

Abstract
The polarity protein Par-3 plays critical roles in axon specification and the establishment of epithelial apico-basal polarity. Par-3 associates with Par-6 and atypical protein kinase C and is required for the proper assembly of tight junctions, but the molecular basis for its functions is poorly understood. We now report that depletion of Par-3 elevates the phosphorylated pool of cofilin, a key regulator of actin dynamics. Expression of a nonphosphorylatable mutant of cofilin partially rescues tight junction assembly in cells lacking Par-3, as does the depletion of LIM kinase 2 (LIMK2), an upstream kinase for cofilin. Par-3 binds to LIMK2 but not to the related kinase LIMK1. Par-3 inhibits LIMK2 activity in vitro, and overexpressed Par-3 suppresses cofilin phosphorylation that is induced by lysophosphatidic acid. Our findings identify LIMK2 as a novel target of Par-3 and uncover a molecular mechanism by which Par-3 could regulate actin dynamics during cell polarization.
Introduction
Polarized mammalian epithelia form tight junctions between adjacent cells to regulate transepithelial permeability (gate function) and to separate the distinct apical and basolateral domains (fence function; Matter and Balda, 2003). The complexity of tight junction functions is reflected in the large number of associated signaling and regulatory molecules, including the polarity proteins Par-3, Par-6, and atypical PKC (aPKC; Macara, 2004). Tight junctions are attached to a cortical actin network through several linker proteins, and modulation of the actin cytoskeleton has a profound impact on both the assembly and functions of tight junctions. Despite intensive studies on the roles of polarity proteins in controlling polarization and cell junction formation (for review see Roh and Margolis, 2003; Munro, 2006), relatively little is known about how they interact with other cellular components to orchestrate these events. We have recently shown by RNA interference (RNAi) that Par-3 is essential for the proper assembly of tight junctions in mammalian epithelial cells (Chen and Macara, 2005) partially through its interaction with Tiam1 to spatially restrict the activity of Rac. We now show that Par-3 can also regulate cofilin phosphorylation by LIM kinase 2 (LIMK2).
Cofilin binds to and severs actin filaments, and it is crucial for numerous fundamental cellular processes such as migration, cytokinesis, and phagocytosis (Bamburg, 1999). Cofilin activity is inhibited by a single phosphorylation on Ser3, which is mediated by LIMKs or testicular protein kinases. Dephosphorylation is executed by protein phosphatases such as Slingshot (Niwa et al., 2002). LIMKs contain two LIM domains and a PDZ domain (Okano et al., 1995) and are activated by members of the Rho family of small GTPases. Recently, cofilin-mediated actin turnover has been shown to contribute to the disassembly of the apical junctional complex, which is induced in epithelial cells by calcium depletion (Ivanov et al., 2004). However, it has not been implicated in tight junction assembly.
Results and discussion
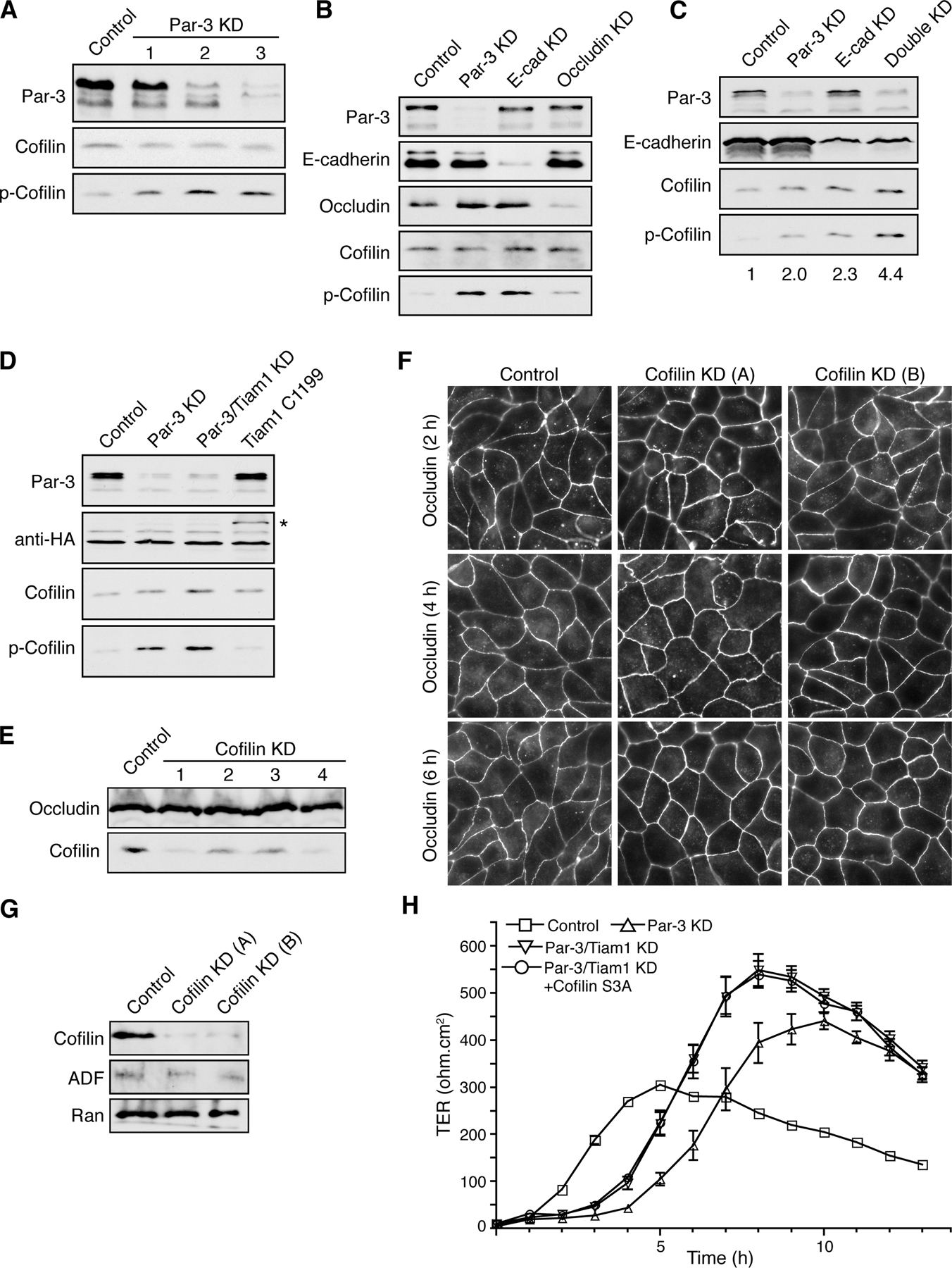
Given the close connections between actin reorganization and tight junction assembly, we examined the effects of reduced Par-3 expression on the phosphorylation of cofilin. The specificity of Par-3 suppression by RNAi has been confirmed previously (Chen and Macara, 2005). Depletion of Par-3 caused a substantial increase in cofilin Ser3 phosphorylation (Fig. 1 A and Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200510061/DC1). In contrast, depletion of the tight junction protein occludin had only a small effect on phospho-cofilin levels (Fig. S1 B). Epithelial (E) cadherin knockdown (KD) also resulted in elevated levels of phospho-cofilin (Fig. S1 B), but depletion of Par-3 did not affect E-cadherin levels or disrupt adherens junctions under normal calcium conditions and only had minor effects on adherens junction assembly during calcium switch (Chen and Macara, 2005). Moreover, when MDCK cells were subjected to calcium depletion overnight to disrupt all cell–cell junctions, the phospho-cofilin levels in Par-3–depleted cells were still higher than in control cells (Fig. 1 B, LCM), indicating that Par-3 can regulate phospho-cofilin levels independently of cell junction status and extracellular calcium levels. In addition, a double KD of Par-3 and E-cadherin led to an additive increase in phospho-cofilin compared with that achieved by a single KD of Par-3 or E-cadherin (Fig. S1 C), further suggesting that Par-3 and E-cadherin regulates cofilin phosphorylation through distinct mechanisms. Transient expression of human Par-3c, which is not recognized by the short hairpin RNA (shRNA) targeting canine Par-3, partially reversed the increase in phospho-cofilin (Fig. 1 B), supporting the argument that the increased phospho-cofilin is caused by Par-3 depletion rather than by off-target effects. Par-3c is a splice variant that lacks the aPKC-binding site (Gao et al., 2002), suggesting that the ability of Par-3 to regulate cofilin phosphorylation does not involve aPKC.
Figure 1.

Loss of Par-3 leads to elevated levels of phospho-cofilin. (A) MDCK cells were transfected with control or pSUPER–Par-3 (Par-3 KD) to suppress Par-3 expression followed by calcium switch. HCM, high calcium medium. Total cell lysates were analyzed by immunoblotting. (B) Ectopic Par-3c reduces phospho-cofilin levels in Par-3–depleted cells. A construct encoding human Par-3c was cotransfected with pSUPER–Par-3 into MDCK cells followed by calcium switch and Western blot analysis of total cell lysates. Numbers indicate relative levels of phospho-cofilin normalized against the total cofilin level. LCM, low calcium medium. (C) Depletion of endogenous Par-3 in HeLa cells increases phospho-cofilin levels. HeLa cells were transfected with human Par-3–specific (Par-3 KD) or control siRNAs. Cell lysates were prepared 48 and 72 h later and blotted with indicated antibodies. (D) NRG treatment abolishes the increase in phospho-cofilin in Par-3–depleted MCF-7 cells. 1 d after transfection with Par-3–specific or control siRNAs, MCF-7 cells were serum starved overnight followed by 45 min of treatment with 50 ng/ml NRG in serum-free medium before cells were lysed.
To determine whether regulation of phospho-cofilin by Par-3 is conserved across different cell types, we suppressed the expression of endogenous Par-3 in HeLa cells and found that cofilin phosphorylation was also enhanced after Par-3 depletion (Fig. 1 C). Similarly, depletion of endogenous Par-3 in MCF-7 breast carcinoma cells also markedly elevated the pool of phospho-cofilin (Fig. 1 D). Neuregulin (NRG)-1β treatment of MCF-7 cells activated the cofilin-specific phosphatase Slingshot (Nagata-Ohashi et al., 2004) and completely blocked the increase in phospho-cofilin in Par-3–depleted cells (Fig. 1 D), indicating that the loss of Par-3 does not interfere with the activation of Slingshot. These data suggest that Par-3 is generally involved in down-regulating cofilin phosphorylation.
Because the depletion of Par-3 results in phosphorylation of cofilin and its inactivation, we investigated whether reduced cofilin activity might contribute to the defects in tight junction assembly caused by Par-3 depletion. Using the transmembrane protein occludin as a marker, we observed, as reported previously, that MDCK cells lacking Par-3 had disrupted tight junctions for many hours after a calcium switch (Fig. 2 A; Chen and Macara, 2005). However, the normal cortical distribution of occludin was partially rescued at later stages of junction assembly by transient expression of a phosphorylation-defective, constitutively active cofilin mutant (S3A; Fig. 2 A). Quantification of the mean length per cell of occludin at cell–cell contacts confirmed the significance of the enhanced cortical localization of occludin (Fig. 2 B). Importantly, the expression of cofilin S3A did not affect the efficiency of Par-3 KD (Fig. 2 C). As shown previously, the loss of Par-3 substantially delayed the development of transepithelial electrical resistance (TER) after calcium readdition, and this was partially reversed by the expression of cofilin S3A (Fig. 2 D; Chen and Macara, 2005). Silencing of cofilin expression alone did not disrupt tight junctions, however, suggesting that cofilin activity is not a limiting regulator of junction assembly (Fig. S1, E–G). Altogether, these results suggest that cofilin activity contributes to tight junction formation during epithelial cell polarization and that one of the functions of Par-3 is to prevent inappropriate cofilin phosphorylation.
Figure 2.

Expression of nonphosphorylatable cofilin S3A mutant promotes tight junction assembly in Par-3 KD MDCK cells. (A) MDCK cells were transfected with control, pSUPER–Par-3 (Par-3 KD), or pSUPER–Par-3 together with a construct expressing cofilin S3A (+cofilin S3A). Cells were subjected to calcium switch and stained for occludin at various times after calcium readdition. (B) Quantification of occludin cortical localization after calcium readdition. Asterisks denote a significant difference from Par-3 KD cells (P < 0.05) by t test. 6-h time point: control, n = 6; Par-3 KD, n = 7; cofilin S3A, n = 6. 7-h time point: control, n = 6; Par-3 KD, n = 7; cofilin S3A, n = 7. (C) Western blot analysis of total cell lysates from A. White lines indicate that intervening lanes have been spliced out. (D) Cofilin S3A expression partially rescues the development of transepithelial electrical resistance (TER) in Par-3 KD MDCK cells during calcium switch. MDCK cells were transfected with indicated constructs, and the kinetics of TER establishment was monitored for 13 h after the readdition of calcium. Each value is the mean of triplicate measurements (n = 3). Error bars represent SD.
We have shown previously that Par-3 binds to Tiam1 to down-regulate Rac activity (Chen and Macara, 2005). However, a double KD of Par-3 and Tiam1 had no effect on the augmented phospho-cofilin levels induced by Par-3 suppression (Fig. S1 D). Furthermore, the expression of a constitutively active fragment of Tiam1 (Tiam1 C1199; Chen and Macara, 2005) did not elevate phospho-cofilin levels (Fig. S1 D), supporting the idea that the elevated phospho-cofilin in Par-3 KD cells is independent of misregulated Tiam1 activity. Interestingly, cofilin S3A expression did not further improve the TER establishment in Par-3 and Tiam1 double KD cells (Fig. S1 H), suggesting that cofilin activity and the Tiam1–Rac pathway may regulate similar aspects or stages of tight junction assembly.
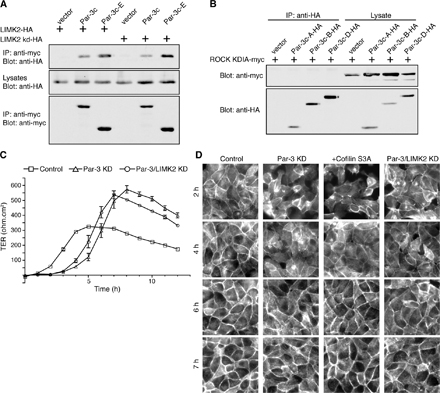
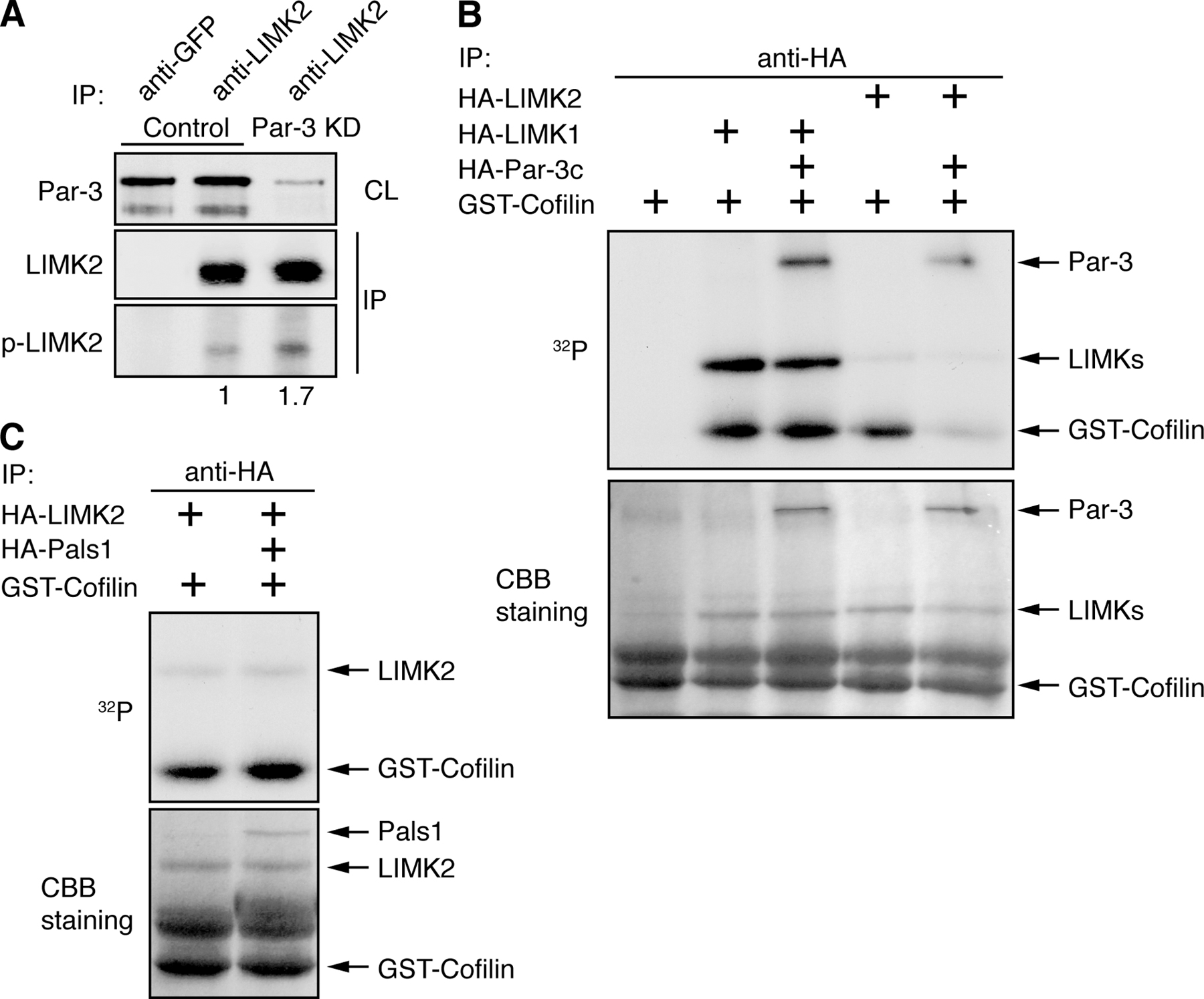
We next asked whether Par-3 might directly or indirectly regulate the activities of LIMKs or phosphatases to influence cofilin phosphorylation. LIMKs are activated by Rho small GTPases through their downstream effectors Rho-associated kinase (ROCK) and p21-activated kinase (Edwards et al., 1999; Sumi et al., 2001). No specific binding of Par-3 to the Slingshot phosphatase was detected (unpublished data), which is consistent with our observation that Slingshot activation is not altered by the loss of Par-3 (Fig. 1 D). However, recombinant Par-3 fragments (Par-3c-B and Par-3c-D) specifically pulled down LIMK2 but not LIMK1 from COS cell lysates (Fig. 3 B). Furthermore, the COOH terminus of Par-3 (Par-3c-E) alone coimmunoprecipitated LIMK2 just as efficiently as a longer fragment of Par-3 (Par-3c-D; Fig. 3 C), suggesting that the COOH terminus of Par-3 is sufficient to mediate the interaction with LIMK2. In contrast, another PDZ domain–containing polarity protein, Pals1, did not coimmunoprecipitate LIMK2. When endogenous LIMK2 from MDCK cells was immunoprecipitated, an association of endogenous Par-3 with LIMK2 was detected (Fig. 3 D). These data demonstrate that Par-3 and LIMK2 interact in vivo.
Figure 3.

Par-3 interacts with LIMK2 in vivo and in vitro. (A) Schematic diagrams of Par-3, LIMK2, and their various deletion fragments. Amino acid residue numbers are shown. Par-3c is a splice variant that lacks the aPKC-binding site. (B) Association of LIMK2 but not LIMK1 with recombinant Par-3 fragments. S-tagged Par-3 fragments immobilized on beads were incubated with COS cell lysates expressing Flag-tagged LIMK1 or LIMK2. Bound proteins were analyzed by Western blotting. (C) The COOH terminus of Par-3 is sufficient to associate with LIMK2. COS cells transfected with the indicated constructs were lysed, and the myc-tagged Par-3 fragments and Pals1 were immunoprecipitated by an anti-myc antibody. The associated LIMK2 was detected with an anti-HA antibody. Arrowhead points to LIMK2-HA. IP, immunoprecipitation. (D) Association of endogenous Par-3 and LIMK2 in MDCK cells. Endogenous LIMK2 was immunoprecipitated with an anti-LIMK2 antibody. Control immunoprecipitations were performed with anti-GFP and anti–β-catenin antibodies. (E) The NH2-terminal region of LIMK2 is required for association with the Par-3 COOH terminus. COS cells were transiently transfected with constructs expressing the COOH terminus of Par-3 (Par-3c-E) together with various LIMK2 deletion fragments. LIMK2 fragments were immunoprecipitated with an anti-HA antibody, and the associated Par-3c-E fragment was detected with an anti-myc antibody. Asterisks indicate LIMK2 fragments. (F) Binding of LIMK2 NH2 terminus to Par-3 fragments in vitro. GST–LIMK2 ΔC or GST-Rac was incubated with S-tagged Par-3 fragments immobilized on beads, and their interactions were analyzed by Western blotting. Asterisks indicate breakdown products. Arrowhead points to nonspecifically associated GST-Rac. White lines indicate that intervening lanes have been spliced out.
To further dissect the region of LIMK2 that is involved in its interaction with Par-3, deletion fragments of LIMK2 were tested for their abilities to coimmunoprecipitate the COOH terminus of Par-3 (Fig. 3 E). Deletion of the entire NH2-terminal half of LIMK2 (LIMK2 ΔN) or of the two LIM domains (LIMK2 ΔLIM) severely abrogated association with the Par-3 COOH terminus, indicating that the LIM domains are required for the interaction. In contrast, LIMK2 ΔC or a kinase-dead mutant of LIMK2 was able to associate with Par-3 (Fig. 3 E and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200510061/DC1). To examine whether Par-3 fragments can bind to the NH2 terminus of LIMK2 in vitro, recombinant Par-3 fragments immobilized on beads were incubated with GST–LIMK2 ΔC or with GST-Rac as a negative control (Fig. 3 F).GST–LIMK2 ΔC interacted very weakly with the Par-3c-A fragment, but it showed more robust interaction with the Par-3c-B and Par-3c-D fragments, which is in agreement with the pull-down experiments.
LIMK2 can be activated by ROCK downstream of RhoA (Sumi et al., 2001). A specific ROCK inhibitor H-1152 greatly reduced the phospho-cofilin level in Par-3 KD MDCK cells (Fig. 4 A), supporting the involvement of LIMK2 in cofilin phosphorylation. To confirm that endogenous LIMK2 mediates cofilin phosphorylation in Par-3–depleted cells, we partially suppressed the expression of LIMK2 in MDCK cells by RNAi without affecting the total levels of Par-3 and cofilin (Fig. 4 B). Double KD of both Par-3 and LIMK2 inhibited the increase in phospho-cofilin caused by Par-3 depletion (Fig. 4 C), suggesting that LIMK2 is important for increasing phospho-cofilin levels after Par-3 depletion. Consistent with the reduction in phospho-cofilin level, the suppression of both Par-3 and LIMK2 also promoted the assembly of tight junctions after a calcium switch, as monitored by the improved localization of occludin to cell–cell borders (Fig. 4, D and E) and by the small but reproducible enhancement in TER establishment (Fig. S2 C).
Figure 4.

Suppressing LIMK2 expression promotes tight junction assembly in Par-3 KD MDCK cells. (A) ROCK activity is required for the elevated phospho-cofilin in Par-3–depleted cells. Control or Par-3 KD MDCK cells were subjected to calcium switch and treated with 10 μM H-1152 for indicated times after the readdition of calcium. Cells without calcium switch (HCM) were treated with H-1152 for 1 h. Total cell lysates were analyzed by immunoblotting. (B) Suppression of LIMK2 level in MDCK cells. MDCK cells were transfected with control vector or two different pSUPER-LIMK2 (LIMK2 KD) constructs targeting distinct regions of canine LIMK2 mRNA. Total cell lysates were probed with the indicated antibodies. pSUPER–LIMK2 #2 reduced LIMK2 levels and was used for subsequent studies. (C) Double KD of LIMK2 and Par-3 blocks the increase in phospho-cofilin levels. MDCK cells were transfected with control vector, pSUPER–Par-3 (Par-3 KD) alone, or pSUPER–Par-3 together with pSUPER-LIMK2 (double KD). Total cell lysates were probed with the indicated antibodies. Numbers indicate relative levels of phospho-cofilin normalized against the total cofilin level. (D) Double KD of LIMK2 and Par-3 partially rescues occludin localization during calcium switch. MDCK cells were transfected as in C followed by calcium switch and staining for occludin at various times after calcium readdition. (E) Quantification of occludin cortical localization after the readdition of calcium. Asterisks denote significant differences from Par-3 KD cells (P < 0.05) by t test. Control, n = 5; Par-3 KD, n = 6; Par-3/LIMK2 KD, n = 6. Error bars represent SD.
RhoA activity is not altered in Par-3 KD cells (Chen and Macara, 2005), and we could not detect any interaction between Par-3 and ROCK (Fig. S2 B). The ability of Par-3 to interact with LIMK2 suggests that Par-3 is acting directly on LIMK2. Interestingly, cells lacking Par-3 showed a small increase in the level of phosphorylation on Thr505 of LIMK2 (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200510061/DC1). Phosphorylation at this site is mediated by ROCK and leads to LIMK2 activation (Sumi et al., 2001). Therefore, Par-3 might be involved in negatively regulating LIMK2 activation by ROCK in vivo.
We next examined whether Par-3 can mediate the inhibition of LIMK2 kinase activity in vitro. Immunoprecipitated LIMK2 specifically phosphorylated GST-cofilin, and the presence of Par-3b inhibited this activity (Fig. 5 A). Par-3b was also phosphorylated during the kinase assay, which was likely caused by endogenous aPKC associated with Par-3b (Gao et al., 2002). However, the Par-3c splice variant also inhibited the kinase activity of LIMK2 (Fig. 5 B). This variant does not bind to aPKC (Gao et al., 2002), and it was minimally phosphorylated compared with Par-3b. To test the specificity of the inhibition, LIMK1 was used, and its activity was not inhibited by Par-3 (Fig. S3 B). Moreover, when we replaced Par-3 with another PDZ domain protein, Pals1, no inhibition of LIMK2 activity was observed (Fig. S3 C).
Figure 5.

Par-3 negatively regulates LIMK2 activity. (A) Par-3 inhibits LIMK2 kinase activity in vitro. HA-tagged LIMK2 or Par-3b was expressed in COS cells and immunoprecipitated. The immunocomplexes were subjected to in vitro kinase assays using GST-cofilin as the substrate or GST-Rac as a control. Coomassie brilliant blue (CBB) staining showed equal amounts of LIMK2 and GST-cofilin in the assays. (B) aPKC is not required for Par-3 to inhibit LIMK2 activity in vitro. HA-tagged Par-3c variant, which does not bind to aPKC, was used in the kinase assay together with Par-3b. (C) Ectopic Par-3 inhibits cofilin phosphorylation after LPA treatment. Control COS cells or cells expressing Par-3c were serum starved for 4 h and briefly treated with 5 μM LPA for indicated times. Total cell lysates were probed with specified antibodies. Numbers on the bottom of the blot indicate relative levels of phospho-cofilin normalized against the total cofilin level. Graph is representative of two independent experiments.
To determine whether Par-3 can inhibit cofilin phosphorylation in vivo, Par-3c was transiently expressed in COS cells followed by treatment with lysophosphatidic acid (LPA), which activates the Rho–ROCK–LIMK2 pathway (Fig. 5 C). Overexpression of Par-3c decreased the basal phospho-cofilin level (0 min) and inhibited the increase in cofilin phosphorylation induced by LPA. Together, our data suggest that Par-3 binding to LIMK2 mediates the inhibition of LIMK2 activity and the down-regulation of cofilin phosphorylation.
LIMK1 and 2 share similar domain structures and have 50% overall amino acid identity (Okano et al., 1995), but they are regulated by distinct mechanisms. LIMK1 is activated specifically by p21-activated kinase and is inhibited by association with bone morphogenetic protein type II receptor, which binds to the NH2-terminal region of LIMK1 (Foletta et al., 2003). LIMK2 is activated specifically by ROCK and, as we show here, is inhibited by association through its NH2-terminal region with Par-3. The two kinases also exhibit different tissue distributions, with LIMK1 highly expressed in the brain and LIMK2 broadly expressed in the epithelia of nonneuronal tissues (Mori et al., 1997), which is consistent with a role for LIMK2 in epithelial polarization.
Although Par-3 is known to be a key regulator of epithelial polarization, its molecular actions have remained elusive. We have now identified a novel function for Par-3 in the regulation of LIMK2, which phosphorylates and inactivates cofilin. Depletion of Par-3 leads to elevated cofilin phosphorylation, and the restoration of cofilin activity by either active cofilin mutant or double KD of Par-3 and LIMK2 partially rescues tight junction assembly, indicating that the suppression of cofilin phosphorylation by Par-3 plays a positive role in junction assembly. Although it is possible that other indirect mechanisms contribute to the increase in phospho-cofilin after Par-3 KD, our data strongly suggest that Par-3 is directly involved in regulating cofilin phosphorylation through its interaction with LIMK2. Together with our previous finding that Par-3 regulates Rac activity through its interaction with Tiam1 (Chen and Macara, 2005), our current study suggests a possible role for Par-3 in modulating actin dynamics to facilitate junction assembly and polarization. On the other hand, actin organization is required for the correct localization of Bazooka (the Drosophila melanogaster homologue of Par-3) in Drosophila embryonic epithelia (Harris and Peifer, 2005). We propose that one of the functions of Par-3 is to coordinate and fine-tune the actin remodeling processes associated with junction assembly through its abilities to interact with and regulate multiple signaling pathways that control actin reorganization. It will be interesting to examine whether this mechanism of Par-3–directed cofilin activation is used in other cellular processes that require polarization, such as cell migration.
Materials and methods
Constructs
The following constructs have been described previously: myc and HA-tagged full-length Par-3b, Par-3c and Par-3c fragments (Par-3c-A, Par-3c-B, Par-3c-D, and Par-3c-E), and S-tagged Par-3c fragments (Joberty et al., 2000; Chen and Macara, 2005). Human cofilin-1 was generated by PCR from a human kidney cDNA library (CLONTECH Laboratories, Inc.) and cloned into the pKH3 and the pGEX-2T vectors. Cofilin S3A was created using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Human Slingshot in pcDNA3.1/myc-His was a gift from T. Uemura (Kyoto University, Kyoto, Japan). Flag-tagged LIMK1 in pEF-BOS was a gift from O. Bernard (The Walter and Eliza Hall Institute of Medical Research,Victoria, Australia). HA-tagged full-length rat LIMK2 and the LIMK2 kinase-dead mutant in pcDNA3 were gifts from T. Nakamura (Osaka University, Osaka, Japan). LIMK2 deletion fragments were generated by PCR and cloned into pKH3 and pGEX-2T. ROCK KD-IA construct was a gift from S. Narumiya (Kyoto University, Kyoto, Japan). Tiam1 C1199 was provided by J.G. Collard (Netherlands Cancer Institute, Amsterdam, Netherlands).
Cell culture and transfection
MDCK, COS-7, HeLa, and MCF-7 cells were grown in DME supplemented with 10% calf serum, 100 U ml−1 penicillin, and 100 U ml−1 streptomycin (Invitrogen). For transfection of MDCK cells, 2–4 μg DNA was introduced into 2–4 × 106 cells by Nucleofection (Amaxa) according to the manufacturer's recommendations. Transfection efficiency was generally >70%. For transfection with pSUPER constructs, enhanced YFP was coexpressed as a marker for transfected cells. Experiments were performed 2–3 d after transfection. COS-7 cells were transiently transfected using the calcium phosphate method.
RNAi
For RNAi in MDCK cells, pSUPER constructs expressing gene-specific shRNA were introduced into MDCK cells by Nucleofection. Generation of pSUPER constructs expressing shRNA targeting canine Par-3 or Tiam1 has been described previously (Chen and Macara, 2005). The target sequences for other pSUPER constructs are as follows: canine occludin, AGAGGAATTATTGCAAGCA; canine E-cadherin, GTCTAACAGGGACAAAGAA; canine LIMK2, TGTTGACAGAGTACATCGA; canine cofilin-1 #1, GGATCAAGCATGAGTTACA; and canine cofilin-1 #4, AGATCCTGGTAGGTGATGT. Empty pSUPER vector or a pSUPER construct targeted against luciferase was used as a negative control. To silence Par-3 in HeLa and MCF-7 cells, a pool of four siRNA duplexes (SMARTpool reagent M-015602-00) was purchased from Dharmacon. As a negative control, we used a siCONTROL nontargeting siRNA. HeLa and MCF-7 cells were transfected using Oligofectamine and LipofectAMINE 2000 (Invitrogen), respectively. Cells were processed 48 or 72 h after transfection.
Calcium switch, immunofluorescence, and quantification of occludin length
Calcium switch experiments were performed as described previously (Chen and Macara, 2005). To detect occludin and YFP, cells were washed and fixed in 1:1 methanol/acetone (vol/vol) for 3 min at −20°C. After washing with PBS, cells were blocked with 5% BSA in PBS for 25 min and incubated with primary and secondary antibodies. Primary antibodies were monoclonal anti-occludin (Zymed Laboratories) and rabbit anti-GFP (Invitrogen). Secondary antibodies were AlexaFluor-conjugated goat anti–mouse and goat anti–rabbit antibodies (Invitrogen). AlexaFluor594-conjugated phalloidin was obtained from Invitrogen. Coverslips were mounted on slides, and images were obtained and processed as previously described (Joberty et al., 2000).
To quantify the mean occludin length per cell, six to seven fields of cells with similar density were randomly selected from each coverslip, and the total length of occludin at cell junctions in each field was measured using the measurement tool in Openlab (Improvision). Only fields with >70% transfected cells (as determined by YFP expression) were measured. Cell numbers were counted for each field by DAPI staining, and the mean occludin length per cell on each field was calculated (Chen and Macara, 2005). Between 300 and 500 cells were counted for each coverslip. Each bar represents the mean value from all of the fields on each coverslip. Statistical analysis was performed using a two-tailed t test.
Imaging
Epifluorescence images were collected using an inverted microscope (Eclipse T200; Nikon) equipped with a 60× NA 1.2 plan Achromatic water immersion lens (Nikon) and a charge-coupled device camera (Orca C4742-95-12NRB; Hamamatsu). Images were collected at a 12-bit depth and 1,024 × 1,280 pixel resolution with 1 × 1 binning using Openlab 4.0 software (Improvision). Images were converted to eight-bit TIFF files and processed in Photoshop 7.0 (Adobe) to increase the gray scale range and to reduce haze using an unsharp mask.
TER measurements
After transfection, 6.6 × 105 MDCK II cells were plated into polycarbonate Transwell TM filters (0.4-μm pore size and 12-mm diameter; Corning Costar). 2 d later, cells were changed into low calcium medium (LCM) for 18–20 h before being switched back to normal growth medium (high calcium medium [HCM]). TER was measured with an Epithelial Voltohmmeter (EVOM; World Precision Instruments). TER values were calculated by subtracting the blank value from an empty filter with HCM and were expressed in ohm.cm2. Three parallel filters from each transfection were measured for each time point.
Immunoprecipitation, Western blot, pull-down assays, and in vitro binding assays
Immunoprecipitation from transiently transfected COS-7 cells was performed as previously described (Chen and Macara, 2005). Anti-HA antibody 12CA5 or anti-myc antibody 9E10 was used. To immunoprecipitate endogenous complexes of Par-3 and LIMK2 from MDCK cells, polyclonal anti-GFP antibody (Invitrogen), polyclonal anti-LIMK2 antibody (Santa Cruz Biotechnology, Inc.), and monoclonal anti–β-catenin antibody (BD Biosciences) were used.
To detect proteins by Western blotting, the following primary antibodies were used: 12CA5 and 9E10 antibodies; polyclonal anti–Par-3 (Upstate Biotechnology); monoclonal anti-Ran (BD Biosciences); polyclonal anti-LIMK2 (Santa Cruz Biotechnology, Inc.); polyclonal anti-cofilin, antiphospho-cofilin (Ser3), and antiphospho-LIMK2 (Thr505; Cell Signaling Technology); anti-destrin/actin-deploymerizing factor (Sigma-Aldrich); and HRP-conjugated S protein (Novagen). The ROCK inhibitor H-1152 (Calbiochem) was used at 10 μM. Cells were pretreated for 30 min in LCM and switched to HCM with H-1152 for indicated times. NRG-1β (Research Diagnostics) was used at 50 ng/ml. Densitometry analysis was performed using ImageJ software (National Institutes of Health) or the Odyssey Infrared Imaging System (LI-COR Biotechnology). For pull-down assays, 5 μg of purified S-tagged Par-3 fragments were bound to S-protein agarose beads and incubated for 40 min at 4°C with lysates from COS cells overexpressing LIMK1 or LIMK2. Bound proteins were eluted with SDS sample buffer, separated by SDS-PAGE, and detected by Western blotting.
For in vitro binding assays, 40 μg GST–LIMK2 ΔC or GST-Rac was incubated with 10 μg S-tagged Par-3 fragments immobilized on S-protein agarose beads in 300 μl of binding buffer (1% Triton X-100, 145 mM NaCl, 10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 2 mM EGTA, 1 mM PMSF, and 1 mM dithiotreitol) for 1 h at 4°C. The beads were washed three times with binding buffer containing 500 mM NaCl and once with regular binding buffer without Triton X-100. Proteins bound to beads were analyzed by SDS-PAGE and Western blotting.
In vitro kinase assays
In vitro kinase assays were performed essentially as described previously (Sumi et al., 2001). Constructs expressing HA-tagged proteins were individually transfected into COS cells. 40 h later, HA-tagged proteins were immunoprecipitated with an anti-HA antibody 12CA5 either alone or together after combining the lysates. After extensive washing with lysis buffer and kinase buffer, immunoprecipitates were incubated for 20 min at 30°C in 16 μl of kinase buffer containing 50 μM ATP, 5 μCi γ-[32P]ATP (4,500 Ci/mmol; MP Biomedicals), and 6 μg GST-cofilin or GST-Rac1. The reactions were terminated by adding 4× SDS sample buffer followed by SDS-PAGE, Coomassie staining, and autoradiography.
Online supplemental material
Fig. S1 shows the effects of Par-3, occludin, and E-cadherin KD as well as Par-3/E-cadherin double KD on phospho-cofilin levels in MDCK cells (A−C). Par-3/Tiam1 double KD does not reduce phospho-cofilin levels (D). It also shows that KD of cofilin alone does not disrupt tight junctions (E−G) and shows the effects of Tiam1 KD and cofilin S3A expression on TER establishment in Par-3–depleted MDCK cells (H). Fig. S2 shows that the kinase activity of LIMK2 is not required for its association with Par-3 (A) and that Par-3 does not associate with ROCK (B). It also shows that LIMK2 KD promotes TER development in Par-3–depleted MDCK cells (C) and the actin organization during calcium switch in control and Par-3 KD cells (D). Fig. S3 shows the elevated levels of Thr505 phosphorylation on LIMK2 in Par-3–depleted MDCK cells (A). It also shows that Par-3 inhibits LIMK2 but not LIMK1 kinase activity in vitro (B) and that Pals1 does not inhibit LIMK2 activity (C). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200510061/DC1.
Supplementary Material
Acknowledgments
We thank J.G. Collard, T. Nakamura, S. Narumiya, T. Uemura, and O. Bernard for plasmids.
This work was supported by grants GM070902 and CA40042 from the National Institutes of Health and Department of Health and Human Services.
Abbreviations used in this paper: aPKC, atypical PKC; HCM, high calcium medium; KD, knockdown; LCM, low calcium medium; LIMK, LIM kinase; LPA, lysophosphatidic acid; NRG, neuregulin; RNAi, RNA interference; ROCK, Rho-associated kinase; shRNA, short hairpin RNA; TER, transepithelial electrical resistance.
References
- Bamburg, J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230. [DOI] [PubMed] [Google Scholar]
- Chen, X., and I.G. Macara. 2005. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat. Cell Biol. 7:262–269. [DOI] [PubMed] [Google Scholar]
- Edwards, D.C., L.C. Sanders, G.M. Bokoch, and G.N. Gill. 1999. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1:253–259. [DOI] [PubMed] [Google Scholar]
- Foletta, V.C., M.A. Lim, J. Soosairajah, A.P. Kelly, E.G. Stanley, M. Shannon, W. He, S. Das, J. Massague, and O. Bernard. 2003. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol. 162:1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L., I.G. Macara, and G. Joberty. 2002. Multiple splice variants of Par3 and of a novel related gene, Par3L, produce proteins with different binding properties. Gene. 294:99–107. [DOI] [PubMed] [Google Scholar]
- Harris, T.J., and M. Peifer. 2005. The positioning and segregation of apical cues during epithelial polarity establishment in Drosophila. J. Cell Biol. 170:813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, A.I., I.C. McCall, C.A. Parkos, and A. Nusrat. 2004. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol. Biol. Cell. 15:2639–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joberty, G., C. Petersen, L. Gao, and I.G. Macara. 2000. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat. Cell Biol. 2:531–539. [DOI] [PubMed] [Google Scholar]
- Macara, I.G. 2004. Parsing the polarity code. Nat. Rev. Mol. Cell Biol. 5:220–231. [DOI] [PubMed] [Google Scholar]
- Matter, K., and M.S. Balda. 2003. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 4:225–236. [DOI] [PubMed] [Google Scholar]
- Mori, T., I. Okano, K. Mizuno, M. Tohyama, and A. Wanaka. 1997. Comparison of tissue distribution of two novel serine/threonine kinase genes containing the LIM motif (LIMK-1 and LIMK-2) in the developing rat. Brain Res. Mol. Brain Res. 45:247–254. [DOI] [PubMed] [Google Scholar]
- Munro, E.M. 2006. PAR proteins and the cytoskeleton: a marriage of equals. Curr. Opin. Cell Biol. 18:86–94. [DOI] [PubMed] [Google Scholar]
- Nagata-Ohashi, K., Y. Ohta, K. Goto, S. Chiba, R. Mori, M. Nishita, K. Ohashi, K. Kousaka, A. Iwamatsu, R. Niwa, et al. 2004. A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J. Cell Biol. 165:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, R., K. Nagata-Ohashi, M. Takeichi, K. Mizuno, and T. Uemura. 2002. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 108:233–246. [DOI] [PubMed] [Google Scholar]
- Okano, I., J. Hiraoka, H. Otera, K. Nunoue, K. Ohashi, S. Iwashita, M. Hirai, and K. Mizuno. 1995. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J. Biol. Chem. 270:31321–31330. [DOI] [PubMed] [Google Scholar]
- Roh, M.H., and B. Margolis. 2003. Composition and function of PDZ protein complexes during cell polarization. Am. J. Physiol. Renal Physiol. 285:F377–F387. [DOI] [PubMed] [Google Scholar]
- Sumi, T., K. Matsumoto, and T. Nakamura. 2001. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276:670–676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}