed on P5 MEFs according to the manufacturer's protocol (Roche), as were annexin V/PI stainings (BD Biosciences). FACS analysis was performed as previously described (Tran et al., 2005). DNA damage survival experiments were performed as described previously (Baker et al., 2004).
SA β-galactosidase staining
MEFs were stained with an SA β-galactosidase kit according to the manufacturer's protocol (Cell Signal Technology). Nuclei were stained with Hoechst for visualization. The percentage of senescent cells is the total number of senescent cells divided by the total number of cells.
Western blot analysis
We performed Western blot analysis as previously described (Kasper et al., 1999). Antibodies for SA proteins were used at a 1:200 dilution (purchased from Santa Cruz Biotechnology, Inc. unless otherwise noted): p16 (M-156), p19 (NB200-106; Novus Biologicals), p21 (M-19), and p53 (FL-393-G). β-actin was used as a loading control (1:50,000 dilution; Sigma-Aldrich).
Time-lapse live microscopy
To allow visualization of chromosomes by fluorescent microscopy on live cells, we used a retrovirus expressing YFP-tagged H2B that was constructed as previously described (Jeganathan et al., 2005). P2 MEFs cells were seeded in T25 flasks at 50% confluence and cultured in DME/10% FBS. The next day, these MEFs were grown for 24 h in medium harvested from EcoPACK cells that produce pMCSV-puro H2B-YFP virus. 24 h after transduction with pMCSV-H2B-YFP retrovirus, MEFs were seeded in 35-mm glass-bottomed culture dishes (MatTek Corporation). The next day, Hepes was added to the culture medium at a final concentration of 20 mM. 4 h later, the dish was placed in a heat-controlled stage of a microscope (Axiovert 200; Carl Zeiss MicroImaging, Inc.). The temperature was maintained at 37°C. CO2 levels were maintained at 10% using a controller (CTI 3700; Carl Zeiss MicroImaging, Inc.). For nocodazole challenge experiments, nocodazole was added to a final concentration of 100 ng/ml. During the next 30 min, 20–30 cells undergoing NEBD were marked by the use of a mark/find module (AV4 MOD; Carl Zeiss MicroImaging, Inc.). Subsequently, time-lapse sequences were captured by using a plan Apo 63× NA 1.4 differential interference contrast (D = 0.18) oil objective (Carl Zeiss MicroImaging, Inc.) and a camera (AxioCam Hrm; Carl Zeiss MicroImaging, Inc.). The exposure times in nocodazole challenge experiments were always 100 ms at 2 × 2 binning. Interframe intervals were 15 min except for wild-type, BubR1+/H, and BubR1+/− MEFs (intervals were 30 min). Imaging WS/20A software (Carl Zeiss MicroImaging, Inc.) was used to determine the time of arrest in mitosis of individual cells. The time of arrest in mitosis was defined as the interval between NEBD (onset of mitosis) and chromatin decondensation (exit from mitosis without cytokinesis). For analysis of mitotic defects, cells entering mitosis were marked, and images were acquired at 2- or 3-min interframe intervals until the completion of mitosis.
Indirect immunofluorescence and staining for poly(A)+ RNA
Indirect immunofluorescence for mAb414 was performed as described previously (Wu et al., 2001), as were stainings for poly(A)+ RNA (Bastos et al., 1996).
Online supplemental material
Fig. S1 shows that Mad2 haploinsufficiency does not accelerate cellular senescence or aging. Fig. S2 shows that Bub3+/−/Rae1+/− MEFs have no overt defects in nuclear pore complex architecture and mRNA export. Fig. S3 shows that BubR1H/H mice are highly susceptible to DMBA-induced tumor formation. Fig. S4 shows examples of BubR1H/H anaphases with lagging chromosomes. Video 1 shows a Bub3+/−/Rae1+/− cell undergoing normal mitosis. Videos 2 and 3 show mitotic Bub3+/−/Rae1+/− cells with one and two lagging chromosomes, respectively. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200507081/DC1.
Supplementary Material
Acknowledgments
We thank Robert Benezra for providing Mad2 heterozygous knockout mice. We are grateful to Rick Bram, Meelad Dawlaty, and Debra Pearson for critical reading of the manuscript and Michael Thompson, Caili Tong, and Jay Hiddinga for assistance. We thank Douglas Cameron, Susan Abraham, and Cynthia Inwards for histopathological evaluations.
This work was supported by grants from the National Institutes of Health to J. van Deursen (CA77262 and CA96985).
Abbreviations used in this paper: APC, anaphase-promoting complex; DMBA, dimethylbenzanthrene; MEF, mouse embryonic fibroblast; MVA, mosaic variegated aneuploidy; NEBD, nuclear envelope breakdown; PI, propidium iodide; PMSCS, premature sister chromatid separation; SA, senescence associated.
References
- Atadja, P., H. Wong, I. Garkavtsev, C. Veillette, and K. Riabowol. 1995. Increased activity of p53 in senescing fibroblasts. Proc. Natl. Acad. Sci. USA. 92:8348–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu, J.R., K.B. Jeganathan, D.J. Baker, X. Wu, N. Kang-Decker, and J.M. van Deursen. 2003. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J. Cell Biol. 160:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D.J., K.B. Jeganathan, J.D. Cameron, M. Thompson, S. Juneja, A. Kopecka, R. Kumar, R.B. Jenkins, P.C. de Groen, P. Roche, and J.M. van Deursen. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36:744–749. [DOI] [PubMed] [Google Scholar]
- Baker, D.J., J. Chen, and J.M. van Deursen. 2005. The mitotic checkpoint in cancer and aging: what have mice taught us? Curr. Opin. Cell Biol. 17:583–589. [DOI] [PubMed] [Google Scholar]
- Bastos, R., A. Lin, M. Enarson, and B. Burke. 1996. Targeting and function in mRNA export of nuclear pore complex protein Nup153. J. Cell Biol. 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath, I., and R.A. Weinberg. 2005. The signals and pathways activating cellular senescence. Int. J. Biochem. Cell Biol. 37:961–976. [DOI] [PubMed] [Google Scholar]
- Brown, J.A., A. Bharathi, A. Ghosh, W. Whalen, E. Fitzgerald, and R. Dhar. 1995. A mutation in the Schizosaccharomyces pombe rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J. Biol. Chem. 270:7411–7419. [DOI] [PubMed] [Google Scholar]
- Burds, A.A., A.S. Lutum, and P.K. Sorger. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc. Natl. Acad. Sci. USA. 102:11296–11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi, J. 2005. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 120:513–522. [DOI] [PubMed] [Google Scholar]
- Chestukhin, A., C. Pfeffer, S. Milligan, J.A. DeCaprio, and D. Pellman. 2003. Processing, localization, and requirement of human separase for normal anaphase progression. Proc. Natl. Acad. Sci. USA. 100:4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., B. Howell, P. Maddox, A. Khodjakov, F. Degrassi, and E.D. Salmon. 2001. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J. Cell Biol. 153:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, W., Q. Wang, T. Liu, M. Swamy, Y. Fang, S. Xie, R. Mahmood, Y.M. Yang, M. Xu, and C.V. Rao. 2004. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 64:440–445. [DOI] [PubMed] [Google Scholar]
- de Boer, J., J.O. Andressoo, J. de Wit, J. Huijmans, R.B. Beems, H. van Steeg, G. Weeda, G.T. van der Horst, W. van Leeuwen, A.P. Themmen, et al. 2002. Premature aging in mice deficient in DNA repair and transcription. Science. 296:1276–1279. [DOI] [PubMed] [Google Scholar]
- Dobles, M., V. Liberal, M.L. Scott, R. Benezra, and P.K. Sorger. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 101:635–645. [DOI] [PubMed] [Google Scholar]
- Draviam, V.M., S. Xie, and P.K. Sorger. 2004. Chromosome segregation and genomic stability. Curr. Opin. Genet. Dev. 14:120–125. [DOI] [PubMed] [Google Scholar]
- Fang, G., H. Yu, and M.W. Kirschner. 1998. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 12:1871–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks, S., K. Coleman, S. Reid, A. Plaja, H. Firth, D. Fitzpatrick, A. Kidd, K. Mehes, R. Nash, N. Robin, et al. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 36:1159–1161. [DOI] [PubMed] [Google Scholar]
- Harris, S.L., and A.J. Levine. 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24:2899–2908. [DOI] [PubMed] [Google Scholar]
- Hasty, P., J. Campisi, J. Hoeijmakers, H. van Steeg, and J. Vijg. 2003. Aging and genome maintenance: lessons from the mouse? Science. 299:1355–1359. [DOI] [PubMed] [Google Scholar]
- Jallepalli, P.V., and C. Lengauer. 2001. Chromosome segregation and cancer: cutting through the mystery. Nat. Rev. Cancer. 1:109–117. [DOI] [PubMed] [Google Scholar]
- Jeganathan, K.B., L. Malureanu, and J.M. van Deursen. 2005. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 438:1036–1039. [DOI] [PubMed] [Google Scholar]
- Kajii, T., T. Ikeuchi, Z.Q. Yang, Y. Nakamura, Y. Tsuji, K. Yokomori, M. Kawamura, S. Fukuda, S. Horita, and A. Asamoto. 2001. Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: report of five infants. Am. J. Med. Genet. 104:57–64. [DOI] [PubMed] [Google Scholar]
- Kalitsis, P., E. Earle, K.J. Fowler, and K.H. Choo. 2000. Bub3 gene disruption in mice reveals essential mitotic spindle checkpoint function during early embryogenesis. Genes Dev. 14:2277–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalitsis, P., K.J. Fowler, B. Griffiths, E. Earle, C.W. Chow, K. Jamsen, and K.H. Choo. 2005. Increased chromosome instability but not cancer predisposition in haploinsufficient Bub3 mice. Genes Chromosomes Cancer. 44:29–36. [DOI] [PubMed] [Google Scholar]
- Kane, G.C., A. Behfar, S. Yamada, C. Perez-Terzic, F. O'Cochlain, S. Reyes, P.P. Dzeja, T. Miki, S. Seino, and A. Terzic. 2004. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes. 53:S169–S175. [DOI] [PubMed] [Google Scholar]
- Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Nathke. 2001. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 3:429–432. [DOI] [PubMed] [Google Scholar]
- Kasper, L.H., P.K. Brindle, C.A. Schnabel, C.E. Pritchard, M.L. Cleary, and J.M. van Deursen. 1999. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol. Cell. Biol. 19:764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J., D.R. Foltz, and D.W. Cleveland. 2004. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA. 101:8699–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 5:773–785. [DOI] [PubMed] [Google Scholar]
- Kraemer, D., and G. Blobel. 1997. mRNA binding protein mrnp 41 localizes to both nucleus and cytoplasm. Proc. Natl. Acad. Sci. USA. 94:9119–9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, N.A., and S.C. Harrison. 2004. Crystal structure of the spindle assembly checkpoint protein Bub3. J. Mol. Biol. 344:885–892. [DOI] [PubMed] [Google Scholar]
- Lengauer, C., K.W. Kinzler, and B. Vogelstein. 1998. Genetic instabilities in human cancers. Nature. 396:643–649. [DOI] [PubMed] [Google Scholar]
- Lombard, D.B., K.F. Chua, R. Mostoslavsky, S. Franco, M. Gostissa, and F.W. Alt. 2005. DNA repair, genome stability, and aging. Cell. 120:497–512. [DOI] [PubMed] [Google Scholar]
- MacIntosh, C., J.E. Morley, and I.M. Chapman. 2000. The anorexia of aging. Nutrition. 16:983–995. [DOI] [PubMed] [Google Scholar]
- Meraldi, P., V.M. Draviam, and P.K. Sorger. 2004. Timing and checkpoints in the regulation of mitotic progression. Dev. Cell. 7:45–60. [DOI] [PubMed] [Google Scholar]
- Michel, L.S., V. Liberal, A. Chatterjee, R. Kirchwegger, B. Pasche, W. Gerald, M. Dobles, P.K. Sorger, V.V. Murty, and R. Benezra. 2001. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 409:355–359. [DOI] [PubMed] [Google Scholar]
- Michel, L., E. Diaz-Rodriguez, G. Narayan, E. Hernando, V.V. Murty, and R. Benezra. 2004. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc. Natl. Acad. Sci. USA. 101:4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung, K., S. Smith, and R.D. Kolodner. 2004. Mitotic checkpoint function in the formation of gross chromosomal rearrangements in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 101:15980–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, T.R., and A.L. Clair. 2000. Precision and accuracy of dual-energy X-ray absorptiometry for determining in vivo body composition of mice. Obes. Res. 8:392–398. [DOI] [PubMed] [Google Scholar]
- Nasmyth, K., and C.H. Haering. 2005. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 74:595–648. [DOI] [PubMed] [Google Scholar]
- O'Cochlain, D.F., C. Perez-Terzic, S. Reyes, G.C. Kane, A. Behfar, D.M. Hodgson, J.A. Strommen, X.K. Liu, W. van den Broek, D.G. Wansink, et al. 2004. Transgenic overexpression of human DMPK accumulates into hypertrophic cardiomyopathy, myotonic myopathy and hypotension traits of myotonic dystrophy. Hum. Mol. Genet. 13:2505–2518. [DOI] [PubMed] [Google Scholar]
- Peters, J.M. 2002. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell. 9:931–943. [DOI] [PubMed] [Google Scholar]
- Plaja, A., T. Vendrell, D. Smeets, E. Sarret, T. Gili, V. Catala, C. Mediano, and J.M. Scheres. 2001. Variegated aneuploidy related to premature centromere division (PCD) is expressed in vivo and is a cancer-prone disease. Am. J. Med. Genet. 98:216–223. [DOI] [PubMed] [Google Scholar]
- Pritchard, C.E., M. Fornerod, L.H. Kasper, and J.M. van Deursen. 1999. RAE1 is a shuttling mRNA export factor that binds to a GLEBS-like NUP98 motif at the nuclear pore complex through multiple domains. J. Cell Biol. 145:237–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan, H., and C. Lengauer. 2004. Aneuploidy and cancer. Nature. 432:338–341. [DOI] [PubMed] [Google Scholar]
- Rieder, C.L., and H. Maiato. 2004. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 7:637–651. [DOI] [PubMed] [Google Scholar]
- Shah, J.V., and D.W. Cleveland. 2000. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell. 103:997–1000. [DOI] [PubMed] [Google Scholar]
- Shin, H.J., K.H. Baek, A.H. Jeon, M.T. Park, S.J. Lee, C.M. Kang, H.S. Lee, S.H. Yoo, D.H. Chung, Y.C. Sung, et al. 2003. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 4:483–497. [DOI] [PubMed] [Google Scholar]
- Sudakin, V., G.K. Chan, and T.J. Yen. 2001. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J. Cell Biol. 154:925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z., R. Bharadawaj, B. Li, and H. Yu. 2001. Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BUBR1. Dev. Cell. 1:227–237. [DOI] [PubMed] [Google Scholar]
- Tran, D.D., C.E. Edgar, K.L. Heckman, S.L. Sutor, C.J. Huntoon, J. van Deursen, D.L. McKean, and R.J. Bram. 2005. CAML is a p56Lck-interacting protein that is required for thymocyte development. Immunity. 23:139–152. [DOI] [PubMed] [Google Scholar]
- Tyner, S.D., S. Venkatachalam, J. Choi, S. Jones, N. Ghebranious, H. Igelmann, X. Lu, G. Soron, B. Cooper, C. Brayton, et al. 2002. p53 mutant mice that display early ageing-associated phenotypes. Nature. 415:45–53. [DOI] [PubMed] [Google Scholar]
- Waizenegger, I., J.F. Gimenez-Abian, D. Wernic, and J.M. Peters. 2002. Regulation of human separase by securin binding and autocleavage. Curr. Biol. 12:1368–1378. [DOI] [PubMed] [Google Scholar]
- Wang, Q., T. Liu, Y. Fang, S. Xie, X. Huang, R. Mahmood, G. Ramaswamy, K.M. Sakamoto, Z. Darzynkiewicz, M. Xu, and W. Dai. 2004. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 103:1278–1285. [DOI] [PubMed] [Google Scholar]
- Weaver, B.A., and D.W. Cleveland. 2005. Decoding the links between mitosis, cancer, and chemotherapy: the mitotic checkpoint, adaptation, and cell death. Cancer Cell. 8:7–12. [DOI] [PubMed] [Google Scholar]
- Wu, X., L.H. Kasper, R.T. Mantcheva, G.T. Mantchev, M.J. Springett, and J.M. van Deursen. 2001. Disruption of the FG nucleoporin NUP98 causes selective changes in nuclear pore complex stoichiometry and function. Proc. Natl. Acad. Sci. USA. 98:3191–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, Y.M., K.H. Baek, S.J. Jeong, H.J. Shin, G.H. Ha, A.H. Jeon, S.G. Hwang, J.S. Chun, and C.W. Lee. 2004. WD repeat-containing mitotic checkpoint proteins act as transcriptional repressors during interphase. FEBS Lett. 575:23–29. [DOI] [PubMed] [Google Scholar]
- Yu, H. 2002. Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14:706–714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}