

Abstract
We show that DNA double-strand breaks (DSBs) induce complex subcompartmentalization of genome surveillance regulators. Chromatin marked by γ-H2AX is occupied by ataxia telangiectasia–mutated (ATM) kinase, Mdc1, and 53BP1. In contrast, repair factors (Rad51, Rad52, BRCA2, and FANCD2), ATM and Rad-3–related (ATR) cascade (ATR, ATR interacting protein, and replication protein A), and the DNA clamp (Rad17 and -9) accumulate in subchromatin microcompartments delineated by single-stranded DNA (ssDNA). BRCA1 and the Mre11–Rad50–Nbs1 complex interact with both of these compartments. Importantly, some core DSB regulators do not form cytologically discernible foci. These are further subclassified to proteins that connect DSBs with the rest of the nucleus (Chk1 and -2), that assemble at unprocessed DSBs (DNA-PK/Ku70), and that exist on chromatin as preassembled complexes but become locally modified after DNA damage (Smc1/Smc3). Finally, checkpoint effectors such as p53 and Cdc25A do not accumulate at DSBs at all. We propose that subclassification of DSB regulators according to their residence sites provides a useful framework for understanding their involvement in diverse processes of genome surveillance.
Introduction
To avoid deleterious consequences of DNA damage, eukaryotic cells activate a signaling network that coordinates rapid detection of the DNA lesions with temporary delay of cell cycle progression and activation of repair machinery (Zhou and Elledge, 2000). One important aspect that determines the effectiveness of these genome surveillance pathways is a carefully orchestrated redistribution of their components to nuclear regions containing the damaged DNA (J. Lukas et al., 2004). The cytological manifestation of nuclear rearrangements in response to ionizing radiation (IR) and/or radiomimetic drugs is the formation of the so-called IR-induced foci (IRIF; Shiloh, 2003).
IRIF are dynamic, microscopically discernible structures containing thousands of copies of proteins involved in various aspects of double-strand break (DSB) metabolism. As such, IRIF are widely used as a convenient marker of DSB location. Apart from various repair-associated DNA transactions (Essers et al., 2002), proteins associated with IRIF also participate in restructuring of large segments of chromatin in the vicinity of the DNA lesions (van Attikum and Gasser, 2005), thereby increasing the accessibility of damaged DNA to the repair factors (Murr et al., 2006). In addition, sustained protein assembly in the DSB-flanking chromatin seems to be required to preserve the integrity of the epigenetic information encrypted in these regions (Koundrioukoff et al., 2004; van Attikum and Gasser, 2005).
Despite the general consensus that IRIF formation signifies an important step in cellular protection against the deleterious effects of DSB-generating insults, the question of how the genome surveillance pathways actually benefit from the increased local concentration of their regulators remains poorly understood. For instance, recent results unmasked an unexpected level of complexity by showing that Chk2, the integral component of the genome surveillance machinery, interacts with DSBs only transiently, without forming cytologically discernible foci. The existence of such a “cryptic” mode of Chk2–DSB interaction indicates that some enzymatic transactions associated with the DNA damage response do not strictly require massive protein concentration at the DSB sites. In addition, relatively little attention has been paid to whether all damage-induced foci, once formed, are structurally similar. Furthermore, it is unclear to which extent the protein assemblies at the DSB sites vary during the cell cycle progression. We performed a systematic survey of protein redistribution after a defined DSB-generating insult and under standardized experimental conditions. Our intention was to subclassify the key components of the DSB network (sensors, signaling components, mediators, repair factors, and checkpoint effectors) according to the residence sites after DNA damage and to determine whether and how the protein assemblies at the DSB sites fluctuate during the cell cycle.
Results
Experimental requirements to study spatial redistribution of DSB regulators
To obtain standardized experimental conditions, we generated DSBs by laser microirradiation (Lukas et al., 2003). The key advantage of the microlaser approach is its ability to target defined nuclear volumes and generate a similar amount of DNA damage both in different areas in the same nucleus and in distinct nuclei within the microirradiated cell population (C. Lukas et al., 2004; Bekker-Jensen et al., 2005). Although several laboratories have used this approach to study various aspects of the DNA damage response (for review see Lukas et al., 2005), we initiated this study by revisiting a key parameter that must be considered when interpreting redistribution of proteins after genotoxic insults: the type and amount of the DNA lesions. It has been shown that, like IR, the laser-induced DNA damage produces single- and double-strand DNA breaks and base modifications (Lukas et al., 2005). However, the relative distribution and the density of these chromosomal alterations are hugely influenced by the laser type and energy output and by the type of photosensitizers. Our choice of a laser line within the UV-A spectrum (λ = 337 nm) and sensitization of cells with halogenated thymidine analogues has been dictated by the need to generate experimental conditions where we can still benefit from targeting the damage to the defined nuclear volumes, yet induce protein redistribution that both qualitatively and quantitatively resembles that of the “classical” IRIF.
To rigorously test this approach, we took advantage of the fact that a subset of DSBs in mammalian cells is repaired by homologous recombination (Essers et al., 1997) and that these lesions could be identified by detection of the single-stranded DNA (ssDNA) intermediates coated by the replication protein A (RPA; the RPA detection method used here recognizes selectively long stretches of ssDNA that result from enzymatic DSB resection). Because these RPA-coated ssDNA regions manifest as distinct, microscopically discernible foci that could be counted, we used this approach to quantify the amount of homologous recombination–repaired DSBs generated by the microlaser and IR, respectively. We exposed cells to the microlaser and/or increasing doses (0–10 Gy) of IR, detected the ssDNA foci by immunostaining with an antibody to RPA, and recorded a series of 3D images to ensure detection of all RPA foci in the entire nuclear volumes. Examination of these images revealed that both laser and IR produced RPA foci of a similar size (Fig. 1 A, left) and, importantly, that the total amount generated by the microlaser (under settings used for all experiments in this study) was very similar to that generated by 3 Gy of IR (Fig. 1 A, right).
Figure 1.

The local impact of laser microirradiation. (A) U2OS cells were either sensitized with BrdU followed by laser microirradiation or cultured without any presensitization followed by exposure to IR. 1 h later, the cells were fixed, immunostained with an antibody to the p32 subunit of RPA, and subject to z stack recording. (left) Representative 3D projections of cells exposed to the microlaser and 3 Gy of IR, respectively, are shown. (right) Quantification of the RPA foci in the microirradiated tracks or in the whole cell nuclei exposed to IR was obtained from 10 independent cells for each treatment. (B) U2OS cells were treated by the microlaser or exposed to increasing doses of IR. 1 h later, the cells were fixed and processed for RPA immunodetection as in A. A region spanning the entire microirradiated nuclear track containing the RPA foci (left) was placed over the maximum nuclear diameter of the IR-treated cells (right). The graph summarizes quantification of the RPA foci in these regions from 10 independent cells for each treatment. All images in this section are 3D projections as in A. (C) U2OS cells were microirradiated as in A. 1 h later, the cells were fixed and coimmunostained with antibodies to γ-H2AX and phospho-serine 15 of p53 (S15-P). The total nuclear fluorescence associated with S15-P was determined and compared with that measured in cells exposed for 1 h to the indicated doses of IR. The blue line marks the nucleus of an unirradiated cell to illustrate the background fluorescence associated with the S15-P antibody. The graph represents quantification of the S15-P fluorescence intensities from at least 50 cells for each treatment. Error bars indicate standard deviation. Bars, 10 μm.
Because the biological effect of DSBs is a combination of their absolute amount and their relative density in a given nuclear volume, we set out to also assess the local impact of both laser microirradiation and IR. We first determined the size of the region spanning the entire nuclear volume exposed to the microlaser (Fig. 1 B, top left) and then placed the same region over the maximum nuclear diameter of cells exposed to increasing doses of IR (Fig. 1 B, right; note that these images are also 3D projections). Subsequent counting of the RPA foci revealed that the DSB density in the microlaser tracks was similar to that produced in comparable nuclear volumes of cells exposed to 10 Gy of IR (Fig. 1 B). This approach allowed us to locate the microlaser response to a dose range of between 3 and 10 Gy of IR.
To further refine the estimate of the damage extent generated by the microlaser, we directly assessed its impact on p53 phosphorylation on Ser15. This DSB-induced, ataxia telangiectasia–mutated kinase (ATM)–mediated phosphorylation event was chosen because of the unique capacity of p53 to become targeted by activated ATM without the sustained focal accumulation of either of these proteins at the DSB sites (Bakkenist and Kastan, 2003). As a result, cells treated with a DSB-generating insult respond by a homogeneous increase of phosphorylated p53 throughout the entire nucleus (Fig. 1 C, left), thereby providing a sensitive surrogate for the extent of DNA damage induced by diverse DSB-generating stimuli. Quantitative measurement of the immunofluorescent signal associated with Ser15 phosphospecific antibody revealed that the amount of DNA damage delivered by the microlaser was comparable to that generated by IR in a dose range of between 4 and 6 Gy (Fig. 1 C, right).
Collectively, these data provide structural and functional evidence that the moderate quanta of focused UV-A light in BrdU-sensitized cells elicit cellular responses similar to those generated by the commonly used doses of IR and indicate that, under these conditions, the microlaser is a suitable tool for an in-depth analysis of spatial organization of the DSB-induced genome surveillance machinery. In the following sections, we provide evidence that proteins involved in the DSB response could be subclassified according to their distinct intranuclear redistribution and that the residence site of a particular DSB regulator helps refine its role in the complex cellular response to chromosomal breakage.
DSB-flanking chromatin
First, a group of proteins assembled within the entire regions of modified chromatin that surrounds the DNA breaks and spans up to a megabase distance from the initial DSB lesion (Table I). The diagnostic cytological manifestation of this pattern was a complete colocalization with the γ-H2AX–decorated chromatin and could be best illustrated on the example of the DSB-induced redistribution of Mdc1 and 53BP1, both in microlaser-generated DSB tracks (Fig. 2 A) and in IRIF induced by moderate doses of IR (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200510130/DC1). Interaction of both proteins with the DSB-modified chromatin was also reproduced in primary human fibroblasts (Fig. S2) and in living cells expressing GFP-Mdc1 and GFP-53BP1 (see Fig. 9). The observed spreading of these so-called checkpoint mediators throughout the entire DSB-flanking “microenvironment” is consistent with the recent findings describing the ability of Mdc1 and 53BP1 to interact with posttranslationally modified histones (Stewart et al., 2003; Huyen et al., 2004; C. Lukas et al., 2004; Stucki et al., 2005).
Table I.
Spatial redistribution of proteins in response to DSB-generating insults
| DSB-flanking chromatina |
ssDNA microcompartmentsb |
No retention at DSBsc |
|---|---|---|
| ATMd | ATRe | DNA-PKf |
| Nbs1e | ATRIPg | Ku70f |
| Mre11 | RPAg | Smc1f |
| Rad50 | Rad17 | Smc3e |
| Mdc1e g | Rad9 | Chk1e |
| 53BP1e g | Rad51g | Chk2e |
| BRCA1 | Rad52e | p53e g |
| BRCA2g | Cdc25A | |
| FANCD2 | ||
| Nbs1e | ||
| Mre11 | ||
| Rad50 | ||
| BRCA1 |
Interactions operating throughout the interphase.
S/G2-restricted interactions.
See text for further subclassification.
Interaction with the ssDNA microcompartments cannot be excluded.
Confirmed on the level of GFP.
Some retention could be observed in extremely dense DSB regions.
Confirmed by more than one antibody.
Figure 2.

Protein interactions with the DSB-flanking chromatin. (A) Exponentially growing U2OS cells were sensitized with BrdU and microirradiated. 1 h later, the cells were fixed and stained with the indicated antibodies. Insets show higher magnifications of the microirradiated fields. (B) Accumulation of checkpoint mediators at the sites of DNA damage can occur throughout interphase. U2OS cells stably expressing GFP-tagged Mdc1 were treated as in A. After fixation, the cells were immunostained for cyclin A and the p32 subunit of RPA to indicate the cell cycle position (schematically illustrated in the right panel). Bars, 10 μm.
Figure 9.

The major spatial patterns of DSB-induced protein redistribution visualized in living cells. (A) U2OS cell lines stably expressing the indicated DSB regulators tagged with GFP and/or the GFP spectral variants were sensitized with BrdU and microirradiated with moderate laser doses, as in Fig. 2. 1 h later, the microirradiated regions were retrieved and the microlaser-induced protein redistribution was recorded in living cells. Insets show higher magnification of the microirradiated fields; arrows indicate the laser direction through the respective nuclei. (B) The same set of cell lines as in A was exposed to 4 Gy of IR, and the living cells were recorded 1 h later. All localization patterns in A and B were maintained from the first signs of their appearance for up to several hours after microirradiation and IR exposure, respectively. Bars, 10 μm.
Other proteins in this category include the ATM kinase and the components of the Mre11–Rad50–Nbs1 nuclease complex (MRN; Fig. 2; see Fig. 5 A for Nbs1). Interaction of all these proteins with the DSB-flanking chromatin makes sense in light of the recent discoveries. We have previously shown (C. Lukas et al., 2004) that the retention of MRN at the DSB sites required direct binding of its Nbs1 component to Mdc1, the latter being the main recognition module of γ-H2AX within the DSB-flanking chromosomal microenvironment (Stucki et al., 2005). In addition, it has been reported that the recruitment of activated ATM to the DSB sites is mediated via the COOH-terminal region of Nbs1 (Falck et al., 2005; You et al., 2005). Thus, the ability of Nbs1 to bind Mdc1 on one hand and Nbs1's potential to recruit ATM on the other provide a basis for a large-scale concentration of these factors around the DSB-containing chromosomal lesions.
Figure 5.

Nbs1 interacts with both chromatin and the ssDNA subcompartments. (A) U2OS cells were microirradiated as in Fig. 2 and coimmunostained with the indicated antibodies. Under these standard conditions, Nbs1 occupies broad areas of γ-H2AX–decorated chromatin. (B) U2OS cells were transfected with control or H2AX-targeting siRNA oligonucleotides as indicated. 4 d later, the cells were microirradiated, incubated for 1 h, preextracted (see Materials and methods), and immunostained with an antibody to Nbs1. Note that in H2AX-depleted cells, Nbs1 assembles at the DSB sites in a form of subchromatin microfoci. (C) U2OS cells with stably down-regulated Mdc1 by short hairpin RNA were treated and immunostained for endogenous Nbs1 as in B. ssDNA compartments were detected by antibodies to RPA (top) or BrdU (bottom). Note that the fraction of Nbs1 that remains assembled at the DSB sites under these conditions is restricted to ssDNA (insets) and could be readily detected only in cells that are able to form these compartments (S/G2 phase). Arrows indicate the direction of the laser line during microirradiation. Bars, 10 μm.
An important denominator shared by all proteins in this category is their ability to assemble at the DSB-flanking chromatin throughout most of the cell cycle. We have consistently seen that all of the ∼200 cells microirradiated in each experiment responded by a robust accumulation of these proteins in the microirradiated tracks (unpublished data). Such a uniform response did not indicate cell cycle–dependent interactions. More specifically, Mdc1 (Fig. 2 B; see Fig. 4 C) and other proteins described in this section (unpublished data) readily accumulated in the microirradiated nuclear regions in cells with various intensities of cyclin A or B1 (cells in S and G2), as well as in cells lacking a detectable amount of both cyclins (cells in G1). Together, these data suggest that the assembly of proteins at the DSB-flanking chromatin can occur throughout the interphase.
Figure 4.

Protein assembly in the ssDNA microcompartments is restricted to the S and G2 phases of the cell cycle. (A) U2OS cells were microirradiated as in Fig. 2. 1 h later, the ssDNA was revealed by immunodetection of BrdU without previous denaturation or nuclease treatment. Cells were coimmunostained with an antibody to γ-H2AX to demonstrate the location of the ssDNA microfoci within the larger regions of the DSB-modified chromatin and to show that generation of ssDNA occurred only in a subset of the microirradiated cells. (B) Cells were treated, and the ssDNA was detected as in A. Coimmunostaining of FANCD2 (shown here as an example) and other proteins from the spatial category (unpublished data) revealed close colocalization with ssDNA. (C) U2OS cells stably expressing GFP-Mdc1 were microirradiated and 1 h later subject to ssDNA detection as in A. In parallel, the cells were immunostained for cyclin B1 to indicate the cell cycle position. Cyclin B1 and ssDNA do not overlap and are therefore displayed in the same channel (red). (D) U2OS cells were treated as in A and coimmunostained with antibodies to Rad51, γ-H2AX (to detect the microirradiated tracks), and cyclin B1 (to reveal the cells in S/G2). The latter two proteins are simultaneously displayed in the same channel (red). Bars, 10 μm.
Interactions restricted to ssDNA-containing microcompartments
Another group of proteins assembled in much smaller areas (“microfoci”) located to the center of the microirradiated tracks and surrounded by relatively vast regions of γ-H2AX–modified chromatin (Fig. 3). This spatial pattern could be resolved on the standard IRIF level (Fig. S1 A), it could be observed in primary human fibroblasts (Fig. S2), and it is discernible in living cells expressing GFP-tagged proteins (see Fig. 9). We also verified that the cytological appearance of these microfoci was not affected by various fixation protocols, the relative affinity of antibodies, and/or the image acquisition conditions (Fig. S1 B; unpublished data). As the proteins in this category typically include nuclear factors involved in DNA repair by homologous recombination (Rad51, Rad52, BRCA2, and FANCD2), these microfoci likely represent either individual DSBs or closely neighboring DSBs assembled in repair centers (Lisby et al., 2003). The same compartments are occupied by the ATR kinase and the protein machinery (ATR interacting protein [ATRIP] and RPA) required for ATR's assembly at the DSB sites (Cortez et al., 2001). Finally, Rad17 and -9, both components of the proliferating cell nuclear antigen–like sliding clamp loaded on DNA after DNA damage, also show similar localization patterns (Table I).
Figure 3.

Protein assemblies restricted to subchromatin microfoci. U2OS cells were microirradiated as in Fig. 2. Immunostaining with target specific antibodies (FANCD2, Rad51, RPA, and ATRIP) and a direct imaging of GFP-ATR revealed that accumulation of these proteins is restricted to nuclear subdomains that are distinct from the DSB-flanking chromatin compartments (the latter marked by γ-H2AX). Bars, 10 μm.
Two prominent features discriminate these DSB-induced microcompartments from the chromatin-mediated interactions. First, accumulation of all proteins in this category is restricted to areas of ssDNA formed after resection of the initial DSB lesions (Fig. 4 A). This is illustrated by a close overlap of FANCD2 with ssDNA (Fig. 4 B), the latter structure being revealed by immunostaining of BrdU without previous DNA denaturation (Raderschall et al., 1999; see Materials and methods). Second, not all microirradiated cells were able to generate detectable stretches of ssDNA (and the corresponding focal protein accumulation), despite the fact that all such cells responded by an equally robust phosphorylation of H2AX (Fig. 4 A) and/or Mdc1 assembly (Fig. 2 B). Indeed, coimmunostaining experiments revealed that the interaction of FANCD2, Rad51, and other proteins from this category with the ssDNA microcompartments could only be detected in cells that expressed cyclin B1 and/or A (Fig. 2 B and Fig. 4, B–D). These findings are consistent with the recent study showing that the DSB resection is cell cycle dependent (Jazayeri et al., 2006) and suggest that, unlike the chromatin-mediated interactions, accumulation of proteins in the ssDNA microcompartments is temporally restricted to S and G2 phases. The latter conclusion is also consistent with a report showing that the recruitment of Rad51 to the laser-generated DSBs and/or IRIF requires postreplicative DNA (Tashiro et al., 2000) and with experiments in yeast that failed to detect Rad51 in IRIF during the G1 phase (Lisby et al., 2004). On the other hand, we note that a recent study reported accumulation of Rad51 in laser-damaged nuclei also during G1 (Kim et al., 2005). However, these authors used a relatively high laser energy in unsensitized cells, resulting in visible morphological destruction of the microirradiated nuclear areas, a condition that could have overpowered the physiological restrictions for Rad51 to attempt DSB repair in the context of prereplicative chromatin (see also the following sections).
Nbs1 and BRCA1 interact with both DSB-flanking chromatin and the ssDNA microcompartments
The components of the MRN complex share the striking feature to interact with both the DSB-flanking chromatin (Fig. 5 A) and the microfoci described in the previous section (Fig. 5, B and C). The ability of Nbs1 to interact with the latter compartments could be revealed by its ability to form microfoci even in cells with reduced levels of H2AX, that is, after disruption of the key step in forming the DSB-induced chromatin microenvironment (Fig. 5 B). These data extend our earlier observation that a very similar pattern of Nbs1 redistribution was observed in cells where the integrity of the DSB-flanking chromatin was disrupted by the down-regulation of the γ-H2AX–binding protein Mdc1 (C. Lukas et al., 2004). To determine whether these microfoci represent the ssDNA compartments, we microirradiated Mdc1-deficient cells and coimmunostained Nbs1 with antibodies to RPA and BrdU (the latter without previous DNA denaturation). Indeed, both of these ssDNA markers showed tight colocalization with a fraction of Nbs1 that assembled in the microirradiated tracks under these conditions (Fig. 5 C). Consistent with the cell cycle–dependent formation of the ssDNA compartments, the retention of Nbs1 in these microfoci was much more pronounced in cells capable of resecting the primary lesions and generating cytologically discernible stretches of ssDNA (Fig. 5 C, bottom). Thus, in addition to its ability to assemble in the large DSB-flanking chromosomal regions, Nbs1 can interact with the ssDNA microcompartments in a chromatin-independent manner. This is consistent with a study detecting an extraction-resistant pool of Mre11 in the center of γ-H2AX–coated chromosomal domains (Aten et al., 2004) and with the recently reported causative role of MRN in the formation of the ssDNA microcompartments (Jazayeri et al., 2006). Furthermore, the ability of Nbs1 to assemble in the ssDNA microcompartments can explain the H2AX-independent accumulation of the MRN components in early stages of the DSB response (Celeste et al., 2003).
The only other protein capable of simultaneous interaction with both ssDNA compartments and the DSB-flanking chromatin is BRCA1. After laser microirradiation, BRCA1 clearly spreads throughout the entire chromatin regions marked by γ-H2AX and/or by retention of typical chromatin binding proteins, such as 53BP1 (Fig. 6, A and B, top). Like in all other chromatin-specific interactions described in the previous sections, this pattern of BRCA1 redistribution could be detected throughout the interphase, although the amount of BRCA1 retained in the microirradiated G1 cells is less pronounced because of the lower abundance of the total BRCA1 protein in this cell cycle stage (Fig. 6 C). Importantly, although disruption of this compartment by Mdc1 down-regulation triggered dissociation of BRCA1 from the DSB-flanking chromatin, it did not impair a productive assembly of BRCA1 at the subchromatin microfoci (Fig. 6, A and B, bottom). Additional evidence for the chromatin-independent role of BRCA1 comes from the observation that reducing BRCA1 levels by siRNA precluded assembly of important repair factors (BRCA2 and Rad51; both are known to function downstream of BRCA1) at the ssDNA microcompartments (Fig. S3 D, available at http://www.jcb.org/cgi/content/full/jcb.200510130/DC1). Collectively, these results add an important spatial dimension to the emerging functional interplay between Mdc1 and BRCA1 (Lou et al., 2003; Stewart et al., 2003) by showing that it is the chromatin bound (but not the ssDNA associated) fraction of BRCA1 whose retention at the DSB sites is controlled by Mdc1.
Figure 6.

Spatial pattern of BRCA1 assembly at the DSB sites. (A) U2OS cells were transfected with control or Mdc1-targeting siRNA oligonucleotides for 4 d. The cells were then microirradiated and 1 h later fixed and coimmunostained with antibodies to γ-H2AX and BRCA1. (B) U2OS cells were treated with the siRNA oligonucleotides as in A. 1 h after microirradiation, the cells were fixed and immunostained with the indicated antibodies. Note that in the absence of Mdc1 (A and B, bottom), BRCA1 is lost from the DSB-flanking chromatin but remains assembled in the microfoci along the microirradiated tracks. The complete loss of 53BP1 from the DSB sites (B, bottom left) serves as a control of efficient Mdc1 down-regulation. (C) U2OS cells were microirradiated and 1 h later fixed and immunostained with antibodies to BRCA1 and cyclin A (the latter to reveal the cells in S/G2 phases). BRCA1 assembly could be detected also in the G1 cell (marked by the green arrow), although the overall abundance of BRCA1 in the nucleus and at in the DSB tracks is reduced compared with the S/G2 cells.
It is important to emphasize that the complex interaction pattern of Nbs1 and BRCA1 described in this section is quite unique, likely reflecting the central position of the MRN complex and BRCA1 in DSB recognition and signaling (Petrini and Stracker, 2003; Greenberg et al., 2006). For instance, uncoupling of the typical chromatin binding proteins 53BP1 (Fig. S3 A) and Mdc1 (Fig. S3 B) from their respective histone residues was sufficient to completely abrogate their ability to accumulate at the DSB sites, despite the fact that, under the same experimental conditions, the ssDNA compartments were clearly formed (Fig. 5, B and C; and Fig. 6, A and B). Furthermore, although down-regulation of Mdc1 impaired stable interaction of 53BP1 with the DSB-flanking chromatin (Fig. 6 B; Bekker-Jensen et al., 2005), it did not prevent productive assembly of ATRIP and Rad51, the typical components of the ssDNA compartments (Fig. S3 C). Thus, the majority of the DSB regulators studied so far tend to interact rather exclusively with either ssDNA or the DSB-flanking chromatin, and the retention of proteins in these respective compartments seems to be regulated by mutually independent mechanisms. Interestingly, one study (Ward et al., 2004) reported that cells deficient in H2AX (a chromatin component) are not able to retain ATR (the ssDNA component) at stalled replication forks. Thus, the rather strict spatial subcompartmentalization of checkpoint regulators described here might be specific for DBSs, likely because of the complexity of the DNA and chromatin rearrangements required for efficient repair of these serious chromosomal lesions.
Chk1 does not stably accumulate at the DSB sites
Some proteins intimately involved in the genome surveillance network do not visibly accumulate at the damage sites, a striking phenomenon that we have previously described for the Chk2 kinase (Lukas et al., 2003). The new addition to this spatial category provided here is Chk1 (Fig. 7 A, top; see Fig. 9 for GFP-Chk1 in living cells). The inability of Chk1 to form cytologically discernible foci was not caused by the lack of its activation after laser microirradiation because the microirradiated cells readily induced phosphorylation of Chk1 on Ser317 (Fig. 7 A, bottom), one of the ATR target sites whose phosphorylation accompanies activation of Chk1 (Zhao and Piwnica-Worms, 2001). An important aspect of this spatial pattern is that phosphorylated Chk1 (Fig. 7 A) and Chk2 (Lukas et al., 2003) do not concentrate around the DNA damage sites but rapidly spread to the entire nucleus. Together with the evidence that Chk1 phosphorylation by ATR requires physical interaction of these two components directly at the sites of damaged DNA and/or stalled replication forks (Chen and Sanchez, 2004; Smits et al., 2006), these results indicate that Chk1 (like Chk2) facilitates signal transduction between focal DNA lesions and relatively immobile effector structures (replication origins, stalled replication forks, and gene promoters) elsewhere in the undamaged parts of the nucleus. The distinction between Chk1 and -2 in this respect is that although Chk2 could be activated throughout the interphase (Lukas et al., 2003), Chk1 function is temporally limited because of the fact that the ssDNA formation and ATR signaling is restricted to S/G2 phases of the cell cycle (Jazayeri et al., 2006).
Figure 7.

DSB responses without cytologically discernible protein retention. (A) U2OS cells were microirradiated as in Fig. 2 and coimmunostained with antibodies to γ-H2AX, total Chk1, or Chk1 phosphorylated on serine 317 (S317-P), as indicated. Although the cells contained DSBs only within the nuclear tracks exposed to the laser (see the γ-H2AX pattern), the activated form of Chk1 was disseminated throughout the nucleus. The dotted yellow line marks the boundary between microirradiated and control cells. (B) U2OS cells were treated as in A and immunostained with antibodies to DNA-PK, Ku70, total Smc1, or Smc1 phosphorylated on serine 957 (S957-P), as indicated. Although the total Smc1 protein did not massively relocate to the DSB sites, it became locally phosphorylated within the microirradiated regions. Bars, 10 μm.
Other DSB interactions that do not readily form cytologically discernible foci
Several other proteins known to function on various levels of the DSB-induced signaling did not readily accumulate at the DSB sites (Table I). The inability of DNA-PK, Ku70, Smc1, and Smc3 to form cytologically discernible foci was not restricted to a single time point (we failed to detect increased local accumulation of any of these proteins between 5 min and 8 h after DNA damage); neither was it influenced by the cell type, DSB insult, fixation, and/or imaging conditions (Figs. 7, 9, S1, and S2). However, we note that the DNA damage–induced redistribution of some of these factors had been studied before and produced conflicting results. We therefore set out to critically reexamine some of these cases and explain the discrepancies.
On one hand, our inability to detect cytologically detectable accumulation of DNA-PK and its regulatory subunit Ku70 both in locally microirradiated cells (Fig. 7 B and 9 A) and after global exposure to the commonly used dose range of IR (Figs. S1 A and S2) is consistent with the results obtained by Jakob et al. (2002), who generated DSBs by irradiating cells with charged ion beams. On the other hand, another group has reported local accumulation of DNA-PK in nuclei exposed to Nd:YAG laser (Kim et al., 2005). To find the reason for these differences, we tried to modify our experimental conditions and test whether any of those would be able to locally increase the concentration of DNA-PK/Ku70 to the levels detectable by light microscopy. Neither variation of the laser dosage in sensitized cells nor the extension of the assay period from several seconds to many hours produced such results, despite generating clear and robust DSB response, including induction of γ-H2AX and recruitment of the DNA-PK–related ATM and ATR kinases (see Figs. 2 and 3 for examples). Only a substantial increase of the laser output in nonsensitized cells was able to induce weak but detectable accumulation of DNA-PK/Ku70 at the microirradiated tracks (Fig. 8 A, left). However, such treatment also produced massive local damage of the overall nuclear structure (never seen after microirradiating the sensitized cells by moderate laser doses and/or after IR), manifest, for instance, by a decrease of DNA staining within the microirradiated areas (Fig. 8 A, inset). Similar signs of general nuclear disruption (areas with markedly altered optical density) were observed also in the previous study describing local accumulation of DNA-PK/Ku70 (Kim et al., 2005).
Figure 8.

Local accumulation of Ku70 and Smc1 in cells exposed to high doses of laser irradiation. (A) U2OS cells cultured without presensitization with halogenated thymidine analogues were locally irradiated with a high laser dose (73% energy output). Although such treatment induced local accumulation of Ku70 (left) and Smc1 (right) in the irradiated tracks, it was also accompanied by a pronounced destruction of the laser-exposed nuclear regions manifest by the decreased DNA staining (see the inset for magnification). (B) U2OS cells were either sensitized by BrdU and microirradiated with moderate laser dose (55% energy output; top) or cultured without presensitization and exposed to high laser dose (73% energy output; bottom). 1 h later, the cells were fixed and coimmunostained with antibodies to RPA and 53BP1 (three independent cells for each treatment are shown). Although exposure to the low laser energy was compatible with local DSB processing, formation of RPA foci and a robust assembly of 53BP1, the high laser dose generated extreme density of the RPA (without a clear resolution into individual repair foci) and impaired assembly of 53BP1 at the DSB-flanking chromatin. Bars, 10 μm.
Interestingly, the same group applied these assay conditions to demonstrate local accumulation of Smc1, the structural component of the multiprotein cohesin complex (J.S. Kim et al., 2002). Also in this case, we were able to reproduce these results but, again, only after exposing the unsensitized cells to very high laser energy that was accompanied by local disruption of the overall nuclear structure (Fig. 8 A, right). Neither IR (up to 10 Gy; Fig. S1) nor moderate laser microirradiation in sensitized cells (Fig. 7 B) showed any signs of cytologically discernible accumulation of Smc1. Similar results (no accumulation after moderate doses of IR and/or laser) were obtained with GFP-Smc3, the heterodimerizing partner of Smc1 (Fig. 9 and Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200510130/DC1).
Hence, despite both DNA-PK/Ku70 and Smc1 being integral components of various facets of DBS response (S.T. Kim et al., 2002; Yazdi et al., 2002; Lieber et al., 2003; Kitagawa et al., 2004; Strom et al., 2004; Unal et al., 2004), local accumulation of these proteins to a degree that could be resolved by light microscopy seems to require enormous local concentration of DSBs. Indeed, when we tested the conditions compatible with Ku70 and/or Smc1 recruitment to DSBs by the same assay described in Fig. 1, we observed that the nuclear areas exposed to the high laser energy were unable to resolve clear RPA foci. Instead, these regions showed signs of a uniform and strong RPA accumulation, indicating extremely high density of DSBs (Fig. 8 B). At the same time, the relative abundance of the typical chromatin binding protein, such as 53BP1, was reduced in these regions compared with moderate laser doses in sensitized cells (Fig. 8 B, right), indicating that the high laser energy output not only generates a massive DNA damage by itself but also triggers local destruction of histones and/or the other chromatin-associated protein complexes. Such extreme density (and complexity) of chromosomal damage likely saturates the cellular capacity to repair the lesions, thereby generating conditions that may stabilize (or aggregate) the templates for the assembly of proteins that specifically interact with DNA ends (Lieber et al., 2003). Based on these results and considerations, we propose that the assembly of the DNA-PK/Ku70 holoenzyme and loading of the Smc1/3-containing cohesin complex is spatially restricted to unprocessed and/or only partially processed DNA breaks and that, within a range of physiologically relevant doses of DSB-generating insults, these microcompartments are beyond the resolution of light microscopy and do not manifest as cytologically discernible nuclear foci.
Local phosphorylation of Smc1 without its large-scale physical recruitment
Importantly, the latter conclusion does not contradict a recent report describing IRIF containing the ATM-phosphorylated Smc1 (Kitagawa et al., 2004). Indeed, we were able to reproduce these results by detecting local phosphorylation of Smc1 on serine 957 (one of the key ATM target sites) after moderate doses of laser microirradiation that did not cause any discernible accumulation of the total Smc1 protein (Fig. 7 B, compare the two bottom panels). Thus, it appears that in addition to cohesin, which assembles at the DSBs de novo (Strom et al., 2004; Unal et al., 2004), and the amount of which (at least in mammalian cells) is likely limited below the levels that could be resolved by light microscopy, there must be a sizable pool of Smc1 throughout the undamaged chromatin as a result of the physiological processes accompanying DNA replication. Indeed, loading of the vertebrate cohesin on chromatin is triggered by formation of the prereplication complexes, an event that occurs early during the cell cycle (telophase in cycling cells and G1 after stimulation from quiescence; Takahashi et al., 2004). The spatial pattern of the Smc1 response to DNA damage reported here (readily detectable local phosphorylation without discernible increase of the total protein concentration) indicates that after DSB generation, this prereplication complex–loaded fraction of cohesin remains bound to chromatin and becomes locally accessible to the ATM-mediated phosphorylation. In this regard, it is important to note that phosphorylated Smc1 spreads throughout the entire regions of γ-H2AX–decorated chromatin (Fig. 7 B, bottom). This is consistent with our findings that all three components required for the DSB-induced Smc1 phosphorylation—ATM, Nbs1, and BRCA1 (S.T. Kim et al., 2002; Kitagawa et al., 2004)—avidly interact with this DSB-generated nuclear subcompartment (Figs. 2, 5, and 6).
Pan-nuclear effectors of the DSB signaling
Some integral components of the DSB-induced genome surveillance network did not accumulate at the damaged areas under any of the conditions explored in this study, including the high-energy laser illumination described in the preceding section (Table I; Lukas et al., 2003). The key proteins in this category, p53 and Cdc25A, share their functional position within the DSB network by serving as the key effectors of the DNA damage–induced genome surveillance pathways (Kastan and Bartek, 2004). Their lack of direct physical engagement with DSBs indicates that the efficient DSB-induced gene expression (p53) and cell cycle arrest (Cdc25A and p53) requires a specific signaling component capable of rapid and efficient connection of these effector molecules with the focal DNA lesions. The spatial properties of activated Chk1, Chk2 (Lukas et al., 2003), and ATM (Bakkenist and Kastan, 2003) render these kinases the most plausible candidates for a “messenger” function.
Discussion
Our study provides evidence that after DSB-generating insults, a mammalian nucleus undergoes a complex compartmentalization reflected by distinct patterns of protein redistribution. The essence of our results is summarized in Table I. For the sake of clarity, the key implications of how the residence sites of the studied proteins help us better understand their roles in the DSB response were systematically discussed while describing the individual spatial categories in the preceding sections. We would like to complement these specific conclusions with more general and conceptual ramifications of the reported results.
In particular, we would like to emphasize that among the diverse modes of protein redistribution after a DSB-generating insult, only the proteins assembled in the DSB-flanking chromatin regions and the ssDNA microcompartments could be readily detected as intranuclear foci. An important implication of this finding is that within the range of physiologically relevant doses of DNA damage, the IRIF formation (and/or the protein assembly at the laser-damaged nuclear tracks) cannot serve as the only criterion for the direct involvement of a given protein in the DSB response. This conclusion is supported by adding several new members to the expanding family of proteins that, although functionally distinct, share the capability of a productive interaction with DSBs without a massive increase in their local concentration. Thus, in addition to signaling kinases whose interaction with the DSB sites is too transient to manifest as foci (Chk1 and -2), other proteins in this category assemble at DSB intermediates whose size is below the resolution of light microscopy (DNA-PK/Ku70). In addition, Smc1 is likely just one example of a larger group of proteins that stably interact with chromatin even in undamaged nuclei and yet become engaged in the DSB signaling and/or repair after local modification by enzymes recruited to the sites of DNA damage.
In conclusion, we hope that the results reported here will provide the necessary framework to add more proteins to the emerging “spatial map” of the DSB-induced genome surveillance network. If coordinated in terms of experimental conditions, subclassification of proteins according to their residence sites before and after DNA damage may help validate, predict, or even exclude their roles in the increasingly complex DSB response. One example illustrating the potential usefulness of the spatial dimension in approaching some lingering questions in the field is the redistribution of the key apical kinases induced by DSBs. Most notably, ATM, ATR, and DNA-PK each occupy distinct nuclear subcompartments (DSB-flanking chromatin, ssDNA microcompartments, and unprocessed DSB ends, respectively). Because deficiency of the respective kinases is accompanied by distinct phenotypes (Abraham, 2004), it is clear that these kinases, despite sharing several downstream substrates, have limited capability to substitute each other. Although the regulatory network determining the exact function of each of these kinases is very complex, their relocation to distinct compartments after DNA damage can, at least partly, explain their overlapping versus nonoverlapping potential.
Materials and methods
Cell culture and generation of DNA damage
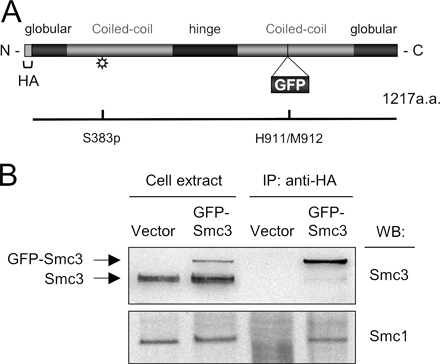
The U2OS cell line and the BJ fibroblasts were seeded onto glass coverslips (Menzel) and grown in DME supplemented with 10% fetal bovine serum and standard antibiotics. The U2OS-derived cell lines stably expressing GFP-Mdc1, -53BP1, -ATR, and -Chk1 were described previously (Kramer et al., 2004; C. Lukas et al., 2004; Bekker-Jensen et al., 2005; Jazayeri et al., 2006). The YFP-Rad52 construct was generated by subcloning the Rad52 cDNA (a gift from R. Kanaar, Erasmus Medical Centre, Rotterdam, Netherlands) to the pEYFP-N expression plasmid (CLONTECH Laboratories, Inc.). Generation of the GFP-Smc3 plasmid is described in detail in Fig. S4. The U2OS cell lines stably expressing YFP-Rad52 and GFP-Smc3 were generated by cotransfecting the respective expression plasmids together with the pBabe-puro plasmid containing the puromycin resistance cassette. Upon selection with 1 μg/ml puromycin (Sigma-Aldrich) for 10 d, resistant clones were tested for the expression and functionality of the GFP/YFP-tagged proteins (see Fig. S4 for characterization of the GFP-Smc3–expressing cells). Laser microirradiation to generate DSBs in defined nuclear volumes was performed essentially as described previously (Lukas et al., 2003; C. Lukas et al., 2004; Bekker-Jensen et al., 2005). In brief, the culture medium was supplied with 10 μM BrdU (Sigma-Aldrich) for 24 h to sensitize the cells to DSB generation by UV-A laser (λ = 337 nm). Before laser treatment, the coverslips were transferred to a phenol red–free CO2-independent medium (Invitrogen). After microirradiation of ∼200 cells (a procedure lasting in total for <10 min), the coverslips were incubated for 1 h in the incubator before fixation. IR was delivered by an x-ray generator (HF160 [Pantak]; 150 kV; 15 mA; dose rate: 2.18 Gy/min) as previously described (Syljuasen et al., 2004).
RNA interference and plasmids
siRNAs against H2AX and Mdc1 were described previously (C. Lukas et al., 2004). The control siRNA was against HSP70B (Bekker-Jensen et al., 2005). Cells were transfected with the siRNA duplexes with oligofectamine (Invitrogen) according to the manufacturer's instructions and incubated for 96 h before further treatment. For generation of stable Mdc1 knockdown, the oligonucleotides 5′-gatccccgtctcccagaagacagtgattcaagagatcactgtcttctgggagacttttt and 5′-agctaaaaagtctcccagaagacagtgatctcttgaatcactgtcttctgggagacggg were annealed and ligated into the pSUPER plasmid digested with HindIII and BglII. For generation of stable 53BP1 knockdown, the oligonucleotides 5′-gatccccgaacgaggagacggtaatattcaagagatattaccgtctcctcgttcttttt and 5′-agctaaaaagaacgaggagacggtaatatctcttgaatattaccgtctcctcgttcggg were annealed and processed as above. The resulting constructs were transfected into U2OS cells and selected with Puromycin as described previously (C. Lukas et al., 2004).
Antibodies and microscopy
Rabbit polyclonal antibodies against the following targets were used: Smc1 (Abcam), Smc3 (Abcam), Smc1-S957P (Novus Biologicals), p53-S15P (Santa Cruz Biotechnology, Inc.), TRF2 (Santa Cruz Biotechnology, Inc.), ATRIP (a gift from R. Abraham, The Burnham Institute, La Jolla, CA), Rad17 (Santa Cruz Biotechnology, Inc.), Rad9 (Santa Cruz Biotechnology, Inc.), Rad51 (Santa Cruz Biotechnology, Inc.), FANCD2 (Novus Biologicals), Nbs1 (Novus Biologicals), ATM (Abcam), 53BP1 (Santa Cruz Biotechnology, Inc.), γ-H2AX (Upstate Biotechnology), cyclin A (Santa Cruz Biotechnology, Inc.), and Chk1-S317P (Cell Signaling). The following mouse monoclonal antibodies were used: DNA-PK (Lab Vision), Ku70 (Lab Vision), Chk1 (DCS-310; Abcam), RPA p32 (Lab Vision), BRCA2 (Calbiochem), Mre11 (GeneTex), BRCA1 (Santa Cruz Biotechnology, Inc.), γ-H2AX (Upstate Biotechnology), and cyclin B1 (Santa Cruz Biotechnology, Inc.). Sheep anti-Mdc1 antibody was a gift from S. Jackson (Wellcome Trust/Cancer Research UK Gurdon Institute, Cambridge, UK) and M. Stucki (University of Zürich, Zürich, Switzerland). Anti-BrdU mouse monoclonal antibody (RPN20AB) to detect ssDNA was obtained from GE Healthcare and was applied without any preceding DNA denaturation or nuclease treatment (Raderschall et al., 1999). Other immunostaining steps were identical to those described in the following section.
Cells were fixed for 15 min in 4% formaldehyde and permeabilized in 0.2% Triton X-100 for 5 min. To facilitate discrimination of the chromatin-associated versus ssDNA bound pools of Nbs1 (Fig. 5, B and C), cells were preextracted for 5 min at 4°C with a buffer containing 25 mM Hepes, pH 7.5, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X-100 as described previously (C. Lukas et al., 2004). Coverslips were incubated with primary antibodies for 1 h followed by secondary antibodies coupled to Alexa 488, 568, or 647 (Invitrogen) for 30 min. Where indicated, the DNA stain ToPro3 (Invitrogen) was added to the last washing solution. Coverslips were mounted onto glass slides (Menzel) with DAPI-containing mounting medium (Vector Laboratories) and subject to two- or three-color confocal microscopy on an LSM-510 (Carl Zeiss MicroImaging, Inc.) mounted on an Axiovert 100M (Carl Zeiss MicroImaging, Inc.) equipped with Plan-Neofluar 40×/1.3 oil-immersion objective, as previously described (Bekker-Jensen et al., 2005). For quantitative assessment of the DNA damage–induced p53 phosphorylation, masks were manually drawn around the individual nuclei, and the mean fluorescence associated with antibody to phosphorylated serine 15 of p53 (S15-P) subtracted for the background fluorescence was determined using the ImageJ software (NIH). The obtained values were exported to Prism4 (GraphPad) software for further data processing.
Online supplemental material
Fig. S1 shows the spatial patterns of DSB-induced protein redistribution resolved on the level of the IRIF and provides evidence that the DSB-induced redistribution of proteins to distinct nuclear subcompartments was assayed under unsaturated conditions for image acquisition. Fig. S2 shows the main spatial patterns of DSB-induced protein localization in primary cells (the BJ strain of human diploid fibroblasts). Fig. S3 provides evidence for autonomous protein interactions with distinct DSB-generated nuclear subcompartments. Fig. S4 describes generation and characterization of the GFP-Smc3 cohesin subunit. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200510130/DC1.
Supplementary Material
Acknowledgments
We thank Robert Abraham, Roland Kanaar, Manuel Stucki, and Stephen Jackson for reagents.
This work was supported by grants from the Danish Cancer Society (DP 03 035), the Danish National Research Foundation, the European Union (integrated projects DNA Repair 512113 and Active p53 503576), the European Science Foundation (EuroDYNA 21-04-280), the John and Birthe Meyer Foundation, and the National Cancer Institute (CA93632 and CA71387).
Abbreviations used in this paper: ATM, ataxia telangiectasia–mutated; ATR, ATM and Rad-3–related; ATRIP, ATR interacting protein; DSB, double-strand break; IR, ionizing radiation; IRIF, IR-induced foci; MRN, Mre11–Rad50–Nbs1 nuclease complex; RPA, replication protein A; ssDNA, single-stranded DNA.
References
- Abraham, R.T. 2004. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst.). 3:883–887. [DOI] [PubMed] [Google Scholar]
- Aten, J.A., J. Stap, P.M. Krawczyk, C.H. van Oven, R.A. Hoebe, J. Essers, and R. Kanaar. 2004. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 303:92–95. [DOI] [PubMed] [Google Scholar]
- Bakkenist, C.J., and M.B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421:499–506. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen, S., C. Lukas, F. Melander, J. Bartek, and J. Lukas. 2005. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 170:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste, A., O. Fernandez-Capetillo, M.J. Kruhlak, D.R. Pilch, D.W. Staudt, A. Lee, R.F. Bonner, W.M. Bonner, and A. Nussenzweig. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5:675–679. [DOI] [PubMed] [Google Scholar]
- Chen, Y., and Y. Sanchez. 2004. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA Repair (Amst.). 3:1025–1032. [DOI] [PubMed] [Google Scholar]
- Cortez, D., S. Guntuku, J. Qin, and S.J. Elledge. 2001. ATR and ATRIP: partners in checkpoint signaling. Science. 294:1713–1716. [DOI] [PubMed] [Google Scholar]
- Essers, J., R.W. Hendriks, S.M. Swagemakers, C. Troelstra, J. de Wit, D. Bootsma, J.H. Hoeijmakers, and R. Kanaar. 1997. Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell. 89:195–204. [DOI] [PubMed] [Google Scholar]
- Essers, J., A.B. Houtsmuller, L. van Veelen, C. Paulusma, A.L. Nigg, A. Pastink, W. Vermeulen, J.H. Hoeijmakers, and R. Kanaar. 2002. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 21:2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck, J., J. Coates, and S.P. Jackson. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 434:605–611. [DOI] [PubMed] [Google Scholar]
- Greenberg, R.A., B. Sobhian, S. Pathania, S.B. Cantor, Y. Nakatani, and D.M. Livingston. 2006. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 20:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen, Y., O. Zgheib, R.A. Ditullio Jr., V.G. Gorgoulis, P. Zacharatos, T.J. Petty, E.A. Sheston, H.S. Mellert, E.S. Stavridi, and T.D. Halazonetis. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 432:406–411. [DOI] [PubMed] [Google Scholar]
- Jakob, B., M. Scholz, and G. Taucher-Scholz. 2002. Characterization of CDKN1A (p21) binding to sites of heavy-ion-induced damage: colocalization with proteins involved in DNA repair. Int. J. Radiat. Biol. 78:75–88. [DOI] [PubMed] [Google Scholar]
- Jazayeri, A., J. Falck, C. Lukas, J. Bartek, G.C. Smith, J. Lukas, and S.P. Jackson. 2006. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8:37–45. [DOI] [PubMed] [Google Scholar]
- Kastan, M.B., and J. Bartek. 2004. Cell-cycle checkpoints and cancer. Nature. 432:316–323. [DOI] [PubMed] [Google Scholar]
- Kim, J.S., T.B. Krasieva, V. LaMorte, A.M. Taylor, and K. Yokomori. 2002. Specific recruitment of human cohesin to laser-induced DNA damage. J. Biol. Chem. 277:45149–45153. [DOI] [PubMed] [Google Scholar]
- Kim, J.S., T.B. Krasieva, H. Kurumizaka, D.J. Chen, A.M. Taylor, and K. Yokomori. 2005. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J. Cell Biol. 170:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.T., B. Xu, and M.B. Kastan. 2002. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 16:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, R., C.J. Bakkenist, P.J. McKinnon, and M.B. Kastan. 2004. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 18:1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, A., N. Mailand, C. Lukas, R.G. Syljuasen, C.J. Wilkinson, E.A. Nigg, J. Bartek, and J. Lukas. 2004. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6:884–891. [DOI] [PubMed] [Google Scholar]
- Koundrioukoff, S., S. Polo, and G. Almouzni. 2004. Interplay between chromatin and cell cycle checkpoints in the context of ATR/ATM-dependent checkpoints. DNA Repair (Amst.). 3:969–978. [DOI] [PubMed] [Google Scholar]
- Lieber, M.R., Y. Ma, U. Pannicke, and K. Schwarz. 2003. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 4:712–720. [DOI] [PubMed] [Google Scholar]
- Lisby, M., U.H. Mortensen, and R. Rothstein. 2003. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 5:572–577. [DOI] [PubMed] [Google Scholar]
- Lisby, M., J.H. Barlow, R.C. Burgess, and R. Rothstein. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 118:699–713. [DOI] [PubMed] [Google Scholar]
- Lou, Z., C.C. Chini, K. Minter-Dykhouse, and J. Chen. 2003. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem. 278:13599–13602. [DOI] [PubMed] [Google Scholar]
- Lukas, C., J. Falck, J. Bartkova, J. Bartek, and J. Lukas. 2003. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 5:255–260. [DOI] [PubMed] [Google Scholar]
- Lukas, C., F. Melander, M. Stucki, J. Falck, S. Bekker-Jensen, M. Goldberg, Y. Lerenthal, S.P. Jackson, J. Bartek, and J. Lukas. 2004. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 23:2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas, C., J. Bartek, and J. Lukas. 2005. Imaging of protein movement induced by chromosomal breakage: tiny ‘local’ lesions pose great ‘global’ challenges. Chromosoma. 114:146–154. [DOI] [PubMed] [Google Scholar]
- Lukas, J., C. Lukas, and J. Bartek. 2004. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst.). 3:997–1007. [DOI] [PubMed] [Google Scholar]
- Murr, R., J.I. Loizou, Y.G. Yang, C. Cuenin, H. Li, Z.Q. Wang, and Z. Herceg. 2006. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8:91–99. [DOI] [PubMed] [Google Scholar]
- Petrini, J.H., and T.H. Stracker. 2003. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 13:458–462. [DOI] [PubMed] [Google Scholar]
- Raderschall, E., E.I. Golub, and T. Haaf. 1999. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. USA. 96:1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 3:155–168. [DOI] [PubMed] [Google Scholar]
- Smits, V.A., P.M. Reaper, and S.P. Jackson. 2006. Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr. Biol. 16:150–159. [DOI] [PubMed] [Google Scholar]
- Stewart, G.S., B. Wang, C.R. Bignell, A.M. Taylor, and S.J. Elledge. 2003. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 421:961–966. [DOI] [PubMed] [Google Scholar]
- Strom, L., H.B. Lindroos, K. Shirahige, and C. Sjogren. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. 16:1003–1015. [DOI] [PubMed] [Google Scholar]
- Stucki, M., J.A. Clapperton, D. Mohammad, M.B. Yaffe, S.J. Smerdon, and S.P. Jackson. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 123:1213–1226. [DOI] [PubMed] [Google Scholar]
- Syljuasen, R.G., C.S. Sorensen, J. Nylandsted, C. Lukas, J. Lukas, and J. Bartek. 2004. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation. Cancer Res. 64:9035–9040. [DOI] [PubMed] [Google Scholar]
- Takahashi, T.S., P. Yiu, M.F. Chou, S. Gygi, and J.C. Walter. 2004. Recruitment of Xenopus Scc2 and cohesin to chromatin requires the pre-replication complex. Nat. Cell Biol. 6:991–996. [DOI] [PubMed] [Google Scholar]
- Tashiro, S., J. Walter, A. Shinohara, N. Kamada, and T. Cremer. 2000. Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J. Cell Biol. 150:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal, E., A. Arbel-Eden, U. Sattler, R. Shroff, M. Lichten, J.E. Haber, and D. Koshland. 2004. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell. 16:991–1002. [DOI] [PubMed] [Google Scholar]
- van Attikum, H., and S.M. Gasser. 2005. The histone code at DNA breaks: a guide to repair? Nat. Rev. Mol. Cell Biol. 6:757–765. [DOI] [PubMed] [Google Scholar]
- Ward, I.M., K. Minn, and J. Chen. 2004. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 279:9677–9680. [DOI] [PubMed] [Google Scholar]
- Yazdi, P.T., Y. Wang, S. Zhao, N. Patel, E.Y. Lee, and J. Qin. 2002. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 16:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, Z., C. Chahwan, J. Bailis, T. Hunter, and P. Russell. 2005. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol. Cell. Biol. 25:5363–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., and H. Piwnica-Worms. 2001. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21:4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, B.B., and S.J. Elledge. 2000. The DNA damage response: putting checkpoints in perspective. Nature. 408:433–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}