

Abstract
Conditional mouse models of polyglutamine diseases, such as Huntington's disease (HD), have revealed that cells can clear accumulated pathogenic proteins if the continuous production of the mutant transgene is halted. Invariably, the clearance of the protein leads to regression of the disease symptoms in mice. In light of these findings, it is critical to determine the pathway responsible for alleviating this protein accumulation to define targets to fight these diseases. In a functional genetic screen of HD, we found that activation of insulin receptor substrate-2, which mediates the signaling cascades of insulin and insulin-like growth factor 1, leads to a macroautophagy-mediated clearance of the accumulated proteins. The macroautophagy is triggered despite activation of Akt, mammalian target of rapamycin (mTOR), and S6 kinase, but still requires proteins previously implicated in macroautophagy, such as Beclin1 and hVps34. These findings indicate that the accumulation of mutant protein can lead to mTOR-independent macroautophagy and that lysosome-mediated degradation of accumulated protein differs from degradation under conditions of starvation.
Introduction
Polyglutamine (polyQ) disorders such as Huntington's disease (HD) are caused by a dominantly heritable expansion mutation of a triplet repeat in the coding region of the gene. The expression of this mutant protein leads to the onset of a slow, progressive disorder that invariably leads to death. Thus far, neither an effective treatment nor viable targets for drug design are available.
A prevalent feature of HD and other polyQ diseases is the accumulation and aggregation of the mutant protein. These changes lead to the formation of cytoplasmic and nuclear inclusion bodies, the appearance of which indisputably signifies the inability of the cell to properly dispose of the mutant protein. Indeed, overexpression of expanded polyQ proteins has been shown to alter proteasome (Bence et al., 2001) and lysosome function (Qin et al., 2003). Over the past several years, animal models of HD (Yamamoto et al., 2000; Regulier et al., 2003; Harper et al., 2005) and spinocerebellar ataxia type 1 (Xia et al., 2004; Zu et al., 2004) revealed that cells have the capacity to clear these products if the continuous production of the mutant transgene is halted. Invariably, clearance of the protein is accompanied by reversal of the disease-like symptoms in the mice. In light of these findings, it is critical to determine the pathway responsible for alleviating this protein accumulation to define targets to fight these diseases.
To determine the pathway responsible for the clearance of mutant huntingtin (htt), we conducted a two-tiered functional genetic screen. We first used gene arrays to quickly assess the transcriptional changes induced by pathogenic polyQ lengths. Although these changes alone can be somewhat informative, it is difficult to determine the functional relevance of these changes. Thus, we next targeted transcripts of genes that were “increased” with chemically synthesized small interfering RNAs (siRNAs) to determine which of these proteins were required for mutant htt clearance. Those specific proteins revealed by the second screen then became the focus of further investigation.
Of the 56 up-regulated transcripts, 23 were required for mutant htt clearance. Interestingly, the pattern of genes revealed that activation of insulin receptor substrate 2 (IRS-2), a scaffolding protein that mediates the signaling cascades of growth factors such as insulin and insulin-like growth factor 1 (IGF-1; White, 2003), leads to a macroautophagy-mediated clearance of the accumulated polyQ proteins. Clearance is present despite the activation of Akt, mammalian target of rapamycin (mTOR), and p70 S6 kinase. This is surprising because activation of mTOR is an inhibitor of the classic, starvation-induced macroautophagy (Meijer and Codogno, 2004). The significance of this is twofold: first, that macroautophagy in the presence of accumulated proteins can also occur in an mTOR-independent manner; and second, that this represents another important pathway through which factors such as insulin and IGF-1 may exert beneficial effects.
Results
Clearance of accumulated proteins in inducible cell lines
To determine if the clearance of mutant protein can be observed in stable cell lines, we designed a series of functional cell-based assays that were similar to the HD94 mouse model (Yamamoto et al., 2000). Cell lines inducibly express exon1 of htt (exon1htt) carrying a polyQ expansion of 25, 65, or 103 residues. Inducibility is conferred using the tet-off system (Gossen et al., 1994). To monitor the state of the proteins, and to ensure that aggregation was mediated primarily by the polyQ repeat, the COOH termini were fused to monomeric enhanced CFP (mCFP; Zacharias et al., 2002). To ensure that our siRNA-based screen can be conducted as efficiently as possible, we first focused on HeLa-based cell lines (Fig. 1). siRNA transfection efficiency in these cells reaches >80% (Elbashir et al., 2001; Pelkmans et al., 2005; unpublished data). To confirm our findings, however, we also used a neuronal background with Neuro2a cell lines (N2a; Fig. 9), which have been previously used to characterize different cellular aspects of HD (Wang et al., 1999; Jana et al., 2000, 2001).
Figure 1.

Stable cell lines with conditional expression of exon1htt-polyQmCFP. (a) Representative images of stable cell lines with conditional expression of exon1htt with 25QmCFP, 65QmCFP, and 103QmCFP. (b) Cell lines clear polyQmCFP inclusions within 6 d upon abolishing protein expression. Data represented as mean ± SEM. n = 6.
Figure 9.

IRS-2 activation-mediated autophagy in a conditional neuronal cell line. (a) N2a stable cell lines inducibly expressing exon1htt-25QmCFP and -103QmCFP. Nuclei are stained with Hoechst 33342. (b) Clearance of accumulated protein occurred in 6 d. Data represented as mean ± SEM. (c) IRS-2 and Beclin 1 are required for IGF-1–mediated clearance. ANOVA revealed a significant effect across groups (F(4,43) = 5.793; P = 0.0008) Fisher's protected least significant difference test for “%Control” revealed a significant difference between control and dox-treated cells (P = 0.0006) and control and IGF-1–treated cells (P = 0.0179). Coadministration of siRNA against beclin1 (P = 0.9611) or IRS-2 (P = 0.4712) with IGF-1 eliminated clearance. Data represented as mean ± SEM. Asterisks indicate statistical significance, as indicated. (d) Quantification of the mCFP-positive inclusions found in LC3-positive autophagosomes. ANOVA revealed a significant effect across group (F(3,16) = 3.873; P = 0.0294). Fisher's protected least significant difference test for “% cells with LC3-positive inclusions” revealed a significant difference between control and IGF1 (P = 0.0097), but no difference with siIRS-2 (P = 0.7881) or siBeclin1 (P = 0.8372). Data represented as mean ± SEM. Asterisks indicate statistical significance, as indicated.
The cell lines demonstrated a polyQ length–dependent increase of intracellular, predominantly cytoplasmic, inclusions (Fig. 1). After transient transfection, we observed acute, polyQ length–dependent cell death, as in other transiently transfected cell–based studies (Saudou et al., 1998; Kim et al., 1999; Waelter et al., 2001; Arrasate et al., 2004). After stable transfection, however, this was no longer observed across three independent cell lines during the duration of our experiment (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200510065/DC1). It is likely that the expression levels achieved were not high enough or that the time of expression was not long enough to elicit acute cell death (Jana et al., 2000). Thus, this assay is well suited to identify regulators of protein degradation because the absence of cell death will allow a clearer interpretation of the hits. Nonetheless, neuronal cell death is a critical aspect of HD and other polyQ diseases, and therefore should always be considered when integrating these findings in the context of the disease.
Next, we determined if the abolition of mutant exon1htt expression would lead to protein clearance. Inhibition of polyQ expression with 100 ng/ml doxycycline (dox) led to clearance of both the soluble and aggregated protein (Fig. 1). Similar to primary cultures derived from the HD94 mice (Martin-Aparicio et al., 2001), within 6 d the inclusions cleared (Fig. 1 and see Fig. 9). Thus, both nonneuronal and neuronal cell lines are capable of clearing the mutant forms of exon1htt. These findings indicate that the elimination of accumulated mutant exon1htt is very slow. Furthermore, because the amount of time required for clearance is similar across cell types, including primary neurons, the process underlying the elimination of this protein may be a general cellular event.
Continuous expression of mutant exon1 htt leads to transcriptional changes
To identify genes that are altered because of expression of mutant exon1htt, we tested our hypothesis that stable expression of 65Q or 103Q leads to transcriptional changes that reflect sequestration and elimination of inclusions. To examine these changes in an unbiased global manner, we determined the genetic profile of the cell lines using Affymetrix gene arrays. Comparisons between exon1htt-65QmCFP (65Q) and 25QmCFP clones (25Q) revealed a total of 70 transcripts increased and 89 transcripts decreased. Comparisons between 103QmCFP (103Q) and 25Q revealed 132 increased and 96 decreased (Fig. 2 a). Common across both pathogenic glutamine lengths was the 56 increased and 42 decreased (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200510065/DC1). Interestingly, increases were seen in only one proteasomal subunit (PSMD8), but in two lysosome-associated membrane proteins (LAMP1 and 2). The profile also revealed changes in vesicle trafficking, signaling proteins, metabolic proteins, and hypothetical proteins.
Figure 2.

Two-tiered screen reveals genes that are required for the clearance of accumulated mutant exon1htt. (a) Gene profiling identifies transcriptional changes that are attributable to polyQ expression. Three Hum133A chips were run per polyQ length. For each chip, mRNA from independent clones was isolated to avoid clonal variability. Cluster and comparison analyses reveal that expression of pathogenic polyQ proteins leads to distinct transcriptional changes. The dendogram (left) is of the genes that differed from 25QmCFP samples. (b) siRNA gene knockdown reveals genes required for clearance of accumulated mutant exon1htt. Three to four siRNA sequences were designed and individually transfected into 65QmCFP or 103QmCFP HeLa cell lines. Each bar represents an individual siRNA. Cells were transfected for 48 h and then given 100 ng/ml dox for 2 d. The accumulation index is the data normalized to scramble siRNA-transfected cells treated with dox. 23 genes with an accumulation index greater than two standard deviations from control (designated by the gray box) were considered genes required for clearance. SiRNAs against 23 genes, including Lamp1, Lamp2, and IRS-2, lead to increased accumulation. Data represented as mean ± SD. Each siRNA was transfected in eight wells per experiment, and each experiment was repeated five to eight times, as described in Materials and methods.
Functional consequence of siRNA-mediated knockdown of transcripts revealed from profiling
It is impossible to determine from profiling alone if the transcriptional changes have any functional relevance in our pathway. Therefore, we used RNA interference to functionally knockdown the transcripts that were increased in the genetic profile. We reasoned that if up-regulated proteins were relevant to our pathway, then their loss would inhibit clearance and the polyQ protein would continue to accumulate despite abolishing expression.
Eight EST transcripts were excluded from the siRNA screen. For the remaining 48 transcripts, three to four siRNA sequences per gene were individually transfected into two different 65Q and 103Q cell lines. 48 h after transfection, cells were exposed to 100 ng/ml dox for another 48 h to shut down production of new protein and permit 50% of clearance to occur. Cells were fixed and analyzed for the number of inclusions per cell using InCell Analyzer (INCA) 3000 software and calculated for an accumulation index, as described in Materials and methods. An example is shown in Fig. 3.
Figure 3.

IRS2 is required for clearance of mutant htt. (a) Representative images generated on the INCA 3000 of control (Ctrl; scramble + 2d dox), siRNA alone, and Dox + siIRS-2. (b) Quantification of percentage of control of the number of aggregates per cell for the three images. The presence of an effective siRNA against IRS-2 (si8H06) abolishes the clearance seen in the presence of dox. Complete quantitative results can be found in Fig 2.
Loss of function of 23 out of the 48 transcripts led to a complete or partial inhibition of clearance, of 9 led to cell death, and of 16 led to no change (Fig. 2). As predicted, genes involved in protein degradation were the most prevalent, including the lysosomal membrane proteins LAMP1 (Fig. 2 b, probes 3H5, 3H6, 3H7, and 3H8) and LAMP2 (Fig. 2 b, probes A7, A8, A9, and A10). LAMP2 has previously been shown as essential for lysosomal function in LAMP2 knockout mice. LAMP1, on the other hand, was not required; however, it was surmised that its function was redundant to LAMP2 (Andrejewski et al., 1999; Tanaka et al., 2000). The relatively acute loss of function offered by siRNA-mediated silencing may prevent the ability of such compensation to occur. Putative knockdown of the only proteasomal subunit that was altered in the gene arrays, PSMD8, led to cell death, and thus its role in clearance could not be elucidated.
IRS-2 is required for the elimination of mutant exon1htt aggregates
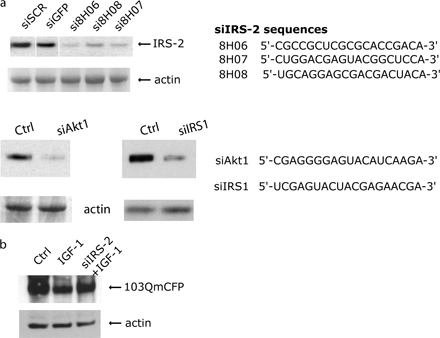
Unexpectedly, knockdown of IRS-2 led to an inhibition of aggregate clearance (Figs. 2 and 3). A scaffolding protein that transmits the phosphatidylinositol 3-kinase (PtdIns3K) signaling of growth factors like IGF-1 and cytokines, IRS-2 knockout mice have also revealed an important role in the brain (Schubert et al., 2003). Western blot analysis confirmed that siRNAs effectively down-regulates the protein (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200510065/DC1). Fig. 3 shows representative images taken from the INCA 3000 of one of the two 103QmCFP clones in the absence of dox, in the presence of dox, and in the presence of dox after transfection with siRNA number 8H06, which is one of the three siRNAs against IRS-2. Quantification using the INCA 3000 software of these images shows that, after transfection of 08H06, the clearance normally observed by abolishing transgene expression is inhibited (Fig. 3 b).
IRS-2 activation leads to enhanced exon1htt clearance
Because elimination of IRS-2 inhibited clearance, IRS-2 activation may stimulate clearance. Therefore, we tested a series of ligands known to activate IRS-2 in the following cells: insulin (Vassen et al., 1999), IGF-1 (Nakamura et al., 2000), and interleukin-4 (IL-4; Loh et al., 1992). All three cells demonstrate a dose-dependent clearance of accumulated polyQ proteins (Fig. 4 a). Unlike our normal clearance paradigm using dox, this clearance occurred despite maintaining continuous expression of exon1htt. As shown in Fig. 4 b, silencing IRS-2 led to a complete inhibition of the enhanced clearance triggered by IGF-1. Western blot analysis also found that IGF-1–mediated clearance is inhibited by small interfereing IRS2 (siIRS-2; Fig. S2). Silencing another IRS family member, IRS-1 (White, 2003), had no effect (Fig. 4 c and Fig. S1). Although this result is compelling, we cannot completely eliminate its role because up-regulation of IRS-2 may mask an effect. Similar dependence on IRS-2, but not -1, was observed with the enhanced protein degradation triggered by insulin and IL-4 (unpublished data).
Figure 4.

IRS-2 activation leads to clearance of accumulated mutant exon1htt. (a) Exon1htt-103QmCFP demonstrates a dose-dependent clearance of accumulated mutant protein after a 3-d treatment of insulin, IGF-1, and IL-4. Expression of the exon1htt-polyQmCFP was maintained throughout treatment. Exon1htt-65QmCFP clones exhibited similar effects (not depicted). (b) Two different siRNAs against hVps34 significantly inhibited clearance by IGF-1 (F(2,253) = 35.576; P < 0.0001) and insulin (not depicted). Direct application of 10 μM dipalmitoyl-PI3P liposomes for 3 d also led to fewer inclusion-positive cells (F(1,30) = 28.718; P < 0.0001). Data represented as mean ± SEM. Asterisks indicate statistical significance, as indicated. (c) Loss of IRS-2 (P = 0.0007) and a regulator of macroautophagy, Beclin1 (P < 0.0001), significantly inhibited IGF-1–mediated clearance. siRNA-mediated silencing of IRS1 (P = 0.3077) and Akt (P = 0.3792) had no effect (ANOVA revealed a significant effect of siRNA on percentage of control. F(5,186) = 4.907; P = 0.003). Data represented as mean ± SEM. Asterisks indicate statistical significance, as indicated.
IRS-2–mediated clearance requires hVps34 and Beclin 1
We next attempted to gain insight into the intracellular pathway by which IRS-2 activation triggers protein clearance. It has been previously shown that activation of IRS-2 can lead to the production of PtdIns[3,4,5]phosphate by turning on the class I PtdIns3K (Saltiel, 2001). However, it has also been shown that IRS-2 phosphorylation may lead to the production of PtdIns[3]phosphate (PI3P; Virbasius et al., 2001; Chaussade et al., 2003; Maffucci et al., 2003). These latter lipid products are predominantly formed by a class III PtdIns3K, called hVps34 (Schu et al., 1993; Backer, 2000).
We first determined whether the presence of PI3P leads to clearance. Synthetic dipalmitoyl-PI3P has been effectively delivered into the cell using liposomes (Franke et al., 1997; Petiot et al., 2000). Administration of these liposomes to the exon1htt-65Q or 103QmCFP cell lines for 3 d led to a significant decrease in the number of inclusions per cell (Fig. 4 c and not depicted). Coadministration of PI3P with IGF-1 or insulin did not significantly enhance this effect, suggesting that they lie in the same pathway (unpublished data). siRNA against hVps34 effectively eliminated the exon1htt clearance caused by IRS-2 activation (Fig. 4 c). Interestingly, similar to IRS-2, the loss of hVps34 also eliminated the clearance revealed by eliminating transgene expression with dox (unpublished data).
The requirement of hVps34, as well as LAMP1 and 2 (Fig. 2), suggests that macroautophagy is being triggered. Macroautophagy, which we will henceforth refer to as autophagy, is a means by which long-lived cytoplasmic proteins are degraded by the lysosome via engulfment and encasement by a multilamellar structure and fusion to the lysosome (Klionsky and Emr, 2000). Indeed, several studies have previously indicated that the induction of autophagy can lead to the clearance of aggregated polyQ proteins (Ravikumar et al., 2002; Iwata et al., 2005a), especially cytoplasmic ones (Iwata et al., 2005b). It has been previously shown that the inhibition of autophagy elicited by 3-methyladenine (3-MA) is caused by the inhibition of hVps34 (Seglen and Gordon, 1982; Blommaart et al., 1997; Petiot et al., 2000). hVps34 acts in a multiprotein complex that includes p150 and the mammalian orthologue of Apg6, Beclin1 (Liang et al., 1999; Kihara et al., 2001; Tassa et al., 2003). As seen in Fig. 4 b, silencing of Beclin1 abolished the clearance stimulated by IRS-2 activation. To determine if this effect is downstream of class I PtdIns3K activation, we next knocked down the serine/threonine kinase Akt. Akt is a survival kinase that was previously shown to be protective in several diseases, such as amyotrophic lateral sclerosis (ALS) and HD (Humbert et al., 2002; Kaspar et al., 2003; Kanekura et al., 2005; Rangone et al., 2005). Although this siRNA against Akt efficiently knocks down the protein (Fig. S2; Jiang et al., 2003), no effect was seen on the IRS-2–mediated clearance (Fig. 4 b). It has previously been shown that Akt phosphorylation of serine 421 on htt leads to fewer aggregates (Humbert et al., 2002; Rangone et al., 2005). Our cell lines express only exon1htt, and thus this residue is not expressed. Perhaps monomeric htt is degraded in an Akt- and proteasome-dependent manner. It is plausible that once an inclusion forms, a different method of protein degradation is required.
IRS-2 activation leads to the activation of autophagy
To further ascertain that IRS-2 activation potentiates autophagy-mediated elimination of the accumulated protein, we examined the effect of established inhibitors of lysosomal degradation and macroautophagy on clearance. As shown in Fig. 5 a, lysosomal inhibitors inhibit the ability of the cell to eliminate the inclusions. 3-MA and wortmannin also eliminated clearance. These findings further demonstrate the importance of lysosomal function in exon1htt clearance and imply a role in autophagy.
Figure 5.

Functional lysosomes are required for the clearance of mutant htt inclusions. (a) Clearance of exon1polyQhtt is inhibited in the presence of 1 μg/mL leupeptin (Leup), 100 nM bafilomycin A1 (Baf A1), 20 nM hydroxychloroquine (Chlor), 10 mM 3-methyladenine (3-MA), and 100 nM wortmannin (Wort). All compounds inhibited the clearance usually observed after 48 h dox, and thus are similar to No dox–treated cells (No dox). Data represented as mean ± SEM. (b) Costaining cells with 65 nM of Lysotracker reveals that inclusions colocalize with acidified compartments.
Next, we examined if the inclusions colocalized with autophagosomes or lysosomes. Administration of 65 nM of Lysotracker, a red fluorescent dye that accumulates in acidic organelles, showed that mCFP-positive inclusions were in acidified compartments (Fig. 5 b). Using the autophagosome marker LC3 (Kabeya et al., 2000; Mizushima et al., 2004) and serial Z-sections, we found mCFP-positive inclusions surrounded by an LC3-positive structure (Fig. 6 and Fig. 7 a). Although most autophagosomes are known to be much smaller than the inclusion, under certain conditions, such as bacterial invasion, large autophagosomes have been shown to occur (Nakagawa et al., 2004). Furthermore, in a cellular model of another polyQ disease, spinal and bulbar muscular atrophy, Taylor et al. (2003) found polyQ inclusion bodies bound by double-membrane structures that were also quite large. Quantifications revealed that ∼10% of the inclusion-positive cells had inclusions within LC3-positive vacuoles (Fig. 7 b). Consistent with the enhanced clearance, activation of IRS-2 using IGF-1 significantly increased the frequency of colocalization. siRNA-mediated silencing of IRS-2 and Beclin1 abolished this increase. Silencing Akt or IRS-1, again, had no effect.
Figure 6.

Exon1htt-polyQmCFP htt inclusions can be found engulfed by an LC3-positive vesicle. Z-sectioned image of an Exon1htt-65QmCFP cell cotransfected with eYFP-LC3. Bar, 100 μM.
Figure 7.

IRS-2 activation leads to macroautophagy-mediated clearance of mutant htt inclusions. (a) Inclusions also colocalize with LC3-positive autophagosomes. Treatment with IGF-1 leads to an increase in the frequency of the colocalization of the mCFP-positive inclusions, whereas knockdown of Beclin1 eliminates this colocalization. (b) Quantification of the percentage of inclusions found in LC3-positive autophagosomes. Administration of IGF-1 leads to an increased frequency of colocalization versus control. P < 0.0001. Loss of Akt had no effect on this outcome. P = 0.0003. Loss of IRS-2 (P = 0.3232) and Beclin1 (P = 0.4281) eliminated this increase. (ANOVA revealed a significant effect of siRNA (F(3,24) = 4.183; P = 0.0161) and treatment (F(1,24) = 29.151; P < 0.0001) on the percentage of cells with LC3-positive inclusions. There was also a significant interaction between siRNA and treatment (F(3,24) = 6.524; P = 0.0022). Data represented as mean ± SEM. Asterisks indicate statistical significance, as indicated.
Class I PtdIns3K pathway is still active in the mutant exon1htt cell lines
Growth factors like insulin are classic signaling molecules that inform the cell of the presence of nutrients. From yeast to mammalian cells, these factors generate PtdIns[3,4,5]phosphate. This leads to a signaling cascade that activates Akt, mTOR, and the translation activator p70S6K (Jacinto and Hall, 2003). A known consequence of this pathway is an inhibition of macroautophagy, which is a catabolic process. This is contrary to our results, in which the same receptor activates degradation of mutant exon1htt inclusions by autophagy. To ensure that our findings are not caused by a disturbance of normal signaling processes, we reexamined the effect of insulin and IGF-1 on both the proper phosphorylation of known downstream kinases and on autophagy caused by amino acid deprivation.
Whole cell lysates were collected from 65QmCFP- or 103QmCFP-expressing cells after exposure to IGF-1 or insulin for 30 min (Fig. 8 b). Phosphospecific antibodies against Akt and p70S6K revealed that, indeed, the targets were rapidly activated in these clones. Therefore, we concluded that the classical autophagic response is intact in our cell lines. Recent work by Scott et al. (2004), however, found that long-term activation of p70S6K can also induce autophagy. To ensure that mTOR signaling is intact in the cell lines, phosphospecific antibodies against mTOR were also used. Phosphorylated mTOR was readily detectable in the presence of mutant exon1htt expression, as well as in the presence or absence of mutant htt expression (Fig. 8 a and not depicted). Amino acid deprivation and rapamycin decreased mTOR phosphorylation in these cells, whereas insulin and IGF-1 inhibited the effect of amino acid withdrawal. Thus, the IGF-1 and insulin lead to the predictive phosphorylation of p70S6K and mTOR.
Figure 8.

Macroautophagy caused by amino acid withdrawal is unchanged in mutant exon1htt-polyQ cell lines. (a) In the presence of mutant exon1htt expression, Akt and p70S6 kinase (S6K) are phosphorylated upon stimulation with IGF-1 and Ins for 30 min. In the presence of mutant exon1, mTOR is phosphorylated and decreases after amino acid withdrawal (-aa; 4 h) or 20 nM rapamycin (1 h). IGF-1 and Ins inhibit the effect of amino acid withdrawal. (b) [14C]valine-labeled long-lived protein degradation in response to 4 h amino acid withdrawal. ANOVA revealed a significant effect of treatment on the percentage of proteolysis (F(5,19) = 7.160; P = 0.0006). 10 μM PI3P-containing liposomes also increased proteolysis in the presence of full serum, as previously shown (P = 0.0010). 100 nM Ins (P = 0.8763) and 10 nM IGF-1 (P = 0.3999) inhibited the enhanced proteolysis caused by amino acid withdrawal (P = 0.0016), as did 10 mM 3-MA (P = 0.3074). Data represented as mean ± SEM.
We next examined proteolysis of long-lived proteins. Proteolysis was measured by [14C]valine-labeled long-lived proteins. 103QmCFP cell lines were under dox suppression for 2 wk to ensure no transgene expression because the presence of these proteins led to high levels of baseline protein degradation, despite the presence of full serum. Insulin and IGF-1 significantly attenuated the amount of proteolysis, whereas PI3P induced proteolysis despite the presence of complete media. Furthermore, the administration of 3-MA also diminished degradation (Fig. 8 b). Thus, the autophagy-mediated clearance of inclusions by IGF-1 and Ins occurs despite proper signaling by mTOR.
IRS-2 activates autophagy in inducible neuronal mutant exon1htt cell lines
IRS-2 is expressed in all insulin-responsive organs, including the brain. We examined if IRS-2 could activate macroautophagy in response to protein accumulation in neuronal cell lines (Fig. 9). Similar to the HeLa cells, elimination of novel polyQ protein production led to clearance over a period of 6 d. We stimulated IRS-2 using IGF-1 to determine if a similar mechanism was at play. IRS-2 activation using IGF-1 also led to an autophagy-mediated clearance of polyQ proteins in cells of a neuronal lineage. The clearance was accompanied by an increased colocalization of mCFP-positive inclusions in YFP-LC3 autophagosomes. Again, knockdown of Beclin1 inhibited the colocalization of inclusions in the autophagosomes.
Discussion
Using a unique two-tiered functional genetic screen, this study revealed an unexpected means by which autophagy-mediated clearance of accumulated mutant protein can be activated. We found that the activation of IRS-2 led to macroautophagy-induced clearance of the accumulated polyQ proteins (Fig. 10). The activation was dependent on class III PtdIns3K activation and occurred despite activation of Akt, mTOR, and p70S6K.
Figure 10.

Working model of IRS-2-mediated activation of autophagy. IRS-2 activation leads to the formation of PI3P via hVps34. Beclin1 and hVps34 allow for the autophagosomes to form and engulf the aggregates. As other studies indicate (gray box) aggregate formation alone may trigger autophagy via a yet unknown pathway, but mTOR inhibition or p70S6 kinase activation (Scott et al., 2004) may play a role.
These findings highlight several points. The first is that activation of IRS-2 can lead to the clearance of accumulated mutant exon1htt. IRS-2 is widely expressed and, together with IRS-1, mediates the signaling of insulin and IGF-1 in most tissue (Sun et al., 1992, 1995). White (2003) found that loss of IRS-2 function in knockout mice led to an accumulation of neurofibrillary tangles containing phosphorylated tau in the absence of changes in the kinase glycogen synthase kinase 3β. In light of our findings, it is possible that the loss of IRS-2 exposed a potential role for these proteins to mediate clearance of these complex proteins. It would be interesting to determine if protein accumulation is occurring in other tissues that are IRS-2 deficient.
Activators of IRS-2, such as insulin and IGF-1, have both been shown to strongly promote neuronal survival through stimulation of Akt. Consequently, its efficacy has been tested as such in other neurodegenerative diseases, such as ALS. For example, retroviral delivery of IGF-1 in a mouse model of ALS led to amelioration of the phenotype, together with a diminishing of the accumulated mutant SOD1 (Kaspar et al., 2003). A placebo-controlled trial in American ALS patients found that the progression of functional impairment significantly slowed in the treated patients, with no adverse side effects (Lai et al., 1997). The outcome suggested an IGF-1 dose-dependent treatment effect. For HD, Humbert et al. (2002) have examined the neuroprotective effect of Akt stimulation by transiently transfecting mutant htt into primary neurons. Similar to the findings in ALS, they found that IGF-1 administration led to both a decrease in the number of aggregates formed and a decrease in cell death. The mechanism through which IGF-1 elicited both effects was believed to be directly downstream of Akt (Rangone et al., 2004, 2005). We were able to determine that the autophagocytic clearance can also occur independent of Akt because its knockdown did not eliminate clearance. Monomeric and aggregated proteins may be degraded differently. These findings indicate that the protection conferred by the insulin signaling pathway in diseases with protein accumulation may be twofold; the classical neuroprotective pathway triggered by Akt and the enhanced clearance stimulated by hVps34 activation. Because cytoplasmic inclusions are more readily degraded by autophagy, it is now critical to determine to what extent these inclusions contribute to pathology.
Another point highlighted by this study is that macroautophagy is indeed capable of degrading large inclusions and is stimulated under conditions previously deemed inhibitory. Activation of IRS-2 in the HeLa cell lines was achieved by using insulin, IGF-1, or IL-4. Signaling through these receptors, however, is also known to inhibit mTOR-mediated autophagy through the activation of class I PtdIns3K and mTOR. Nonetheless, there is evidence that autophagy may occur despite mTOR activation. For example, transgenic expression of the autophagosomal marker LC3 demonstrates that in certain tissue autophagosomes constitutively form, even in the absence of starvation (Mizushima et al., 2004). Consistent with these findings, immortalized cells of certain lineages also have a higher basal activity of macroautophagy (Mizushima, 2004). More recently, Iwata et al. (2005a) found that proteasome inhibition was a potent activator of autophagy, although the role of mTOR in the response was not explored. In this study, our findings indicate that macroautophagy is the mechanism by which the aggregates are cleared, despite mTOR activation. This could be achieved if the autophagy regulators downstream of mTOR, such as hVps34, could be activated. Therefore, the activity observed in our results may represent a competition between the inhibitory effects of activating class I versus III kinases. Moreover, Scott et al. (2004) clearly demonstrated in the Drosophila melanogaster fat body that the regulation of autophagy may occur differently from what was previously believed. They found that constitutive activation of p70S6K was indeed required for autophagy, rather than inhibitory as previously described (Klionsky et al., 2005). The presence of expanded polyQ proteins may offset the balance between p70S6K and mTOR, thus, allowing for IRS-2 activation to perpetuate a signal to drive autophagy.
Another possibility is that inclusion formation itself triggers autophagy (Taylor et al., 2003) in an mTOR-dependent fashion, and IRS-2 activation perpetuates this response downstream of mTOR regulation. Indeed, a previous study showed that inclusions can sequester mTOR and, thus, could activate autophagy (Ravikumar et al., 2004). However, in our cell lines, we did not find mTOR sequestration. Nonetheless, differentiation between mTOR dependence and independence is difficult because the activation of IRS-2 is upstream of both the inhibitory and stimulatory signaling. To test this hypothesis, we must first understand how mTOR negatively regulates autophagy in mammalian cells. This is not yet fully defined. In yeast, TOR has been shown to negatively regulate the activation of the Apg13–Apg1 complex, a kinase required for autophagy, but it is not certain if this is attributable to direct phosphorylation by TOR (Kamada et al., 2000). Furthermore, in mammalian cells, the orthologues of neither Apg13 nor Apg1 have yet to be identified. mTOR-dependent autophagy has mainly been confirmed in mammalian systems through the use of chemical inhibitors such as rapamycin. Chemical compounds that directly activate mTOR are not available.
In conclusion, by starting with a nonhypothesis-driven approach, we were able to discover that the regulation underlying degradation of accumulated proteins differs from the regulation underlying conditions of starvation. Furthermore, this work demonstrates that we can no longer assume that regulatory mechanisms studied under nonpathogenic conditions are static; cells compensate when confronted with toxic conditions. Accordingly, we have found that the insulin-signaling pathway may be an important avenue through which this might be achieved. Indeed, activation of IRS-2 has been an attractive target for the treatment of type II diabetes, and thus this line of research may also benefit other disorders. In any case, it is clear from these studies that more information regarding the importance of lysosomal degradation pathways such as autophagy in neuronal systems is absolutely crucial. Controlling these pathways that degrade mutant htt will allow us to finally begin to treat this terrible disease.
Materials and methods
Materials
N2a cells were purchased from American Type Culture Collection. Insulin, IGF-1, IL-4, and dox were purchased from Sigma-Aldrich. Antibodies were purchased from Upstate Biotechnology (anti–IRS-2, anti–phospho Akt, and anti–phospho S6 kinase), BD Biosciences (Akt, S6 kinase, and IRS-1), Cell Signaling Technology (anti–phospho-mTOR and anti-mTOR), and Roche (anti-GFP). htt-exon1 (CAGCAA) constructs were obtained from A. Kazantsev (Massachusets General Hospital, Charlestown, MA). pYFP-LC3 was obtained from T. Yoshimori (National Institute of Genetics, Mishima, Shizuoka, Japan).
Creation of cell line
N2a were selected to be tTA-positive by transfection with PCMV-tTA-IRES-neo and selection with 800 μg/ml G418. PolyQ cell lines were created by cotransfecting Hela and N2a with tetO-htt (25Q, 65Q, or 103Q) exon1-mCFP and PTk-hygro (CLONTECH Laboratories, Inc.) and then selected with hygromycin using 800 and 200 μg/ml, respectively. 100 ng/ml dox was also maintained in the culture media during selection to maintain suppression of transgene expression. HeLa cells were maintained in DME with 10% FCS, whereas N2a cells were maintained in 50% DME/50% Optimem in 10% FCS.
RNA preparation and gene arrays
Cells were plated in a 100-mm dish, harvested using 100 μL TRIzol reagent (Invitrogen) and isolated per the manufacturer's instructions. RNA was resuspended and further purified using the RNAeasy kit (QIAGEN). RNA was labeled and hybridized onto human U133A chips at the Genome Core Facility at Memorial Sloan Kettering Cancer Center (MSKCC). Array results were analyzed using GeneSpring 2.0 (Agilent Technologies) and an Affymetrix software package 5.0.
Transfection
siRNAs were designed using an algorithm designed by Jagla et al. (2005). siRNA were created at either Integrated DNA Technologies or the Functional Proteonomics Project at MSKCC. Scramble siRNA sequences were purchased from Dharmacon. A final concentration of 10 or 20 nM of siRNAs was used for silencing. Cells were transfected using OligofectAMINE per the manufacturer's instructions. 7.5 × 103 cells were plated in 96-well ViewPlates (Packard Instrument Co.) and transfected the next day. 48 h after transfection, cells were split across two wells and treated with dox. Cells were examined 48 h later. Transfection of plasmid DNA was accomplished using LipofectAMINE per the manufacturer's instructions. Compounds were administered 48 h after transfection unless otherwise noted.
Image analysis with the INCA 3000
For the high throughput screen, images were collected on the INCA 3000 (GE Healthcare). Cells in 96-well ViewPlates were fixed for 10 min with 4% PFA, and the nuclei were stained using 1 μM Hoechst 333342 for 30 min. After scanning the plates, images were analyzed using the granularity analysis module on the accompanying software. In brief, the granularity analysis quantifies the number of inclusions (grains) within a cell, using a two-color strategy to identify individual cells and to analyze associated grains. After recognizing the objects, in this case the Hoechst-positive nuclei, the algorithm next identifies, using a specified size range (in pixels) and fluorescent intensity gradient, the grain in the proximity of the object. Both the Ngrains (number of qualifying grains per cell) and the fraction of fluorescent within the qualifying grains gave similar results. To calculate the accumulation index used in Fig. 2, values were first normalized as a percentage of control for scramble siRNA–transfected cells treated with 2 d of 100 ng/ml dox, and set to 0 by subtracting 100%. This would permit comparison across all of the experiments conducted and a quick assessment of the direction of the change—an increase in accumulation (>0) or an increase in clearance (<0). A gene was considered required for clearance when the absolute value of accumulation index was greater than two standard deviations of the Scramble siRNA + 2 d dox. Each siRNA was transfected on 8 wells per 96-well plate, and each experiment was repeated five to eight times. Cell viability was determined through several measures. First, we examined the number of cells per well scanned. If a drug or siRNA led to significantly fewer cells, they were initially considered toxic. If this toxicity consistently appeared across experiments, we next confirmed toxicity using an inclusion/exclusion assay of cell death, known as a LIVE/DEAD assay (Invitrogen).
Other image analysis
Cells were grown on glass (HeLa) or poly-d-lysine–coated (N2a) coverslips in 24-well plates. Cells were fixed for 10 min with 4% PFA. Nuclei were stained with Hoechst 33342 for 30 min, and membranes were stained using Alexa Fluor 633–labeled cholera toxin subunit B obtained from Invitrogen. Images were acquired using a confocal microscope (TCS SP2; Leica) at 63× magnification, along with the accompanying software package. Data acquisition was performed using National Institutes of Health Image 4.0. Analysis of variance (ANOVA) and post hoc analyses were conducted using Statview 5.0 (SAS Institute, Inc.).
PI3P liposomes
Synthetic dipalymitoyl-PI3Ps (Matreya) were dried together with phosphatidylserine at a 1:1 concentration under argon and vacuum and resuspended in 25 mM Hepes, 100 mM KCl, and 1 mM EDTA to a total lipid concentration of 800 μM. Liposomes were freeze-thawed, then manually extruded through two 50-nm polycarbonate membranes. PI3P/phosphatidylserine liposomes were administered to cells at 20 μM for 3 d, fixed, and assessed for clearance.
Measuring degradation of long-lived proteins in response to amino acid deprivation
This protocol was adapted from previously reported protocols (Talloczy et al., 2002; Scarlatti et al., 2004). Cells were incubated for 18 h at 37°C with 0.5 μCi/ml L-[14C]valine–supplemented media. Cells were rinsed with HBSS to remove unincorporated radioisotopes and then chased in fresh media overnight to allow degradation of short-lived proteins. Cells were rinsed in HBSS + 10 mM Hepes and incubated for 4 h with either full media ± rapamycin, 20 mM PI3P liposomes, or HBSS + 10 mM Hepes ± 3-MA,20 μM PI3P liposomes, 100 nM insulin, or 100 nM IGF-1. Cells were scraped and, using TCA, protein was precipitated from both the incubation media and the cells. Proteolysis was assessed as the acid-soluble radioactivity divided by the radioactivity maintained in the precipitate.
Online supplemental material
Fig. S1 shows cell lines that were monitored for cell death using the LIVE/DEAD assay. Fig. S2 shows whole cell lysates from transfected cells that were run on SDS-PAGE gels, transferred to PVDF membranes, and probed with the antibodies as noted. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200510065/DC1.
Supplementary Material
Acknowledgments
We acknowledge the generosity of Dr. Alexei Kazantsev for the htt-exon1(CAGCAA) constructs, Dr. Tamotsu Yoshimori for pYFP-LC3, and the group of Dr. Agnes Viale at the Memorial Sloan Kettering Genome Core Laboratory for the Affymetrix gene arrays. Special thanks to Dr. Tom Melia for critical reading of this manuscript; Drs. Tom Neufield, Matthew Beard, and David Tareste for helpful discussion; Drs. Bernd Jagla, Ouathek Ouerfelli, Andrzej Zatorski, and their respective groups for the design and creation of the siRNAs; and Drs. Nathalie Aulner and Peter D. Kelly for assistance.
We thank the Mathers Foundation (J.E. Rothman), the Helen Hay Whitney Foundation (A. Yamamoto), and the High Q. Foundation and the Hereditary Disease Foundation for their generous support.
Abbreviations used in this paper: ALS, amyotrophic lateral sclerosis; ANOVA, analysis of variance; dox, doxycycline; HD, Huntington's disease; htt, huntingtin protein mutant; IGF-1, insulin-like growth factor 1; IL, interleukin; INCA, InCell Analyzer; IRS, insulin receptor substrate; LAMP, lysosome-associated membrane protein; MA, methyladenine; mCFP, monomeric enhanced CFP (L221K mutation); mTOR, mammalian target of rapamycin; N2a, Neuro2a cell lines; P70S6K, p70 s6 kinase; PI3P, PtdIns[3]phosphate; polyQ, polyglutamine; PtdIns3K, phosphatidylinositol 3-kinase; siIRS, small interfering IRS; siRNA, small interfering RNA.
References
- Andrejewski, N., E.L. Punnonen, G. Guhde, Y. Tanaka, R. Lullmann-Rauch, D. Hartmann, K. von Figura, and P. Saftig. 1999. Normal lysosomal morphology and function in LAMP-1-deficient mice. J. Biol. Chem. 274:12692–12701. [DOI] [PubMed] [Google Scholar]
- Arrasate, M., S. Mitra, E.S. Schweitzer, M.R. Segal, and S. Finkbeiner. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 431:805–810. [DOI] [PubMed] [Google Scholar]
- Backer, J.M. 2000. Phosphoinositide 3-kinases and the regulation of vesicular trafficking. Mol. Cell Biol. Res. Commun. 3:193–204. [DOI] [PubMed] [Google Scholar]
- Bence, N.F., R.M. Sampat, and R.R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 292:1552–1555. [DOI] [PubMed] [Google Scholar]
- Blommaart, E.F., U. Krause, J.P. Schellens, H. Vreeling-Sindelarova, and A.J. Meijer. 1997. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243:240–246. [DOI] [PubMed] [Google Scholar]
- Chaussade, C., L. Pirola, S. Bonnafous, F. Blondeau, S. Brenz-Verca, H. Tronchere, F. Portis, S. Rusconi, B. Payrastre, J. Laporte, and E. Van Obberghen. 2003. Expression of myotubularin by an adenoviral vector demonstrates its function as a phosphatidylinositol 3-phosphate [PtdIns(3)P] phosphatase in muscle cell lines: involvement of PtdIns(3)P in insulin-stimulated glucose transport. Mol. Endocrinol. 17:2448–2460. [DOI] [PubMed] [Google Scholar]
- Elbashir, S.M., J. Harborth, W. Lendeckel, A. Yalcin, K. Weber, and T. Tuschl. 2001. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 411:494–498. [DOI] [PubMed] [Google Scholar]
- Franke, T.F., D.R. Kaplan, L.C. Cantley, and A. Toker. 1997. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 275:665–668. [DOI] [PubMed] [Google Scholar]
- Gossen, M., A.L. Bonin, S. Freundlieb, and H. Bujard. 1994. Inducible gene expression systems for higher eukaryotic cells. Curr. Opin. Biotechnol. 5:516–520. [DOI] [PubMed] [Google Scholar]
- Harper, S.Q., P.D. Staber, X. He, S.L. Eliason, I.H. Martins, Q. Mao, L. Yang, R.M. Kotin, H.L. Paulson, and B.L. Davidson. 2005. From the cover: RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc. Natl. Acad. Sci. USA. 102:5820–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert, S., E.A. Bryson, F.P. Cordelieres, N.C. Connors, S.R. Datta, S. Finkbeiner, M.E. Greenberg, and F. Saudou. 2002. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves huntingtin phosphorylation by Akt. Dev. Cell. 2:831–837. [DOI] [PubMed] [Google Scholar]
- Iwata, A., J.C. Christianson, M. Bucci, L.M. Ellerby, N. Nukina, L.S. Forno, and R.R. Kopito. 2005. a. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA. 102:13135–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, A., B.E. Riley, J.A. Johnston, and R.R. Kopito. 2005. b. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280:40282–40292. [DOI] [PubMed] [Google Scholar]
- Jacinto, E., and M.N. Hall. 2003. Tor signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol. 4:117–126. [DOI] [PubMed] [Google Scholar]
- Jagla, B., N. Aulner, P.D. Kelly, D. Song, A. Volchuk, A. Zatorski, D. Shum, T. Mayer, D.A. De Angelis, O. Ouerfelli, et al. 2005. Sequence characteristics of functional siRNAs. RNA. 11:864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana, N.R., M. Tanaka, G. Wang, and N. Nukina. 2000. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet. 9:2009–2018. [DOI] [PubMed] [Google Scholar]
- Jana, N.R., E.A. Zemskov, G. Wang, and N. Nukina. 2001. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum. Mol. Genet. 10:1049–1059. [DOI] [PubMed] [Google Scholar]
- Jiang, Z.Y., Q.L. Zhou, K.A. Coleman, M. Chouinard, Q. Boese, and M.P. Czech. 2003. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA. 100:7569–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya, Y., N. Mizushima, T. Ueno, A. Yamamoto, T. Kirisako, T. Noda, E. Kominami, Y. Ohsumi, and T. Yoshimori. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada, Y., T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi, and Y. Ohsumi. 2000. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150:1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura, K., Y. Hashimoto, Y. Kita, J. Sasabe, S. Aiso, I. Nishimoto, and M. Matsuoka. 2005. A Rac1/phosphatidylinositol 3-kinase/Akt3 anti-apoptotic pathway, triggered by AlsinLF, the product of the ALS2 gene, antagonizes Cu/Zn-superoxide dismutase (SOD1) mutant-induced motoneuronal cell death. J. Biol. Chem. 280:4532–4543. [DOI] [PubMed] [Google Scholar]
- Kaspar, B.K., J. Llado, N. Sherkat, J.D. Rothstein, and F.H. Gage. 2003. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 301:839–842. [DOI] [PubMed] [Google Scholar]
- Kihara, A., Y. Kabeya, Y. Ohsumi, and T. Yoshimori. 2001. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M., H.S. Lee, G. LaForet, C. McIntyre, E.J. Martin, P. Chang, T.W. Kim, M. Williams, P.H. Reddy, D. Tagle et al. 1999. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J. Neurosci. 19:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., and S.D. Emr. 2000. Autophagy as a regulated pathway of cellular degradation. Science. 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky, D.J., A.J. Meijer, P. Codogno, T.P. Neufeld, and R.C. Scott. 2005. Autophagy and p70S6 Kinase. Autophagy. 1:59–61. [DOI] [PubMed] [Google Scholar]
- Lai, E.C., K.J. Felice, B.W. Festoff, M.J. Gawel, D.F. Gelinas, R. Kratz, M.F. Murphy, H.M. Natter, F.H. Norris, and S.A. Rudnicki. 1997. Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology. 49:1621–1630. [DOI] [PubMed] [Google Scholar]
- Liang, X.H., S. Jackson, M. Seaman, K. Brown, B. Kempkes, H. Hibshoosh, and B. Levine. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. [DOI] [PubMed] [Google Scholar]
- Loh, J.E., C.H. Chang, W.L. Fodor, and R.A. Flavell. 1992. Dissection of the interferon gamma-MHC class II signal transduction pathway reveals that type I and type II interferon systems share common signalling component(s). EMBO J. 11:1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci, T., A. Brancaccio, E. Piccolo, R.C. Stein, and M. Falasca. 2003. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 22:4178–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Aparicio, E., A. Yamamoto, F. Hernandez, R. Hen, J. Avila, and J.J. Lucas. 2001. Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J. Neurosci. 21:8772–8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer, A.J., and P. Codogno. 2004. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 36:2445–2462. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. 2004. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 36:2491–2502. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., A. Yamamoto, M. Matsui, T. Yoshimori, and Y. Ohsumi. 2004. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, I., A. Amano, N. Mizushima, A. Yamamoto, H. Yamaguchi, T. Kamimoto, A. Nara, J. Funao, M. Nakata, K. Tsuda, et al. 2004. Autophagy defends cells against invading group A Streptococcus. Science. 306:1037–1040. [DOI] [PubMed] [Google Scholar]
- Nakamura, K., A. Hongo, J. Kodama, Y. Miyagi, M. Yoshinouchi, and T. Kudo. 2000. Down-regulation of the insulin-like growth factor I receptor by antisense RNA can reverse the transformed phenotype of human cervical cancer cell lines. Cancer Res. 60:760–765. [PubMed] [Google Scholar]
- Pelkmans, L., E. Fava, H. Grabner, M. Hannus, B. Habermann, E. Krausz, and M. Zerial. 2005. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature. 436:78–86. [DOI] [PubMed] [Google Scholar]
- Petiot, A., E. Ogier-Denis, E.F. Blommaart, A.J. Meijer, and P. Codogno. 2000. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275:992–998. [DOI] [PubMed] [Google Scholar]
- Qin, Z.H., Y. Wang, K.B. Kegel, A. Kazantsev, B.L. Apostol, L.M. Thompson, J. Yoder, N. Aronin, and M. DiFiglia. 2003. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 12:3231–3244. [DOI] [PubMed] [Google Scholar]
- Rangone, H., G. Poizat, J. Troncoso, C.A. Ross, M.E. MacDonald, F. Saudou, and S. Humbert. 2004. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 19:273–279. [DOI] [PubMed] [Google Scholar]
- Rangone, H., R. Pardo, E. Colin, J.A. Girault, F. Saudou, and S. Humbert. 2005. Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J. Biol. Chem. 280:22021–22028. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., R. Duden, and D.C. Rubinsztein. 2002. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11:1107–1117. [DOI] [PubMed] [Google Scholar]
- Ravikumar, B., C. Vacher, Z. Berger, J.E. Davies, S. Luo, L.G. Oroz, F. Scaravilli, D.F. Easton, R. Duden, C.J. O'Kane, and D.C. Rubinsztein. 2004. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36:585–595. [DOI] [PubMed] [Google Scholar]
- Regulier, E., Y. Trottier, V. Perrin, P. Aebischer, and N. Deglon. 2003. Early and reversible neuropathology induced by tetracycline-regulated lentiviral overexpression of mutant huntingtin in rat striatum. Hum. Mol. Genet. 12:2827–2836. [DOI] [PubMed] [Google Scholar]
- Saltiel, A.R. 2001. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell. 104:517–529. [DOI] [PubMed] [Google Scholar]
- Saudou, F., S. Finkbeiner, D. Devys, and M.E. Greenberg. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 95:55–66. [DOI] [PubMed] [Google Scholar]
- Scarlatti, F., C. Bauvy, A. Ventruti, G. Sala, F. Cluzeaud, A. Vandewalle, R. Ghidoni, and P. Codogno. 2004. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem. 279:18384–18391. [DOI] [PubMed] [Google Scholar]
- Schu, P.V., K. Takegawa, M.J. Fry, J.H. Stack, M.D. Waterfield, and S.D. Emr. 1993. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 260:88–91. [DOI] [PubMed] [Google Scholar]
- Schubert, M., D.P. Brazil, D.J. Burks, J.A. Kushner, J. Ye, C.L. Flint, J. Farhang-Fallah, P. Dikkes, X.M. Warot, C. Rio, et al. 2003. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 23:7084–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, R.C., O. Schuldiner, and T.P. Neufeld. 2004. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell. 7:167–178. [DOI] [PubMed] [Google Scholar]
- Seglen, P.O., and P.B. Gordon. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA. 79:1889–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X.J., M. Miralpeix, M.G. Myers Jr., E.M. Glasheen, J.M. Backer, C.R. Kahn, and M.F. White. 1992. Expression and function of IRS-1 in insulin signal transmission. J. Biol. Chem. 267:22662–22672. [PubMed] [Google Scholar]
- Sun, X.J., L.M. Wang, Y. Zhang, L. Yenush, M.G. Myers Jr., E. Glasheen, W.S. Lane, J.H. Pierce, and M.F. White. 1995. Role of IRS-2 in insulin and cytokine signalling. Nature. 377:173–177. [DOI] [PubMed] [Google Scholar]
- Talloczy, Z., W. Jiang, H.W. Virgin, D.A. Leib, D. Scheuner, R.J. Kaufman, E.L. Eskelinen, and B. Levine. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA. 99:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y., G. Guhde, A. Suter, E.L. Eskelinen, D. Hartmann, R. Lullmann-Rauch, P.M. Janssen, J. Blanz, K. von Figura, and P. Saftig. 2000. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 406:902–906. [DOI] [PubMed] [Google Scholar]
- Tassa, A., M.P. Roux, D. Attaix, and D.M. Bechet. 2003. Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 376:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.P., F. Tanaka, J. Robitschek, C.M. Sandoval, A. Taye, S. Markovic-Plese, and K.H. Fischbeck. 2003. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 12:749–757. [DOI] [PubMed] [Google Scholar]
- Vassen, L., W. Wegrzyn, and L. Klein-Hitpass. 1999. Human insulin receptor substrate-2 (IRS-2) is a primary progesterone response gene. Mol. Endocrinol. 13:485–494. [DOI] [PubMed] [Google Scholar]
- Virbasius, J.V., X. Song, D.P. Pomerleau, Y. Zhan, G.W. Zhou, and M.P. Czech. 2001. Activation of the Akt-related cytokine-independent survival kinase requires interaction of its phox domain with endosomal phosphatidylinositol 3-phosphate. Proc. Natl. Acad. Sci. USA. 98:12908–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter, S., A. Boeddrich, R. Lurz, E. Scherzinger, G. Lueder, H. Lehrach, and E.E. Wanker. 2001. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell. 12:1393–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G.H., K. Mitsui, S. Kotliarova, A. Yamashita, Y. Nagao, S. Tokuhiro, T. Iwatsubo, I. Kanazawa, and N. Nukina. 1999. Caspase activation during apoptotic cell death induced by expanded polyglutamine in N2a cells. Neuroreport. 10:2435–2438. [DOI] [PubMed] [Google Scholar]
- White, M.F. 2003. Insulin signaling in health and disease. Science. 302:1710–1711. [DOI] [PubMed] [Google Scholar]
- Xia, H., Q. Mao, S.L. Eliason, S.Q. Harper, I.H. Martins, H.T. Orr, H.L. Paulson, L. Yang, R.M. Kotin, and B.L. Davidson. 2004. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 10:816–820. [DOI] [PubMed] [Google Scholar]
- Yamamoto, A., J.J. Lucas, and R. Hen. 2000. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 101:57–66. [DOI] [PubMed] [Google Scholar]
- Zacharias, D.A., J.D. Violin, A.C. Newton, and R.Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]
- Zu, T., L.A. Duvick, M.D. Kaytor, M.S. Berlinger, H.Y. Zoghbi, H.B. Clark, and H.T. Orr. 2004. Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice. J. Neurosci. 24:8853–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}