

Abstract
Understanding the mechanisms controlling cancer cell invasion and metastasis constitutes a fundamental step in setting new strategies for diagnosis, prognosis, and therapy of metastatic cancers. LIM kinase1 (LIMK1) is a member of a novel class of serine–threonine protein kinases. Cofilin, a LIMK1 substrate, is essential for the regulation of actin polymerization and depolymerization during cell migration. Previous studies have made opposite conclusions as to the role of LIMK1 in tumor cell motility and metastasis, claiming either an increase or decrease in cell motility and metastasis as a result of LIMK1 over expression (Zebda, N., O. Bernard, M. Bailly, S. Welti, D.S. Lawrence, and J.S. Condeelis. 2000. J. Cell Biol. 151:1119–1128; Davila, M., A.R. Frost, W.E. Grizzle, and R. Chakrabarti. 2003. J. Biol. Chem. 278:36868–36875; Yoshioka, K., V. Foletta, O. Bernard, and K. Itoh. 2003. Proc. Natl. Acad. Sci. USA. 100:7247–7252; Nishita, M., C. Tomizawa, M. Yamamoto, Y. Horita, K. Ohashi, and K. Mizuno. 2005. J. Cell Biol. 171:349–359). We resolve this paradox by showing that the effects of LIMK1 expression on migration, intravasation, and metastasis of cancer cells can be most simply explained by its regulation of the output of the cofilin pathway. LIMK1-mediated decreases or increases in the activity of the cofilin pathway are shown to cause proportional decreases or increases in motility, intravasation, and metastasis of tumor cells.
Introduction
The ability of cancer cells to disseminate from primary tumors (and metastases) gives rise to a growing tumor burden that is distributed in multiple sites in the body, resulting in death for many cancer patients. Understanding the steps at the cellular level that are used by cancer cells during invasion and metastasis can form the basis for new diagnostic, prognostic, and therapeutic approaches that allow control of cancer malignancy.
Microarray-based expression analysis of whole tumors has been used to identify genes involved in metastasis and patterns of gene expression that would give an indication of the likelihood of metastasis of breast tumors (van't Veer et al., 2002; Ramaswamy et al., 2003). These studies have successfully identified patterns of gene expression that correlate with metastasis; however, these patterns have not been informative with regard to the mechanisms contributing to metastasis because the expression patterns of all cells of the tumor are averaged (Wang et al., 2005). We have developed an in vivo invasion assay that provides an opportunity to collect primary tumor cells that are actively in the process of invasion. The in vivo invasion assay has been combined with array-based gene expression analyses to investigate the gene expression patterns of carcinoma cells in primary mammary tumors during invasion (Wang et al., 2004). The expression of genes involved in cell division, survival, and motility were most dramatically changed in invasive cells, indicating a population that is neither dividing nor apoptotic but intensely motile (Goswami et al., 2004; Wang et al., 2004). In particular, the genes coding for the three end stage effectors (Arp2/3 complex, capping protein, and cofilin) of the minimum motility machine that regulates actin polymerization at the leading edge and, therefore, the motility and chemotaxis of carcinoma cells were up-regulated. Interestingly, in the cofilin pathway, LIM kinase 1 (LIMK1) and cofilin were coordinately up-regulated in invasive cells (Wang et al., 2004). Chemotaxis to EGF is an important determinant in the haematogenous metastasis of mammary tumors (Wyckoff et al., 2004), and cofilin is required for chemotaxis to EGF (Mouneimne et al., 2004). Thus, it is essential to understand how the LIMK–cofilin pathway contributes to metastasis.
LIMK1 is a member of a novel class of serine–threonine protein kinases that contain two tandem LIM domains at the amino terminus, a PDZ domain in the central region, and a protein kinase domain at the carboxy terminus (Okano et al., 1995). Cofilin is the only known physiological substrate of LIMK1. LIMK1 phosphorylates cofilin, which inactivates it. This inhibits cofilin's actin-severing and depolymerization activities (Arber et al., 1998; Yang et al., 1998; Zebda et al., 2000).
Cofilin activity is required for tumor cell motility and invasion. Local activation of cofilin by uncaging induces lamellipod formation and sets the direction of cell motility (Ghosh et al., 2004). Inhibition of cofilin activity in carcinoma cells with either siRNA (Hotulainen et al., 2005) or the expression of constitutively active LIMK domain (Zebda et al., 2000) inhibits cell motility. The suppression of cofilin expression with siRNA reduces the invasion of carcinoma cells by reducing the assembly and stability of invadopodia (Yamaguchi et al., 2005a). The overexpression of cofilin increases the velocity of cell migration in Dictyostelium (Aizawa et al., 1996) and in human glioblastoma cells (Yap et al., 2005). The spontaneous overexpression of cofilin has been detected in the invasive subpopulation of tumor cells in mammary tumors (Wang et al., 2004). Cofilin is overexpressed in the highly invasive C6 rat glioblastoma cell line (Gunnersen et al., 2000), and the amount of phosphorylated, inactive cofilin is decreased in cell lines derived from T lymphoma (Jurkat) and carcinomas from the cervix (HeLa), colon (KM12), liver (HepG2), and kidney (COS1; Nebl et al., 1996). These results suggest that cofilin activity enhances, but LIMK activity inhibits, cell motility and metastasis.
In previous studies, LIMK1 has been shown to affect cell motility and invasion, but there is confusion about whether LIMK increases or decreases invasion. The expression of a constitutively active LIMK1 that increases the amount of phospho-cofilin in vivo inhibits actin polymerization and motility in response to EGF in mammary carcinoma cells (Zebda et al., 2000). In addition, the overexpression of LIMK suppresses motility in neuroblastoma, and dominant-negative LIMK increases it (Nishita et al., 2005). However, the overexpression of LIMK1 in prostate epithelial cells increased their invasiveness in vitro (Davila et al., 2003) and the formation of osteolytic lesions in nude mice (Yoshioka et al., 2003). Furthermore, a reduction in LIMK1 expression in metastatic prostate cell lines decreased their ability to invade matrigel in vitro (Davila et al., 2003). Clearly, these studies do not agree as to the consequences of LIMK1 expression on invasion. Therefore, it is likely that LIMK1 expression alone is not involved directly in determining the motility and invasion status of carcinoma cells.
Cofilin, the substrate of LIMK1, is directly responsible for severing actin filaments and regulating actin polymerization and depolymerization during cell migration (Desmarais et al., 2005). It is required for cell motility because reduced cofilin expression levels correlate with the inhibition of locomotion (Hotulainen et al., 2005), whereas increased expression levels correlate with enhanced motility (Yap et al., 2005). Hence, cofilin is arguably the key effector in the cofilin pathway that determines cell migration and invasion, whereas LIMK1 may only serve to modify the level of cofilin activity. We propose that the output of the cofilin pathway—the generation of barbed ends during the first transient after EGF stimulation by cofilin, which is required for chemotaxis to EGF (Mouneimne et al., 2004)—is the key determinant of whether a carcinoma cell will be more or less motile and invasive. This hypothesis is based on results demonstrating that the activation of cofilin is sufficient to stimulate cell protrusion and motility and determine the direction of locomotion (Ghosh et al., 2004), that cofilin activity is required for the chemotaxis of carcinoma cells to EGF (Ghosh et al., 2004; Mouneimne et al., 2004), and that invasive cells isolated from mammary tumors overexpress cofilin and its upstream regulatory proteins (Wang et al., 2004). Therefore, if the activity status of the cofilin pathway can be altered by varying the expression of LIMK1, perhaps the metastatic potential of tumor cells might be related to LIMK1 expression.
In this study, we test this hypothesis by varying the expression of LIMK1 and its dominant-negative kinase domain in the rat MTLn3 carcinoma cell line to examine the effects on the activity of the cofilin pathway and invasion and metastasis of primary mammary tumors grown from these cells. The full-length LIMK1 and dominant-negative kinase domain were introduced into MTLn3 cells, and stable cell lines were obtained. The output of the cofilin pathway (barbed end production in response to EGF) was then measured and correlated with invasion and metastasis. Although other research groups have suggested that LIMK1 may play important roles in regulating breast tumor growth independently of cofilin (Davila et al., 2003; Roovers et al., 2003) and that overexpression of LIMK1 causes increased invasion directly (Yoshioka et al., 2003), our results indicate that the effects of LIMK1 on invasion, intravasation, and metastasis of cancer cells can be explained most simply by LIMK1's regulation of cofilin activity and cofilin-dependent cell motility.
Results
We have used a model of breast cancer that allows the direct examination of the chemotaxis and invasion of individual carcinoma cells in live metastatic primary tumors (Wang et al., 2004; Wyckoff et al., 2004). The metastatic tumor cell line used to create these model tumors is the MTLn3 cell line. The invasive cells isolated from rat mammary tumors overexpress cofilin and its upstream regulatory proteins in a pattern suggesting increased activation of the cofilin pathway (Wang et al., 2004). Therefore, we propose that the cofilin pathway is one of the key determinants of whether a carcinoma cell will be more or less invasive and metastatic. To test this hypothesis, we assessed the EGF-induced early barbed end transient in invasive cells, which are cells that were collected by means of the in vivo invasion assay using microneedles from the primary tumor (Wyckoff et al., 2004). The level of barbed ends in tumor cells after 1 min of EGF stimulation has been shown previously to depend on the output of the cofilin pathway (Mouneimne et al., 2004). Therefore, the number of barbed ends in invasive tumor cells was compared with that in FACS-sorted tumor cells from the primary tumor, a population composed primarily of noninvasive cells (Wang et al., 2004). The results show that the EGF-induced cofilin-dependent early transient of barbed ends is increased in the invasive tumor cell population, indicating that the cofilin pathway regulating the early transient has increased activity (Fig. 1, A and B).
Figure 1.
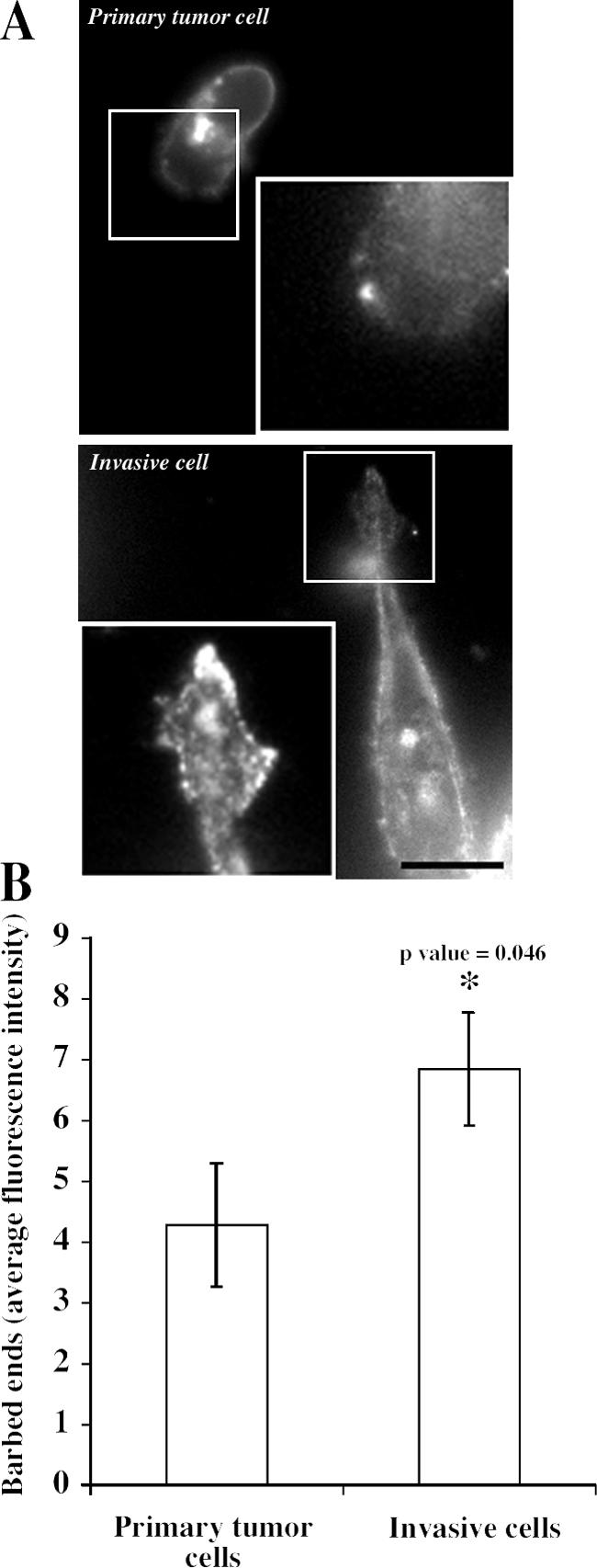
EGF-induced and cofilin-dependent early transient of barbed ends is increased in invasive tumor cells. (A) F-actin staining in control resident tumor cells isolated from the primary tumor by FACS sorting (primary tumor cells) compared with invasive tumor cells isolated from the primary tumor using the in vivo invasion assay. Cells are shown 1 min after EGF stimulation. Insets show the barbed end staining for the cropped areas. Bar, 10 μm. (B) Quantification of the barbed end staining at 1 min after EGF stimulation. Error bars represent SEM from at least 15 cells collected in three independent experiments.
To further test the hypothesis that the output of the cofilin pathway (the generation of barbed ends during the first transient of actin polymerization after EGF stimulation by cofilin) is the key determinant of whether a carcinoma cell will be more or less invasive and metastatic, four LIMK1 constructs were transfected separately into the parental MTLn3 cells. Stable transfectants were obtained for the full-length GFP-LIMK1 and dominant-negative GFP-KS (Fig. 2 A), which grew normally compared with control cells (MTLn3 cells transfected with pGreenLantern-1 vector). However, no stable cell lines were obtained from GFP-FS (inactive GFP-tagged LIMK1 Short), which is consistent with an involvement of LIMK1 in regulating cell cycle dynamics independently of cofilin phosphorylation (Roovers et al., 2003). In addition, no stable cell lines were obtained with GFP-K (constitutively active GFP-tagged kinase domain of LIMK1), which is consistent with (1) the ability of the constitutively active LIMK domain to completely inhibit cofilin activity measured as cofilin-mediated barbed end formation and protrusion (Zebda et al., 2000) and (2) the requirement of cofilin activity for cell viability (Gunsalus et al., 1995). Western blot analysis (Fig. 2 B) confirmed the expression of GFP-LIMK1 and GFP-KS at approximately two to four times the levels of the endogenous LIMK1. These cell lines will hereafter be called GFP (GFP-expressing control), F (full-length LIMK-expressing cells), and KS (LIMK dead truncated). Although the total cofilin levels remained unchanged in all of the transfectants (Fig. 2 B), the phosphorylation of cofilin was greatly increased in F cells, decreased in KS cells, and unchanged in GFP cells (Fig. 2 B).
Figure 2.

Expression of LIMK constructs alters the phosphorylation of cofilin in vivo. (A) The constructs that were used to generate stable expression in F (LIMK overexpressers) and in KS (LIMK dead domain) cells. (B, left) Western blot showing the expression levels of the different LIMK1 constructs, cofilin, and β-actin in the different cell lines. (right) IEF gel showing the ratio of phospho-cofilin to cofilin in the three different cell lines. Cofilin phosphorylation was either unchanged (GFP), elevated (F), or suppressed (KS) in the three cell lines. The level of expression of total cofilin (both phospho-cofilin and nonphospho-cofilin) was unchanged between GFP, F, and KS cells. Error bars represent SEM.
The early barbed end transient involved in chemotactic sensing is suppressed in F cells and enhanced in KS cells
To study the effects of LIMK expression on the EGF-induced free barbed end production, we examined the levels of free barbed ends in GFP, F, and KS cells at 1 min after EGF stimulation. This time corresponds to the cofilin-dependent early barbed end transient in MTLn3 cells that is required for chemotaxis (Mouneimne et al., 2004). Overexpression of the full-length LIMK (F) in MTLn3 suppressed the early transient, whereas expression of the kinase-dead LIMK1 kinase domain (KS) induced a significant increase (P < 0.05) in the levels of barbed ends during the early transient (Fig. 3).In addition, resting unstimulated KS cells (Fig. 3 B, 0 min) showed a significant elevation of free barbed ends as compared with F and GFP cells (P < 0.05). Knockdown of the exogenously expressed LIMK (F) and KS domain using siRNA that targets GFP (F and KS are GFP tagged) returned phospho-cofilin levels for both F (from 66 ± 6% in F to 17.3 ± 0.9% after siRNA knockdown) and KS (from 11.0 ± 1.7% in KS to 19.6 ± 1.8% after siRNA knockdown) to that of control GFP cells (18.0 ± 1.5%) and rescued the barbed ends to control levels (Fig. 3 B). GFP knockdown in the cells expressing GFP alone did not have any effect on EGF-induced barbed ends (Fig. 3 B).
Figure 3.

LIMK overexpression (F) suppresses the early barbed end transient, whereas inactive kinase domain (KS) increases the barbed end output of the cofilin pathway. (A) Representative images of GFP, F, and KS cells before and 1 min after EGF stimulation show that the barbed end staining at the leading edge is elevated in KS and suppressed in F cells. Bar, 10 μm. (B) The relative number of barbed ends (arbitrary units of fluorescence intensity) at the leading edge (between 0 and 0.22 μm inside the cell edge) in GFP, GFP RNAi, F, F RNAi, KS, and KS RNAi cells at 0 and 1 min after EGF stimulation (black bars represent 0 min, and gray bars represent 1 min after EGF stimulation) showing that barbed end staining at the leading edge is elevated in KS and suppressed in F cells, whereas siRNA suppression of F and KS expression returns barbed ends to control levels seen in GFP cells. Error bars represent SEM of ∼25 cells pooled from three independent experiments.
These results are consistent with the elevated phospho-cofilin levels seen in F cells and the decreased phospho-cofilin observed in KS cells (Fig. 2 B) because the phosphorylation of cofilin inhibits its severing activity, which is responsible for the early transient of barbed ends at 1 min after EGF stimulation (Mouneimne et al., 2004; Desmarais et al., 2005).
EGF-induced membrane protrusion and locomotion is inhibited in F but not KS cells
Chemotaxis toward EGF is a key step in the invasion and intravasation of carcinoma cells in the primary tumor in vivo (Wyckoff et al., 2004), and these behaviors involve the protrusion of lamellipods and invadopods (Mouneimne et al., 2004; Yamaguchi et al., 2005a). To determine the effects of LIMK-dependent cofilin phosphorylation on membrane protrusion, EGF-stimulated GFP, F, and KS cells were examined for membrane protrusion activity and motility. The EGF-induced membrane protrusion was dramatically inhibited in F but not KS cells (Fig. 4 A, i and ii). Knockdown of F with siRNA directed to GFP rescued the inhibition of protrusion such that F cells transfected with GFP-directed siRNA protruded identically to control cells (Fig. 4 A, i). Interestingly, when wild-type cofilin was stably overexpressed in F cells (Fig. 4 B, F + CF), which overexpress LIMK, the inhibition of protrusion was also rescued, indicating that the elevation of cofilin expression is sufficient to restore protrusion in cells overexpressing LIMK (Fig. 4 A, iii). This demonstrates that the effect of F and KS overexpression on barbed ends and protrusion is caused by the direct perturbation of the cofilin pathway and is mediated by cofilin.
Figure 4.

LIMK but not KS overexpression inhibits EGF-induced membrane protrusion and locomotion in a cofilin-dependent manner. (A) The average fold increase in membrane protrusion (area standardized over 0-s values) versus time after EGF addition in seconds shows that (i) F cells are dramatically suppressed in their ability to protrude lamellipods in response to EGF, whereas GFP-directed RNAi transfection rescues the observed phenotype to normal; (ii) KS has no effect on protrusion; and (iii) overexpression of cofilin in the F cells also rescues the defect in protrusion as a result of F expression. Y axes are normalized axes where 1.0 = no activity. (B) When wild-type cofilin was stably overexpressed in F cells, the level of total cofilin (both phospho-cofilin and nonphospho-cofilin) was increased to 2.5 times the level seen in the G and F cells, as determined by Western blotting. The proportional phosphorylation of cofilin was greatly increased in F cells (66%) and dropped to 34% in the F + CF cells. (C) Instantaneous velocity of GFP, F, and KS cells as assessed by DIAS analysis of 1-h time-lapse videos of cells maintained in serum-supplied media (constitutive motility) demonstrates a lower velocity in F cells and an increased velocity in KS cells. Error bars represent SEM of ∼30 cells. *, P < 0.05; **, P < 0.01.
Next, we determined the effects of LIMK-dependent cofilin phosphorylation on random cell motility. Under normal growth conditions, the velocity of F cells was significantly slower than that of GFP cells (P < 0.05), whereas the velocity of KS cells was elevated compared with control cells (P < 0.01; Fig. 4 C).
Chemotaxis, invasion, intravasation, and metastasis depend on cofilin activity
During mammary tumor progression, tumors fill with collagen-rich extracellular matrix. Tumor cells are induced by macrophages to migrate on collagen fibers to blood vessels, where they intravasate (Condeelis and Segall, 2003; Wyckoff et al., 2004). We used an in vitro invasion assay that reconstitutes the macrophage-induced invasion of collagen by tumor cells (Goswami et al., 2005) to examine the invasive properties of GFP, F, and KS cells. When cultured alone, <5% of the GFP, F, and KS cells invade the collagen gel, whereas in the presence of BAC1.2F5 macrophages, >20% of the GFP and KS cells invade the collagen, and F cells showed significantly reduced invasion (13%; Fig. 5 A).
Figure 5.
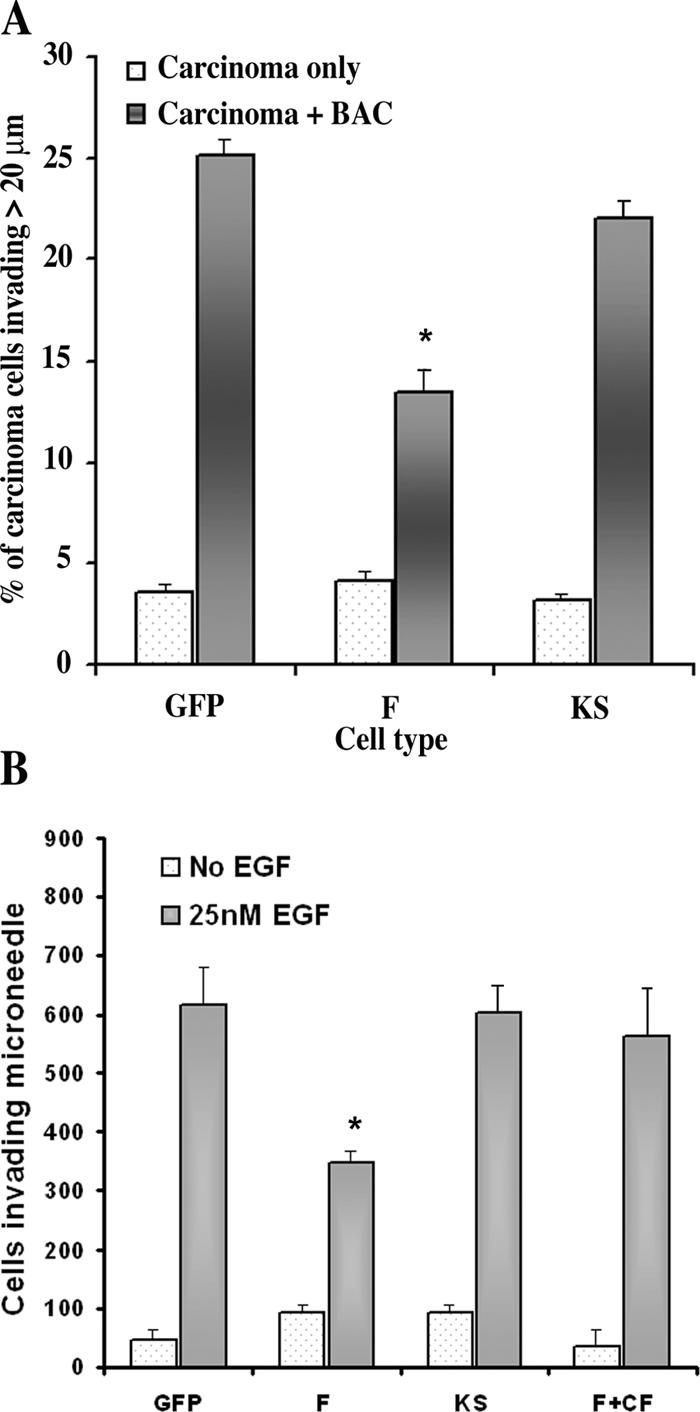
LIMK1 expression inhibits chemotaxis and invasion in vitro and in vivo. (A) The invasion of collagen by tumor cells in response to macrophages is suppressed in F cells. (B) The ability of carcinoma cells to invade microneedles containing matrigel and EGF when placed into primary tumors derived from GFP, F, and KS cells was measured. Tumors derived from F cells were reduced in invasion ability, and invasion was rescued by the overexpression of cofilin. Needles that do not contain EGF represent background. Error bars represent SEM. *, P < 0.05.
To investigate the chemotactic and invasive properties of tumor cells in tumors derived from the GFP, F, and KS cells, the ability of the carcinoma cells to chemotax and invade in the in vivo invasion assay was measured (Wang et al., 2004; Wyckoff et al., 2004). Although the chemotaxis of GFP and KS was similar to each other and was unchanged from wild-type cells in primary tumors, tumors generated from F cells had invasion scores that were significantly reduced, indicating a reduction in chemotaxis and invasion in vivo. However, when wild-type cofilin was stably overexpressed in F cells, invasion was rescued, indicating that the decrease in invasion in F cells was caused by a decrease in the activity of the cofilin pathway (Fig. 5 B).
The amount of intravasation and metastasis of mammary tumors derived from GFP, F, and KS cell lines was characterized by measuring the number of viable tumor cells present in the circulating blood (Fig. 6 A) and the number of spontaneously occurring lung metastatic tumors (Fig. 6 B). The results indicate that F tumors have reduced intravasation (approximately threefold) and metastasis (twofold), whereas KS tumors have significantly enhanced intravasation and metastatic activity when compared with GFP tumors.
Figure 6.
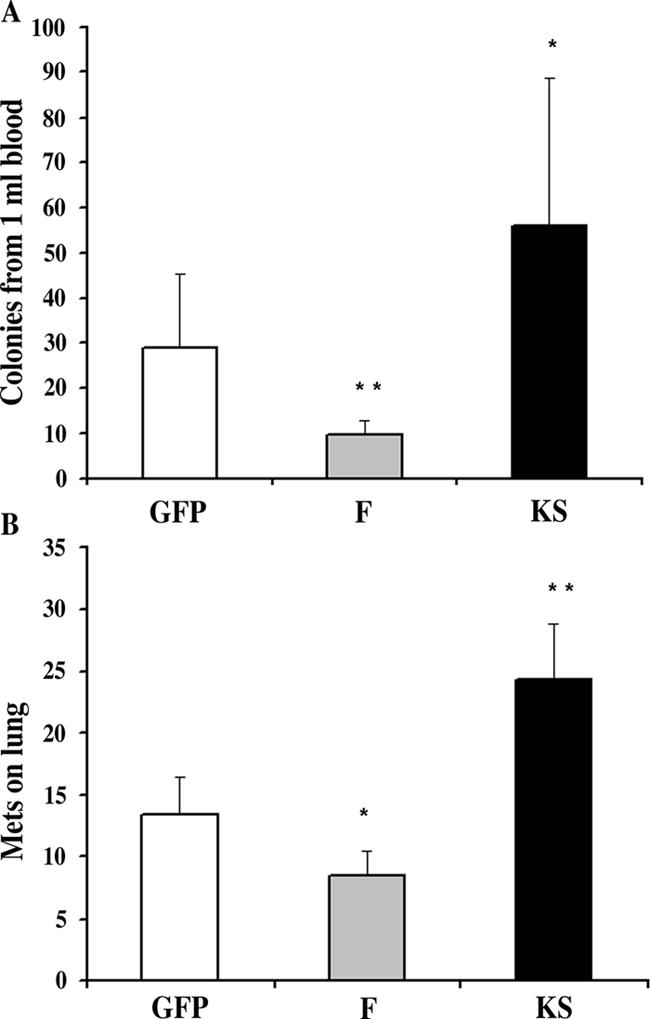
Intravasation and metastasis are determined by the activity of cofilin. (A) The efficiency of intravasation measured as the number of tumor cells present in circulating blood. (B) The number of lung metastatic tumors was reduced in animals with tumors prepared with F cells and increased in tumors prepared with KS cells. Error bars represent SEM. *, P < 0.05; **, P < 0.01 by Mann-Whitney test.
The survival of animals with F and KS tumors is different
The consequence of increased metastatic spread of breast tumors is decreased survival of the host. Therefore, we measured the survival of animals with mammary tumors derived from GFP, F, and KS cells. Fig. 7 A shows the survival curves of the populations of tumor-bearing animals. Animals with F tumors in which LIMK1 is overexpressed in tumor cells, resulting in lower cofilin activity, displayed significantly increased survival, whereas animals with KS tumors in which the amount of phosphorylated cofilin is decreased and, therefore, cofilin activity is increased had dramatically shortened survival. The ability of LIMK1 expression to increase survival was not significantly enhanced in animals bearing tumors that were generated from cells expressing sixfold more LIMK1 when compared with cells expressing threefold more LIMK1 (unpublished data).
Figure 7.
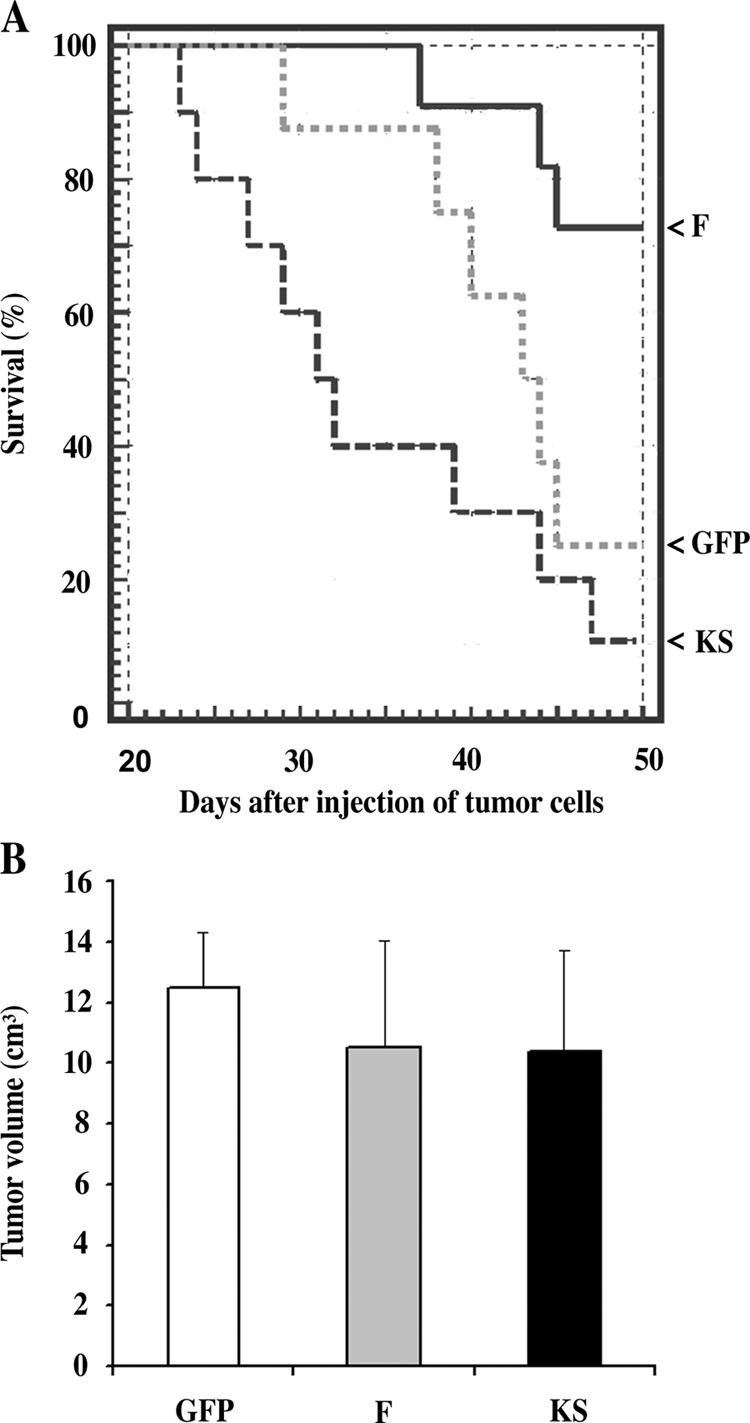
Overexpression of LIMK1 increases and KS decreases the survival of animals with mammary tumors. (A) The 50% survival times for animals bearing GFP and KS tumors are 44 and 32 d, respectively. In the case of F (LIMK1), at least 70% of the animals were still alive after 49 d. (B) Tumor growth was not affected in tumors prepared from F, KS, and GFP cells. Error bars represent SEM.
Finally, tumor growth was not significantly affected by altering the expression of F and KS (Fig. 7 B), indicating that the changes in metastatic potential and survival resulting from F and KS expression were not caused by changes in tumor mass.
LIMK expression is suppressed in tumor cells that successfully metastasize to the lung
To investigate why some animals with F tumors developed lung metastases and died (Figs. 6 and 7), we determined the expression status of LIMK in lung metastases that were spontaneously formed from mammary tumors. Total LIMK1 expression (both the endogenous and exogenous genes) in the primary tumors and lung metastases of animals with GFP, F, and KS tumors was examined by real-time PCR (Fig. 8). As shown in Fig. 8, tumor cells in lung metastases that formed spontaneously from primary tumors derived from F cells had lost the elevated expression of LIMK and now had wild-type levels of LIMK expression. To determine whether elevated LIMK may not be permissive for tumor cell growth in the lung, an experimental metastasis method was used in which the GFP, F, or KS cells were injected directly into the bloodstream to form lung metastases, thereby bypassing invasion and intravasation. In these experimentally derived lung metastases, the expression of LIMK remained elevated, as seen in tumors derived from F cells. These results indicate that elevated levels of LIMK are not permissive for metastasis to lung but are permissive for the growth of metastases in the lung.
Figure 8.

Expression of LIMK1 is suppressed in invading and metastatic tumor cells but not in tumor cells growing in the lung. Resident total primary tumor cells were isolated by FACS-based GFP expression in the cells. Invading cells were collected with the in vivo invasion assay. Lung metastases were isolated from single cell suspensions of lung by FACS based on the GFP expression in the tumor cells. RNA was isolated from the aforementioned cells and analyzed by quantitative real-time PCR for the level of LIMK1 mRNA. Invasive tumor cells as well as tumor cells that have spontaneously metastasized to the lung from the primary tumor have reduced LIMK expression. LIMK levels in tumor cells growing in the lung after intravenous injection of tumor cells (experimental metastasis) remained high, indicating that elevated LIMK is permissive for growth in the lung but not spontaneous metastasis. Error bars represent SEM.
To determine whether elevated LIMK expression is maintained in tumor cells during invasion, the in vivo invasion assay was performed in GFP, F, and KS tumors, and the collected tumor cells were subjected to real-time PCR as described in Materials and methods. In GFP tumors, the LIMK1 expression (along with cofilin expression; unpublished data) in the invading cells is increased compared with all tumor cells of the primary tumor, as observed previously in invasive cells from MTLn3-derived primary tumors (Wang et al., 2004). However, in F tumors, although the expression of total LIMK1 in the primary tumor is high as a result of expression from the exogenous LIMK1 gene, the expression of total LIMK1 in the invading cells was reduced to levels observed in lung metastases from F tumors (Fig. 8). In KS tumors, the expression of total LIMK1-related genes (in this case, endogenous LIMK1 plus exogenous KS) was high in the primary tumor, remained elevated in the invading tumor cells, but was equal to total LIMK expression in GFP and F in the lung metastases. These results indicate that elevated levels of LIMK in F tumors are not permissive for invasion in the primary tumor, whereas elevated KS expression is tolerated.
Discussion
As discussed above, previous studies have come to contradictory conclusions as to the role of LIMK1 in tumor cell motility, invasion, and metastasis, claiming both increases and decreases in cell motility and invasion as a result of LIMK1 overexpression. Therefore, it is unlikely that LIMK1 expression alone directly determines the motility, invasion, and metastasis status of carcinoma cells. LIMK1 is part of the cofilin pathway that regulates barbed end formation during tumor cell chemotaxis in response to EGF (Ghosh et al., 2004; Mouneimne et al., 2004) and is one of the genes up-regulated, along with cofilin, Rho-associated coil-containing protein kinase, and PKCζ, in invasive tumor cells during invasion and metastasis (Goswami et al., 2004; Wang et al., 2004). These results suggest that it is the overall activity of the cofilin pathway and not that of LIMK1 alone that determines the invasive and metastatic phenotype of tumor cells. In this study, we tested this hypothesis by varying the expression of LIMK1 and its dominant-negative kinase domain in the rat MTLn3 carcinoma cell line to examine the effects of LIMK on the activity of the cofilin pathway and, consequently, on the invasion and metastasis of primary mammary tumors grown from these cells. Our results demonstrate that the effect of LIMK1 expression on invasion, intravasation, and metastasis of cancer cells can be most simply explained by the regulation and activity status of the cofilin pathway. When LIMK1 overexpression (F cells) decreases the activity of the cofilin pathway (the EGF-induced early barbed end transient), the invasion and metastasis of tumor cells is proportionately decreased, whereas if the activity of the cofilin pathway is unchanged or increased, as in the GFP and KS cells, the chemotaxis, invasion, and metastasis of the tumor cells is either unaffected or increased. Furthermore, our study indicates that the increased expression of LIMK1 in carcinoma cells significantly reduces their cell motility as the phosphorylation of cofilin by LIMK1 abolishes EGF-stimulated actin nucleation, protrusion, and chemotaxis in vitro and invasion, intravasation, and metastasis in vivo. The in vitro results in particular are consistent with previous studies linking the activity of the cofilin pathway to EGF-stimulated directional sensing, protrusion, and chemotaxis (Zebda et al., 2000; Ghosh et al., 2004; Mouneimne et al., 2004).
The results reported in this study are consistent with a previous study demonstrating that the expression of the kinase domain of LIMK1, which results in the near total phosphorylation of cofilin, inhibits EGF-stimulated actin nucleation, protrusion, and motility in tumor cells (Zebda et al., 2000). This phenotype is caused by cofilin inhibition because these effects could be rescued by the expression of S3A cofilin, which cannot be phosphorylated by LIMK1 (Zebda et al., 2000), whereas the similar results reported in this study for F cells could be rescued by the overexpression of wild-type cofilin. The results reported in this study are also consistent with the inhibition of cell motility by the overexpression of LIMK1 in Ras-transformed Swiss 3T3 fibroblasts (Sahai et al., 2001).
It is interesting to note that in cells expressing KS, although barbed ends are slightly elevated, protrusion and invasion in vitro and in vivo are not significantly affected compared with GFP control cells. However, intravasation, metastasis, and morbidity are significantly increased in tumors derived from KS cells. This observation is consistent with a recent study (Yamaguchi et al., 2005a) showing that cofilin affects the assembly and stability of invadopods that are required for intravasation. Furthermore, it is known that tumor cells exhibit two different types of cell motility in the primary tumor during invasion and intravasation: (1) high velocity amoeboid movement guided by the linear geometry of the extra cellular matrix; and (2) slow, directed invadopod-mediated extracellular matrix degradation at blood vessels where intravasation occurs (Condeelis and Segall, 2003; Yamaguchi et al., 2005b). Therefore, it is expected that metastasis is the sum of two different types of cell motility during invasion and intravasation. Cofilin can affect both types but may do so unequally. In particular, our results indicate that the slightly higher activity of cofilin in KS cells does not significantly affect protrusion and invasion of amoeboid cells but affects invadopod-dependent intravasation (Yamaguchi et al., 2005a). These results suggest that the effect of KS on metastatic phenotype is mainly on intravasation, which is the rate-limiting step for metastasis (Condeelis and Segall, 2003; Wyckoff et al., 2004).
A key feature of the cofilin pathway is the generation of actin filament barbed ends after stimulation with EGF. Cofilin-generated actin filament barbed ends are required for chemotaxis to EGF (Mouneimne et al., 2004), a property essential for invasion and metastasis (Condeelis and Segall, 2003; Wyckoff et al., 2004). The important role of barbed ends generated by the cofilin pathway in chemotaxis and invasion makes it essential to examine the output of this pathway in cells that have been manipulated for the expression of LIMK1. Other studies in which the overexpression of LIMK1 in tumor cell lines was reported to increase their motility and invasiveness in vitro (Davila et al., 2003; Yoshioka et al., 2003) might be resolved with our studies by measuring cofilin-dependent barbed ends during chemotaxis to determine whether the activity of the cofilin pathway is elevated in their models. Furthermore, the stability of LIMK expression in these experimental models should be checked to determine whether LIMK1 expression was suppressed during invasion and metastasis as observed in our study. For example, the inhibitory effect of the overexpression of LIMK1 on metastasis is emphasized by our observation that successful metastatic lung tumors derived from F tumors overexpressing LIMK1 have been selected by the tissue microenvironment for decreased LIMK1 expression independently of tumor growth. This observation may explain why our results appear inconsistent with previous studies in which the overexpression of LIMK1 in tumor cells has been reported to increase their ability to metastasize to bone in an experimental metastasis model (Yoshioka et al., 2003). To resolve this inconsistency, it would be necessary to determine the expression status of LIMK1 in the bone metastases.
Other research groups have shown that LIMK1 may play a role in regulating cell division in a prostate cancer cell line PC3 and mouse embryo fibroblasts in vitro (Davila et al., 2003; Roovers et al., 2003). However, our results demonstrate that tumor growth was not significantly affected by altering the expression of LIMK1 in F- and KS-derived mammary tumors during growth in vivo, indicating that the changes in invasive and metastatic potential resulting from F and KS expression were not caused by changes in tumor growth and/or mass. The different results from our group and others may result from the different cell types used in these studies (for example, mammary vs. prostate cancer cells) and differences between growth in the local tumor microenvironment in vivo as used in our study and growth observed in vitro in the fibroblast and PC3 studies (Davila et al., 2003; Roovers et al., 2003).
Microarray analyses of invasive tumor cells collected in the in vivo invasion assay indicate that LIMK1 is more highly expressed in the most invasive cells compared with the less invasive cells that are residents of the primary tumor (Wang et al., 2004). This observed increase in LIMK1 expression could lead to the incorrect conclusion that the overexpression of LIMK1 is sufficient for increased invasion and metastasis. The most important implication of our study is that looking at the expression status of a single gene can be misleading because it is the collective activity of the pathway of which that gene product is a part that determines the metastatic phenotype of a tumor. In fact, in the microarray study, the elevated expression of LIMK1 was coupled with increased levels of the expression of cofilin, which occurred spontaneously (Wang et al., 2004). In this study, the experimental overexpression of both cofilin and LIMK is required for the activation of the cofilin pathway resulting in EGF-stimulated protrusion, cell motility, invasion, and metastasis. Our results indicate that in invasive cells collected from primary tumors, the stimulatory and inhibitory pathways regulating cofilin must be in balance in order for cells to be invasive and metastatic. The application of this idea to the analysis of microarrays of other pathways may lead to more accurate predictions of metastatic outcome in breast tumors. Our results also suggest that the phosphorylation status of cofilin in tumor cells within the primary tumor in situ may have value in predicting the metastatic potential of a breast tumor.
Materials and methods
LIMK constructs, cell culture, transfection, and generation of stable expression cell lines
Rat MTLn3 cells were maintained and grown to log phase before being used as described previously (Arber et al., 1998; Zebda et al., 2000). GFP-F (active GFP-tagged full-length LIMK1), GFP-FS (inactive GFP-tagged LIMK1 Short), GFP-K (active GFP-tagged kinase domain of LIMK1), and GFP-KS (inactive GFP-tagged kinase domain of LIMK1 Short) were described previously (Fig. 2; Arber et al., 1998). Rat nonmuscle cofilin cDNA was subcloned into the EcoRI/BamHI sites of pBabe-Puro, a retroviral vector with puromycin selection marker (Morgenstern and Land, 1990). To account for any effects that might arise from the introduction of EGFP into cells, MTLn3 cells transfected with pGreenLantern-1 vector (Life Technologies) were used as controls. The different constructs were introduced into cells by transfection using FuGene (Roche). Transfected MTLn3 cells were grown in the presence of 500 μg/ml geneticin or 1 μg/ml puromycin (Invitrogen) for 3–4 wk to select stable expressers. Cells expressing higher amounts of LIMK1 were further selected by increasing the concentration of geneticin to 4 mg/ml.
Phospho-cofilin and immunoblotting
Cells were cultured at low density (30%) and starved free of serum for at least 3 h before stimulation with EGF to assure that the cells at time = 0 were resting. Total cell lysates were resolved in 10% SDS-PAGE and subjected to immunoblotting using goat polyclonal antibody against the COOH terminus of LIMK1 (Santa Cruz Biotechnology, Inc.) so that both long and short forms of LIMK1 could be detected. Chicken polyclonal antibody against rat full-length cofilin was used for total cofilin. To measure the absolute value of phosphorylated cofilin, IEF gels (pH 3–10; Bio-Rad Laboratories) were used to separate phospho-cofilin from cofilin. Anti–β-actin antibody (Sigma-Aldrich) was simultaneously used as an internal control for loading. Three separated experiments were performed for all of the Western blotting, and statistical analysis was performed with a t test. It should be noted that this absolute method is different from the relative method used by Zebda et al. (2000) and gave different numerical values.
Imaging and measurements of free barbed ends and motility
All pictures were taken of live cultures at 37°C and of all cells using a 60× NA 1.4 infinity-corrected objective on a microscope (IX170; Olympus) supplemented with a computer-driven cooled CCD camera (SensiCam QE; Cooke) operated by IPLab Spectrum software (VayTek). Digital images were linearly converted in ImageJ software (National Institutes of Health [NIH]) and analyzed using macro analysis.
Cell culture and barbed end assay.
MTLn3 cells were maintained and cultured for the light microscopy experiments and quantization of free barbed ends as previously described (Mouneimne et al., 2004).
Membrane protrusion assay.
MTLn3 cells were starved for 3 h, stimulated with 5 nM EGF at 37°C, and time-lapse series were recorded for up to 10 min after the addition of EGF. Area fold change was quantitated by tracing cells and measuring their area in ImageJ at 1-min intervals after the EGF addition. Area measurements of each cell were standardized over the area of the corresponding cell at time = 0 and were averaged and plotted versus time after EGF addition.
Average velocity analysis.
Time-lapse series of 60 min (one frame per minute) were recorded for MTLn3 cells maintained in serum-supplied media. Cells were traced throughout all of the frames. Velocities from frame to frame were measured in Dynamic Image Analysis System (DIAS) for at least 20 cells pooled from three independent experiments.
GFP RNAi transfection.
GFP siRNA was purchased from QIAGEN, and transfection was performed as previously described (Mouneimne et al., 2004). The siRNA knockdown efficiency was determined by measuring the fluorescence of the residual GFP.
In vitro invasion assay
The in vitro invasion assay measures the ability of macrophages to induce the invasion of a dense collagen gel by tumor cells, mimicking a paracrine-induced invasion as observed in primary mammary tumors (Wyckoff et al. 2004). This assay was performed as described previously (Goswami et al., 2005). In brief, MTLn3-GFP (n = 80,000) were plated on a 35-mm dish (∼80 cells/mm2; MatTek) in the presence or absence of 200,000 BAC1.2F5 cells in 2 ml α-MEM with 10% FBS and 36 ng/ml CSF-1. After 16 h, cells were overlaid with a 750–1,000-μm layer of 5–6 mg/ml collagen I, which was allowed to gel for 90 min before the addition of 1 ml α-MEM with 10% chemically defined lipid mix (Invitrogen) and insulin-transferrin-selenium (Invitrogen). After 24 h, the assay was fixed with 4% formaldehyde and analyzed by confocal microscopy; optical z sections were taken every 5 μm starting at the base of the dish and extending at least 50 μm into the collagen gel. To quantify the invasion of MTLn3-GFP cells, GFP fluorescence in the z sections from 20 μm into the collagen and above was added and divided by the sum of GFP fluorescence in all of the z sections. The data shown represent the analysis of ≥200 cells with data collected from at least three independent experiments.
In vivo invasion assay
We used MTLn3-derived mammary tumors in rats and the in vivo invasion assay described previously (Wyckoff et al., 2004) to study cancer cell invasion. In brief, the in vivo invasion assay uses microneedles filled with matrigel and growth factors (EGF in this case) to collect the invasive tumor cells from primary tumors. Microneedles are held in a clamping devise and positioned in the primary tumor with a micromanipulator. Cell collection takes 2.5–4 h, and the number of tumor cells collected was counted as described previously (Wyckoff et al., 2004). For barbed end staining, the needle-collected invasive tumor cells and total resident tumor cells collected from the primary tumor by FACS sorting of GFP were plated on collagen-coated dishes (MatTek) for ∼12 h and were starved for 4 h before EGF stimulation. Control experiments include the exposure of FACS-sorted tumor cells to EGF to mimic the collection conditions used for invasive cells (Wang et al., 2004).
Blood burden, tumor cells in the lung, and metastases
MTLn3 cells were injected into the mammary fat pads of female Fischer 344 rats to form primary tumors. After tumor growth, tumor cell blood burden was determined as described previously (Wang et al., 2004). For the measurement of spontaneous metastases, after blood removal and euthanization of the rat, the lungs were removed, and the visible metastatic tumors near the surface of the lungs were counted as described previously (Wang et al., 2004). In a separate experiment, animals bearing MTLn3-LIMK1 (F), MTLn3-KS (KS), or MTLn3-GFP (GFP) tumors were used to observe the effect of LIMK1 on animal survival. At least nine animals were used in each group. The animals were dissected after the animal died, and the metastases were scored by checking the visible metastatic tumors on the lung surface. After 7 wk, all of the animals were killed and dissected to check the metastases.
To study experimental lung metastasis, 7.5 × 105 cells were injected into the lateral tail veins of 7-wk-old female rats. 2.5 wk after injection, the rats were killed, and the lungs were removed for isolation of carcinoma cells using the method described below.
Real-time PCR
To check the expression level of LIMK1 in the resident tumor and lung metastases, quantitative real-time PCR analysis was performed by using a sequence detection system (ABI PRISM 7900; Applied Biosystems) with sequence-specific primer pairs for LIMK1 as described previously (Wang et al., 2004). To isolate the carcinoma cells from primary tumor or lung, a small piece of tumor or lung tissue was minced and filtered twice through a nylon filter to obtain a single cell suspension. FACS was performed on the resulting single cell suspensions based on their GFP expression in tumor cells using a cell sorter (FACSVantage; Becton Dickinson). SYBR Green was used for real-time monitoring of amplification. Results were evaluated with ABI PRISM SDS 2.0 software (Applied Biosystems). The normalization of all samples was performed using at least two housekeeping genes (β-actin and glyceraldehyde-3-phosphate dehydrogenase).
Acknowledgments
We are grateful to members of the Analytical Imaging Facility for support and advice.
This work was supported by grants from the NIH (CA 100324 to A.R. Bresnick and GM38511 to J.S. Condeelis).
W. Wang and G. Mouneimne contributed equally to this paper.
Abbreviation used in this paper: LIMK, LIM kinase.
References
- Aizawa, H., K. Sutoh, and I. Yahara. 1996. Overexpression of cofilin stimulates bundling of actin filaments, membrane ruffling, and cell movement in Dictyostelium. J. Cell Biol. 132:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber, S., F.A. Barbayannis, H. Hanser, C. Schneider, C.A. Stanyon, O. Bernard, and P. Caroni. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809. [DOI] [PubMed] [Google Scholar]
- Condeelis, J., and J.E. Segall. 2003. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer. 3:921–930. [DOI] [PubMed] [Google Scholar]
- Davila, M., A.R. Frost, W.E. Grizzle, and R. Chakrabarti. 2003. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J. Biol. Chem. 278:36868–36875. [DOI] [PubMed] [Google Scholar]
- Desmarais, V., M. Ghosh, R. Eddy, and J. Condeelis. 2005. Cofilin takes the lead. J. Cell Sci. 118:19–26. [DOI] [PubMed] [Google Scholar]
- Ghosh, M., X. Song, G. Mouneimne, M. Sidani, D.S. Lawrence, and J.S. Condeelis. 2004. Cofilin promotes actin polymerization and defines the direction of cell motility. Science. 304:743–746. [DOI] [PubMed] [Google Scholar]
- Goswami, S., W. Wang, J.B. Wyckoff, and J.S. Condeelis. 2004. Breast cancer cells isolated by chemotaxis from primary tumors show increased survival and resistance to chemotherapy. Cancer Res. 64:7664–7667. [DOI] [PubMed] [Google Scholar]
- Goswami, S., E. Sahai, J.B. Wyckoff, M. Cammer, D. Cox, F.J. Pixley, E.R. Stanley, J.E. Segall, and J.S. Condeelis. 2005. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 65:5278–5283. [DOI] [PubMed] [Google Scholar]
- Gunnersen, J.M., V. Spirkoska, P.E. Smith, R.A. Danks, and S.S. Tan. 2000. Growth and migration markers of rat C6 glioma cells identified by serial analysis of gene expression. Glia. 32:146–154. [PubMed] [Google Scholar]
- Gunsalus, K.C., S. Bonaccorsi, E. Williams, F. Verni, M. Gatti, and M.L. Goldberg. 1995. Mutations in twinstar, a Drosophila gene encoding a cofilin/ADF homologue, result in defects in centrosome migration and cytokinesis. J. Cell Biol. 131:1243–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen, P., E. Paunola, M.K. Vartiainen, and P. Lappalainen. 2005. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell. 16:649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern, J.P., and H. Land. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18:3587–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouneimne, G., L. Soon, V. DesMarais, M. Sidani, X. Song, S.C. Yip, M. Ghosh, R. Eddy, J.M. Backer, and J. Condeelis. 2004. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J. Cell Biol. 166:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebl, G., S.C. Meuer, and Y. Samstag. 1996. Dephosphorylation of serine 3 regulates nuclear translocation of cofilin. J. Biol. Chem. 271:26276–26280. [DOI] [PubMed] [Google Scholar]
- Nishita, M., C. Tomizawa, M. Yamamoto, Y. Horita, K. Ohashi, and K. Mizuno. 2005. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J. Cell Biol. 171:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano, I., J. Hiraoka, H. Otera, K. Nunoue, K. Ohashi, S. Iwashita, M. Hirai, and K. Mizuno. 1995. Identification and characterization of a novel family of serine/threonine kinases containing two N-terminal LIM motifs. J. Biol. Chem. 270:31321–31330. [DOI] [PubMed] [Google Scholar]
- Ramaswamy, S., K.N. Ross, E.S. Lander, and T.R. Golub. 2003. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 33:49–54. [DOI] [PubMed] [Google Scholar]
- Roovers, K., E.A. Klein, P. Castagnino, and R.K. Assoian. 2003. Nuclear translocation of LIM kinase mediates Rho-Rho kinase regulation of cyclin D1 expression. Dev. Cell. 5:273–284. [DOI] [PubMed] [Google Scholar]
- Sahai, E., M.F. Olson, and C.J. Marshall. 2001. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 20:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Veer, L.J., H. Dai, M.J. van de Vijver, Y.D. He, A.A. Hart, M. Mao, H.L. Peterse, K. van der Kooy, M.J. Marton, A.T. Witteveen, et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415:530–536. [DOI] [PubMed] [Google Scholar]
- Wang, W., S. Goswami, K. Lapidus, A.L. Wells, J.B. Wyckoff, E. Sahai, R.H. Singer, J.E. Segall, and J.S. Condeelis. 2004. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 64:8585–8594. [DOI] [PubMed] [Google Scholar]
- Wang, W., S. Goswami, E. Sahai, J.B. Wyckoff, J.E. Segall, and J.S. Condeelis. 2005. Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol. 15:138–145. [DOI] [PubMed] [Google Scholar]
- Wyckoff, J., W. Wang, E.Y. Lin, Y. Wang, F. Pixley, E.R. Stanley, T. Graf, J.W. Pollard, J. Segall, and J. Condeelis. 2004. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64:7022–7029. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, H., M. Lorenz, S. Kempiak, C. Sarmiento, S. Coniglio, M. Symons, J. Segall, R. Eddy, H. Miki, T. Takenawa, and J. Condeelis. 2005. a. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168:441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, H., J. Wyckoff, and J. Condeelis. 2005. b. Cell migration in tumors. Curr. Opin. Cell Biol. 17:559–564. [DOI] [PubMed] [Google Scholar]
- Yang, N., O. Higuchi, K. Ohashi, K. Nagata, A. Wada, K. Kangawa, E. Nishida, and K. Mizuno. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 393:809–812. [DOI] [PubMed] [Google Scholar]
- Yap, C.T., T.I. Simpson, T. Pratt, D.J. Price, and S.K. Maciver. 2005. The motility of glioblastoma tumour cells is modulated by intracellular cofilin expression in a concentration-dependent manner. Cell Motil. Cytoskeleton. 60:153–165. [DOI] [PubMed] [Google Scholar]
- Yoshioka, K., V. Foletta, O. Bernard, and K. Itoh. 2003. A role for LIM kinase in cancer invasion. Proc. Natl. Acad. Sci. USA. 100:7247–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebda, N., O. Bernard, M. Bailly, S. Welti, D.S. Lawrence, and J.S. Condeelis. 2000. Phosphorylation of ADF/cofilin abolishes EGF-induced actin nucleation at the leading edge and subsequent lamellipod extension. J. Cell Biol. 151:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]