

Abstract
The human genome encodes ∼70 Rab GTPases that localize to the surfaces of distinct membrane compartments. To investigate the mechanism of Rab localization, chimeras containing heterologous Rab hypervariable domains were generated, and their ability to bind seven Rab effectors was quantified. Two chimeras could bind effectors for two distinctly localized Rabs; a Rab5/9 hybrid bound both Rab5 and Rab9 effectors, and a Rab1/9 hybrid bound to certain Rab1 and Rab9 effectors. These unusual chimeras permitted a test of the importance of effector binding for Rab localization. In both cases, changing the cellular concentration of a key Rab9 effector, which is called tail-interacting protein of 47 kD, moved a fraction of the proteins from their parental Rab localization to that of Rab9. Thus, relative concentrations of certain competing effectors could determine a chimera's localization. These data confirm the importance of effector interactions for Rab9 localization, and support a model in which effector proteins rely on Rabs as much as Rabs rely on effectors to achieve their correct steady state localizations.
Introduction
Human cells encode ∼70 Rab GTPases that are localized to distinct membrane-bound compartments (Pereira-Leal and Seabra, 2000, 2001; Colicelli, 2004). These proteins regulate transport vesicle formation, motility, docking, and fusion via interaction with so-called effector proteins that bind with preference to Rabs in their GTP-bound conformations (Zerial and McBride, 2001). The number of identified Rab effector proteins is growing steadily, yet little is known about how Rabs are localized correctly within cells (Pfeffer and Aivazian, 2004).
COOH-terminal prenylation contributes to the stable membrane association of Rab proteins. Rabs also interact with numerous effectors to form microdomains on organelle surfaces (Zerial and McBride, 2001). For example, Rab5 binds to early endosome antigen-1 (EEA1), which binds to early endosomes via Rab5 and also by binding to phosphatidylinositol-3-phosphate. Rab5 recruits the kinase that generates this lipid, thereby catalyzing the generation of a membrane microdomain. The Rab9 GTPase recruits a cytosolic protein, tail-interacting protein of 47 kD (TIP47), which binds both to Rab9 and to the cytoplasmic domains of two mannose 6-phosphate receptors (MPRs; Pfeffer, 2003). In both cases, combinatorial recognition of a Rab and a membrane constituent enhances the selective recruitment of a cytosolic effector protein. Thus, there are many examples of Rabs that serve as determinants of effector–membrane binding.
But how are Rabs themselves localized? Prenylated Rabs are delivered to membranes by a protein named GDP dissociation inhibitor (GDI; Pfeffer and Aivazian, 2004). Complexes of Rabs bound to GDI bear all of the information needed to accomplish their specific membrane delivery (Soldati et al., 1994; Ullrich et al., 1994). Proteins called GDI displacement factors (GDFs) may facilitate Rab recruitment; e.g., Yip3 protein was recently shown to be able to release Rab9 from GDI and lead to its membrane association (Sivars et al., 2003). But Yip3 is not a Rab receptor, and it acted catalytically to permit Rab9 to associate with membranes. Yip3 catalyzes the release of endocytic, but not exocytic, Rabs from GDI (Sivars et al., 2003). Thus, although GDFs likely contribute to Rab delivery, we know little about how they distinguish between Rab types or the breadth of their substrate recognition. Their partial specificity cannot by itself, explain the sequestration of this category of Rabs into early endosomes, recycling endosomes, or late endosomes. Moreover, steady-state localization of Rab proteins is likely to include interactions of Rabs with other constituents, after they are delivered to a membrane surface.
Early work on Rab localization suggested that COOH-terminal Rab hypervariable domains specified their distinct localizations (Chavrier et al., 1991; Brennwald and Novick, 1993; Stenmark et al., 1994). But more recent analyses suggest a more complex scenario. Ali et al. (2004) found that several Rabs were correctly localized, despite bearing significant alterations in their hypervariable domains. As described herein, we obtained similar findings for a different set of Rab chimeras, and we used this as a starting point to explore the mechanisms of Rab localization.
In this study, we present evidence that certain effectors may play a special role in Rab9 localization. Hints of this came from a recent study in which the Rab9 effector TIP47 was depleted from cells using RNAi (Ganley et al., 2004). Loss of most of the predominantly cytosolic TIP47 protein led to the destabilization of Rab9; its half-life decreased from 32 to 8 h. This was unexpected, because we think of prenylated Rabs as independent entities residing on organelle surfaces or as a complex with GDI in the cytosol. We show that TIP47 is a “key” effector, in that it controls Rab9 stability (Ganley et al., 2004), as well as its steady-state localization. In addition, TIP47 can compete with Rab1 and Rab5 effectors to relocalize Rab1 and Rab5 chimeras to late endosomes.
Results
We generated and purified hypervariable domain chimeras based on late endosome–localized Rab9, early endosome–localized Rab5, and Golgi-localized Rab1 (Fig. 1). The hypervariable domain junction was selected based on sequence alignments (Chavrier et al., 1990; Pereira-Leal and Seabra, 2000) and on the three-dimensional structures of Rab3A (Ostermeier and Brunger, 1999), Rab5C (Merithew et al., 2001), and Rab9 (Chen et al., 2004). Junctions were placed near the end of helix 5 at a dibasic repeat that is conserved in Rab9, Rab5, and Rab1 sequences (Fig. 1 A, blue residues). Most of the hypervariable domain is unstructured and extends beyond the GTPase fold (and is not included in the structures shown). By splicing sequences at or near the end of the structured portion, we hoped to avoid significant alterations in Rab structure.
Figure 1.
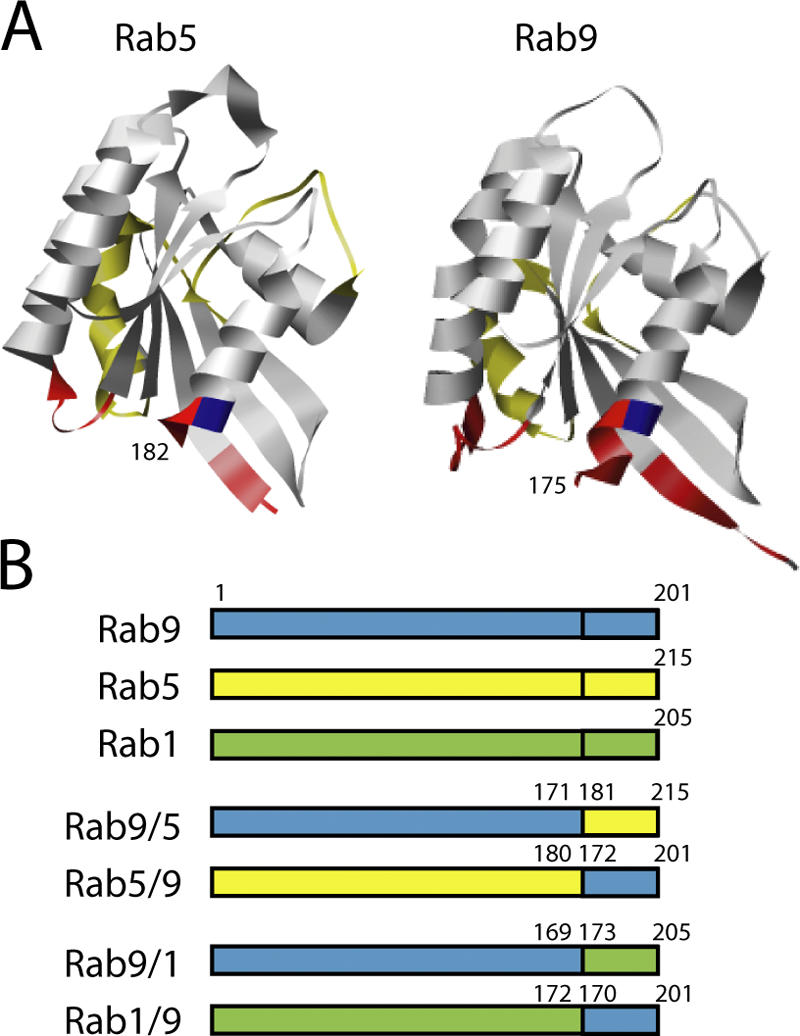
Rab chimeras studied. (A) Ribbon diagrams of the Rab5c-GppNHp (residues 19–182, left; Merithew et al., 2001) and Rab9-GDP (residues 2–175, right; Chen et al., 2004) structures (Pettersen et al., 2004). The so-called complementarity determining regions that are believed to be important for effector interaction (Ostermeier and Brunger, 1999) are highlighted in red. The first conserved basic residue that was used as the site of hypervariable domain exchange is highlighted in blue; the presumed Rab switch regions (Ostermeier and Brunger, 1999; Merithew et al., 2001) are highlighted in yellow. (B) Diagrams of the hypervariable domain chimeras described herein. Residue numbers indicate composition of the constructs; Rabs differ in length at both NH2 and COOH termini, so the junction numbers differ and are shown to represent the precise junction identities from the parental Rab sequences.
When expressed as GFP fusions in cells, the chimeras were efficiently prenylated (Fig. 2, A and B). In all cases, prenylation efficiency was ∼50%, as determined by comparative analysis of membrane and cytosolic fractions (Fig. 2, A and B). Membrane-associated forms migrated at a different mobility upon SDS-PAGE, which is consistent with prenylation. This suggests that the chimeras were folded well enough to be recognized by Rab prenyltransferase.
Figure 2.

Chimeric Rabs are prenylated and active for GTP binding. (A and B) Analysis of GFP-Rab membrane association in HeLa cells. HeLa cells were transiently transfected for GFP-Rab expression, and lysates were separated into membrane and cytosol fractions for analysis by SDS-PAGE and anti-GFP immunoblot. Bands shown in A were quantitated by ImageJ for display in B. (C) Nucleotide (GTPγS) binding to recombinant, purified GST-Rabs. Rabs were incubated with radiolabeled GTPγS, and the extent of binding was quantitated by nitrocellulose filter binding and scintillation counting. Error bars represent the SEM.
In addition, all of the purified chimeras were highly active for nucleotide binding and, in most cases, effector binding (Figs. 3 and 4 and Table I), which are both important tests of proper protein folding. Fig. 2 C shows the ability of the purified GST chimeras to bind radiolabeled GTP in a nucleotide exchange reaction. On average, these chimeras were ∼75% active in terms of their abilities to release bound GDP and to bind added GTP. Although there was slight variability between each purified Rab protein preparation, the 20% maximum differences observed could not account for the differences in effector binding shown in Figs. 3 and 4.
Figure 3.

The Rab9 hypervariable domain is necessary and sufficient for Rab9 effector binding. (A) Binding of purified, untagged Rab9, Rab1A, Rab9/1, and Rab1/9 to the Rab9 effectors His6-TIP47(152–434; closed bars) and His6-p40 (open bars). Data are normalized relative to the binding seen for parental Rabs. (B) Same as in A, with untagged Rab9, Rab5a, Rab9/5, and Rab5/9. (C) Binding of [35S]GTPγS- and [3H]GDP-loaded Rab9, Rab5/9, and Rab1/9 to His6-TIP47(152–434). 100% binding to p40 or TIP47 represented 3.9 or 1.5 pmol Rab9, respectively, in A and B. This assay measures active Rab molecules only; untagged Rab proteins were all ∼100% active, except for Rab9/5, which contained ∼30% active molecules. Error bars represent the SEM.
Figure 4.

Some, but not all, Rab1 and Rab5 effectors require hypervariable domain sequences for binding. (A) Binding of GST versions of Rab9, Rab5, Rab9/5, and Rab5/9 to Rab5 effectors EEA1 and rabaptin-5 from cytosol. (B) Binding of GST versions of Rab9, Rab1, Rab9/1, and Rab1/9 to Rab1 effectors GM130, golgin-84, and p115 from rat liver Golgi detergent extracts. Data are normalized relative to the binding seen for parental Rabs. Error bars represent the SEM.
Table I.
Rab effector binding correlates with localization
| Rab | Localization | Rab9 effector binding |
Rab5 effector binding |
Rab1 effector binding |
|---|---|---|---|---|
| Rab9 | Late endosomes | + | − | − |
| Rab9/5 | Golgi1,2 | − | − | nd |
| Rab9/1 | Golgi1,3 | − | nd | − |
| − | ||||
| Rab5 | Early endosomes | − | + | nd |
| Rab5/9 | Early endosomes4–6 | + | + | nd |
| Rab1 | Golgi | − | nd | + |
| Rab1/9 | Golgi1,7 | + | nd | + GM130, Golgin-84 |
Localizations of CFP- or YFP-tagged Rab chimeras were determined in HeLa cells by comparison with endogenous organelle markers, and in Bs-C-1 cells by comparison with overexpressed CFP- or YFP-tagged Rab GTPases or CFP-Golgi. Markers (superscripted in table) were as follows: 1, p115; 2, CFP-galactosyltransferase; 3, anti-TGN46; 4, CFP-Rab5; 5, anti-EEA1; 6, anti-rabaptin-5; 7, CFP-Rab1. nd, no data.
Fig. 3 shows the ability of each of the chimeras to bind to the Rab9 effectors TIP47 (Diaz and Pfeffer, 1998; Carroll et al., 2001) and p40 (Diaz et al., 1997). Pure, nonepitope-tagged Rab proteins were prebound to [35S]GTPγS; unbound nucleotide was removed by gel filtration, and the active proteins were incubated with His-tagged effectors, and then immobilized on an Ni-NTA resin. As expected, Rab9 bound specifically to TIP47 and p40; Rab1 and Rab5 showed very low binding ability (Fig. 3, A and B). The Rab9 hypervariable domain was required both for TIP47 and p40 binding, as neither Rab9/1 (Fig. 3 A) nor Rab9/5 (Fig. 3 B) bound significantly to either effector (Fig. 3). Surprisingly, Rab5/9 and Rab1/9 bound well to Rab9 effectors compared with the parental Rabs 1 and 5. Therefore, the Rab9 hypervariable domain is not only required for binding TIP47 and p40, but it is also sufficient for Rab9 effector binding (especially to TIP47) within the context of a Rab GTPase structure.
Although the hypervariable domain was sufficient for TIP47 binding to Rab5/9 and Rab1/9 chimeras, binding also required the presence of a GTPase domain. This was clear from experiments in which we tested the nucleotide dependence of the interaction (Fig. 3 C). For both Rab5/9 and Rab1/9, interaction with TIP47 showed the same nucleotide dependence of binding that is seen with Rab9 (Fig. 3 C), with a preference for the GTP-bound state. Thus, interaction with TIP47 requires more than just the Rab9 hypervariable domain, and likely involves the switch domains of Rab5/9 and Rab1/9, as these domains are the only parts of a Rab that change conformation between GTP- and GDP-bound states. In addition, Rab recognition by p40 appears to require more significant interaction with nonhypervariable domain sequences than Rab protein recognition by TIP47.
The same Rab proteins were next tested for binding to the Rab5 effectors rabaptin-5 (Stenmark et al., 1995) and EEA1 (Simonsen et al., 1998; Christoforidis et al., 1999), and to the Rab1 effectors GM130, golgin-84, and p115 (Fig. 4; Allan et al., 2000; Moyer et al., 2001; Satoh et al., 2003). In contrast to what was observed for Rab9 effector interactions, Rab5 binding to two of its effectors did not require Rab5 hypervariable domain sequences. As shown in Fig. 4 A, GST-Rab5, but not GST-Rab9, bound to the Rab5 effectors EEA1 and rabaptin-5 from bovine brain cytosol. GST-Rab5/9 was fully functional for binding both proteins, whereas active GST-Rab9/5 did not bind either effector. These data confirm that the Rab5 hypervariable domain is neither required nor sufficient for EEA1 (Merithew et al., 2003) or rabaptin-5 interaction. The published structure of COOH-terminal–truncated (and hypervariable domain–truncated) Rab5 bound to the coiled coil region of rabaptin-5 (Zhu et al., 2004) is consistent with the hypervariable domain independence of rabaptin-5 binding shown in this study.
Rab1 effector binding was studied using GST-Rab proteins and detergent-extracted rat liver Golgi membranes as an effector protein source (Fig. 4 B). As with the Rab5 effectors rabaptin-5 and EEA1, GM130 did not require the Rab1 hypervariable domain for binding, as it bound well to both GST-Rab1 and GST-Rab1/9 (Fig. 4 B). In contrast, the Rab1 hypervariable domain was more important for binding to both golgin-84 and p115 (Fig. 4 B). Rab1/9 failed to bind p115, and showed weaker binding to golgin-84. Although the Rab9 hypervariable domain was sufficient for TIP47 and p40 interaction (Fig. 3, A and B), the Rab1 hypervariable domain was necessary, but not sufficient, for golgin-84 and p115 interaction (Fig. 4 B). Therefore, these two Rab1 effectors also require nonhypervariable domain determinants for Rab recognition. The differences in the Rab-binding profiles of GM130, golgin-84, and p115 show that these Rab1 effectors rely, to different extents, on distinct binding determinants in the Rab1 structure.
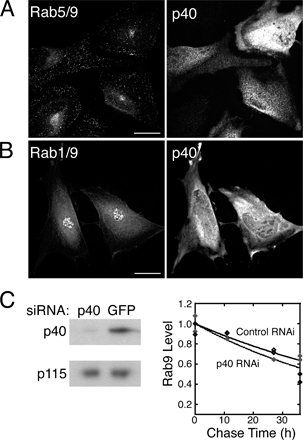
We determined the intracellular localizations of each of the chimeras as GFP-fusions in mammalian cells; the results are summarized in Table I.Rab5/9 was clearly present in early endosomes, as judged by colocalization with EEA1 (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200510010/DC1). Very little Rab5 or Rab5/9 was present in late endosomes (Fig. S2).
For the chimeras studied, the Rab5 hypervariable domain was dispensable for both effector binding (Fig. 4 A) and for early endosome localization (Fig. 5; Fig. S2). Rab5/9 and Rab5/7 (not shown) were present on numerous peripheral early endosomes, many located near the plasma membrane (Fig. 5 A; Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200510010/DC1).
Figure 5.

Colocalization of Rab5/9 with EEA1. (A) Colocalization of CFP-Rab5/9 with the early endosome marker EEA1 in fixed HeLa cells stained with anti-EEA1 and anti-GFP antibodies. The square in A is enlarged for both markers (right); arrows are included to facilitate comparison. Bar, 10 μm. (B) Quantitation of CFP-Rab5/9, CFP-Rab5, and YFP-Rab9 colocalization. Venn diagrams are shown as follows: CFP-Rab5/9, CFP-Rab5, and YFP-Rab9 versus EEA1; CFP-Rab5/9 and CFP-Rab5 versus endocytosed anti–CI-MPR IgG; and CFP-Rab5/9 and CFP-Rab5 versus YFP-Rab5. The number of positive structures is indicated below each protein name; overlap is indicated as described in the Results section.
Quantitation of Rab5/9 colocalization with endogenous EEA1, which is a marker for early endosomes, confirmed that Rab5/9 has a localization profile identical to that of Rab5. 71% of EEA1-positive structures contained exogenous Rab5/9; in a parallel experiment, 74% of EEA1 structures contained exogenous Rab5 (Fig. 5 B). In contrast, only 25% of EEA1-positive structures contained exogenous Rab9 (Fig. 5 B).
Rab5/9 and Rab5 colocalized significantly when coexpressed; 86% of CFP-Rab5/9–positive structures (1,384/1,618) were also positive for YFP-Rab5. Given 85% colocalization between coexpressed CFP- and YFP-Rab5 in control experiments (Fig. 5 B), we conclude that Rab5/9 shows identical localization to Rab5. Finally, Rab5/9 was not found on late endosomes any more than Rab5 was, as both Rabs showed low colocalization (11 and 12%, respectively) with endocytosed anti–cation-independent (CI) MPR (CI-MPR) IgG that had been chased into late endosomes (Fig. 5 B).
These data demonstrate that the Rab5 hypervariable domain is dispensable for the early endosome localization of Rab5/9. A lack of a requirement for certain hypervariable domain sequences has also been reported by Ali et al. (2004). Together, these data confirm that Rab targeting is more complex than originally believed.
In contrast to Rab5, the Rab9 hypervariable domain was required for late endosome localization. Replacement of the Rab9 hypervariable domain in Rab9/5 and Rab9/1 chimeras disrupted late endosome targeting and relocalized both proteins to the Golgi (Table I). Rab9/5 and Rab9/1 were clearly Golgi localized, as determined by their colocalization with p115, but not with endocytosed anti–CI-MPR antibody, in late endosomes (Fig. S1). The Golgi localization of Rab9/5 was not expected if hypervariable domain sequences were key; the protein should have been present on early endosomes. Although it has been proposed that the ER and Golgi may be sites for Rab mislocalization (Ali et al., 2004), we favor an alternative possibility. This laboratory has recently identified two new Rab9 effectors that are localized at the TGN. It is possible that one of these proteins interacts with Rab9/5 preferentially and retains the protein at that site. The Golgi localization of Rab9/1 may also be attributable to its hypervariable domain, as at least one Rab1-binding protein recognizes Rab1 hypervariable domain sequences (Preisinger et al., 2005). Rab1 contains effector binding information in nonhypervariable domain sequences (Fig. 4), and accordingly, Rab1/9 localized to the Golgi (Table I and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200510010/DC1). Although we cannot yet fully explain the localizations of Rab9/5 and Rab9/1, we conclude that the Rab9 hypervariable domain is required for proper late endosome localization of the Rab9 GTPase.
Competition between effectors can relocalize Rab chimeras
These experiments reveal a correlation between effector binding and proper subcellular localization (Table I). Yet, the ability of a chimera to bind a given effector in vitro was not sufficient to dictate cellular localization. Rab5/9 and Rab1/9 could bind TIP47 (Fig. 2), but were not localized to late endosomes. In this regard, it is important to note that endogenous Rab5 is more than twofold more abundant than Rab9 in HeLa cell extracts; 4.2 versus ≥9.4 pmol/mg HeLa cells postnuclear supernatant protein. Rab5 effectors may be present at similarly higher levels, and by simple mass action, direct Rab5/9 to early endosomes.
The ability of the Rab5/9 protein to interact with both Rab5 and Rab9 effectors (Figs. 3 and 4) enabled us to test if effector binding is the primary determinant of Rab localization. If effector interaction is key to Rab localization, overexpression of the late endosomal Rab9 effector TIP47 should be sufficient to move Rab5/9 from early (Fig. 5, Fig. 6 A, and Fig. S2) to late endosomes. As shown in Fig. 6 A (middle), coexpression of GFP-Rab5/9 with wild-type TIP47 relocalized the chimeric Rab5/9 to late endosomes. Relocalization required the ability of TIP47 to bind Rab9 sequences. Coexpression of a mutant TIP47 (TIP47SVV-AAA) that is deficient in Rab9 binding (Hanna et al., 2002) failed to relocalize the Rab5/9 protein (Fig. 6 A, right). The perinuclear, TIP47-relocalized Rab5/9 chimera was present in late endosomes and not the Golgi, as a significant fraction of it colocalized with endocytosed, late endosomal anti–CI-MPR antibody (Fig. 6 B), but not the Golgi marker p115 (Waters et al., 1992; Fig. 6 C). In contrast, overexpression of p40 did not alter the distribution of Rab5/9 from perinuclear early endosomes (Supp. Fig. 4 A).
Figure 6.

TIP47 binding to Rab5/9 triggers relocalization from early to late endosomes. (A) Wide-field light microscopy. GFP-Rab5/9 localization in fixed HeLa cells (left). The overexpression of wild-type TIP47 (middle), but not of mutant TIP47SVV-AAA (Hanna et al., 2002), which is deficient for Rab9 binding (right), causes GFP-Rab5/9 localization to perinuclear late endosomes. (middle) The cell is outlined in white. Levels of TIP47 wild-type and mutant protein overexpression were approximately ninefold. (B) TIP47-relocalized perinuclear Rab5/9 is present on late endosomes. Deconvolution microscopy of cells overexpressing TIP47 protein; perinuclear GFP-Rab5/9 colocalized with late endosomal, endocytosed anti–CI-MPR antibody. (C) Deconvolution microscopy shows TIP47- relocalized perinuclear Rab5/9 is not present on the Golgi, as it did not colocalize with the Golgi marker p115. In the merge images in B and C, Rab5/9 is shown in green, whereas the organelle markers are shown in red. In B and C, the TIP47 is shown as the mean of the summation of total Z-sections; other images are 0.2-μm Z-sections. Bars: (A) 10 μm; (B and C) 5 μm.
67% of cells displayed an entirely perinuclear staining pattern for Rab5/9 in the presence of wild-type TIP47 (Fig. 6 A, middle) compared with <30% in cells expressing Rab5/9 alone. This increase is very likely caused by a direct interaction between Rab5/9 and TIP47, as only 29% of cells expressing TIP47SVV-AAA showed perinuclear Rab5/9 staining (Fig. 6 A, right). Therefore, the interaction of the chimeric Rab with TIP47 was both necessary and sufficient to drive its steady-state localization.
Rab1/9 also interacted with both Rab1 and Rab9 effectors, although slightly less well than Rab5/9 (Fig. 3 A and Fig. 4 B). Therefore, we tested if Rab1/9 could also be relocalized from the Golgi (Fig. S3) to late endosomes upon TIP47 overexpression (Fig. 7 A).Indeed, in 75% of cells expressing low levels of Rab1/9 protein together with exogenous TIP47, we detected colocalization of a portion of Rab1/9 with late endosomal, endocytosed anti–CI-MPR antibody (Fig. 7 A). As with Rab5/9, coexpression of mutant TIP47 (TIP47SVV-AAA) failed to relocalize Rab1/9 to late endosomes (Fig. 7 B). Perinuclear staining of Rab1/9 in the presence of wild-type TIP47 was more punctate than that seen upon coexpression with mutant TIP47 (compare Fig. 7 A with B). Therefore, even in the context of the nonendosomal Rab1 GTPase, binding of TIP47 to the Rab9 hypervariable domain was sufficient for late endosome relocalization. Little Rab1/9 colocalized with the TGN marker GCC185 in cells expressing TIP47 (Fig. 7 C).
Figure 7.

TIP47 binding to Rab1/9 triggers relocalization from the Golgi to late endosomes. (A and B) Deconvolution microscopy shows CFP-Rab1/9 (left) and endocytosed anti–CI-MPR antibody (middle) localizations in HeLa cells in the presence of overexpressed wild-type (A) or Rab-binding–deficient mutant (B) TIP47 (right). The bottom images show the perinuclear region of the same cell. (C) Deconvolution microscopy shows CFP-Rab1/9 (left) and p115 (right) localizations in HeLa cells in the presence of overexpressed wild-type TIP47 (right). In the merge images, Rab1/9 is shown in green, whereas the organelle markers are shown in red. TIP47 is shown as the mean of the summation of total Z-section images; other images are 0.2-μm Z-sections. Bars, 5 μm.
It is possible that GDI delivered prenylated Rab1/9 directly to the endocytic pathway and that it was then stabilized there by TIP47 interaction. Alternatively, Rab1/9 may have cycled from the Golgi through endosomes and been trapped there by TIP47. In any event, effector interaction was sufficient to dictate the steady-state localizations of two different Rab chimeras.
Like Rab5/9, Rab1/9 was not relocalized by overexpression of p40 (Fig. S4 B, available at http://www.jcb.org/cgi/content/full/jcb.200510010/DC1), but remained in a tight, ribbon-like, perinuclear pattern (compare with Fig. S3), in contrast to the punctuate, perinuclear pattern of relocalized Rab1/9 protein. In addition, although siRNA depletion of TIP47 destabilizes Rab9, increasing its half-life from 32 to 8 h (Ganley et al., 2004), depletion of p40 using RNA interference did not affect Rab9 degradation (Fig. S4 C). Rab9 half-life was 54 h in control cells and 43 h in p40 siRNA-treated cells. Thus, TIP47, but not p40, is a key effector for both Rab9 stability and late endosome localization.
Discussion
We have presented a test of the ability of Rab effectors to direct the localization of Rab proteins to distinct membrane compartments. Such an analysis was only possible because of our serendipitous generation of two Rab chimeras that were able to bind two distinct classes of Rab effector proteins. A Rab5 protein containing the Rab9 hypervariable domain (Rab5/9) bound well to the Rab5 effectors rabaptin-5 and EEA1, and also showed significant binding to the Rab9 effectors TIP47 and p40. Similarly, a chimera comprised of Rab1 linked to the Rab9 hypervariable domain bound to the Rab1 effectors GM130 and golgin-84. This Rab1/9 hybrid also bound strongly to the Rab9 effector TIP47 and somewhat less strongly to another Rab9 effector, p40. Upon expression in cultured cells, the chimeric Rabs localized, together with their Rab backbone parental counterparts, on early endosomes and the Golgi complex, respectively. Yet, when the Rab9 effector TIP47 was coexpressed, the Rab5/9 and Rab1/9 chimeras moved to late endosomes, together with endogenous Rab9. These data demonstrate that effector binding can relocalize a Rab from one membrane compartment to another. Only TIP47 had the capacity to relocalize the Rabs; another Rab9 effector (p40) did not. Thus, certain Rab effectors can play a dominant role in Rab9 localization.
Our analyses confirmed the importance of Rab9 hypervariable domain sequences for interaction with the effector proteins TIP47 and p40; in both cases, this domain was necessary and sufficient for Rab–effector interaction, within the context of a Rab GTPase. A recent study has shown a role for hypervariable domain sequences in a Rab–effector interaction; Rab7 COOH-terminal residues are required for binding to the Rab7 effector RILP (Wu et al., 2005). In addition, polo-like kinase, which is a Rab1-binding protein, interacts with the phosphorylated Rab1 hypervariable domain (Preisinger et al., 2005). In the structure of Rab3 bound to Rabphilin-3A, residues in α helix 5 that are part of the hypervariable domain contribute to the Rab-binding interface (Ostermeier and Brunger, 1999). In contrast, two Rab5 effectors and two out of three Rab1 effectors that were tested failed to require the presence of hypervariable domain sequences for binding. Indeed, in the case of EEA1, biochemical experiments ruled out a significant role for the Rab5 hypervariable domain in EEA1 binding (Merithew et al., 2003). In addition, the crystal structure of Rab5 missing its hypervariable domain with a fragment of rabaptin-5 suggested that the hypervariable domain would not contribute to this Rab–effector pair (Zhu et al., 2004). Rab5 hypervariable domain sequences may contribute to effector interactions not tested in this study, but they were not required for the localization of Rab5/9. Thus, for Rab9, hypervariable domain interactions with TIP47 do direct localization, which is consistent with the original proposals of Chavrier et al. (1991) and Brennwald and Novick (1993). For the Rab5/9 and Rab1/9 chimeras, nonhypervariable domain–dependent interactions were sufficient for their initial cellular localizations, which were early endosomes and the Golgi complex, respectively.
Our working model for how effectors contribute to Rab localization is shown in Fig. 8. Rabs are delivered to membranes by GDI in their GDP-bound forms (Soldati et al., 1994; Ullrich et al., 1994). Release of Rabs from GDI may be catalyzed by GDFs (Pfeffer and Aivazian, 2004). If nucleotide exchange occurs, the Rab will be able to bind to effectors. This may involve recruitment of cytosolic effectors (Fig. 8, option A) or binding to effectors that are already membrane associated (Fig. 8, option B).
Figure 8.

Model for how effectors contribute to membrane localization of Rab proteins. GDP-bound Rabs are recruited to membranes from cytosolic complexes with GDI through the catalytic action of a GDF. A GEF catalyzes nucleotide exchange. Interaction of the Rab-GTP with an effector that has been recruited to the same membrane by a Rab (option A) or by an effector-binding partner (option B) will stabilize both the Rab and the effector on the membrane. If not bound to the effector, a GTPase-activating protein (GAP) can activate the Rab to hydrolyze its GTP to GDP, thus, allowing for potential membrane extraction of the Rab by GDI. Thus, the Rab GTPase is dependent on its effectors, and vice versa, for stable interaction with the membrane. The effector-binding partner need not be a protein and can be particular phospholipids, such as phosphoinositides, that are specific to a given compartment (Zerial and McBride, 2001).
Effector binding often stabilizes a Rab in its GTP-bound form, enhancing both Rab–membrane and –effector association. Rab guanine nucleotide exchange factors (GEFs) associate with Rab effectors (Zerial and McBride, 2001), thus, activation of Rabs occurs in the vicinity of specific Rab-binding partners. If a Rab is delivered to the wrong compartment, it will not be a substrate for nucleotide exchange, and GDI can remove it. In this manner, a microdomain can form, containing active Rabs bound to an array of effector proteins. Importantly, the overall specificity of Rab localization is contributed to by multiple components, such as GDFs, the proximity of Rab-specific GEFs, and subsequent effector interactions. For every Rab, there will be numerous potential effector interactions that will depend upon the relative affinity and abundance of each effector. These multiple interactions will contribute to the steady-state localization of a Rab that we score by immunofluorescence microscopy.
What is the mechanism by which TIP47 stabilizes and localizes Rab9? TIP47 is a predominantly cytosolic protein (Diaz and Pfeffer, 1998) that interacts with Rab9 and the cytosolic domains of the two MPRs via two distinct binding sites (Krise et al., 2000; Orsel et al., 2000; Carroll et al., 2001; Hanna et al., 2002; Nair et al., 2003; Burguete et al., 2004). TIP47 binding to Rab9 enhances its affinity for MPR cytoplasmic domains (Carroll et al., 2001). Cytosolic TIP47 occurs as a homohexamer or larger oligomer, and may further oligomerize when it is membrane associated (Sincock et al., 2003). Thus, a TIP47 hexamer has the capacity to interact with multiple Rab9 molecules and multiple MPRs. This combinatorial requirement for both a Rab and MPR cytoplasmic domains enhances the specificity of TIP47 membrane association; the protein binds preferentially to membranes containing both Rab9 and MPRs.
Once TIP47 has found a membrane containing both Rab9 and MPRs, it will bind there and drive transport from late endosomes to the TGN (Carroll et al., 2001; Hanna et al., 2002). If a hybrid Rab is nearby, either as a consequence of GDI-mediated delivery or because of membrane trafficking events, it has the potential to become part of a TIP47–Rab9 microdomain. All Rabs are in equilibrium with their own effectors, and TIP47-mediated localization of the chimeras was only observed when it could predominate in relation to Rab1 and Rab5 effectors by overexpression in cells. Our ability to relocalize Rab5/9 from early to late endosomes and Rab1/9 from the Golgi to late endosomes is most easily explained by a binding competition between Rab5, Rab1, and Rab9 effectors.
The “key” effector concept
We have shown that TIP47, but not p40, can relocalize Rab5/9 and Rab1/9 to late endosomes. Although TIP47 has the capacity to stabilize Rab9 on late endosomes (Ganley et al., 2004), p40 does not. This has led us to propose that each Rab has its own key effectors that are essential for steady-state localization and Rab protein stabilization. Key effectors may or may not recognize hypervariable domain sequences; nevertheless, their identities will be important to determine. This may be facilitated by systematic depletion of every Rab effector and monitoring the corresponding Rab protein turnover. Such a determination is not a trivial matter; Rab5 alone has been estimated to have 30 specific effector proteins (Zerial and McBride, 2001). It seems quite reasonable to us that many (if not most) effectors will lack the capacity to localize Rabs, and more than one effector may localize a Rab.
Another distinguishing feature of TIP47 is its real (although twofold weaker) ability to bind Rab9-GDP (Carroll et al., 2001; Ganley et al., 2004). This attribute may contribute to Rab9 membrane recruitment because TIP47 may retain Rab9 on the membrane until a Rab9 GEF is encountered (Fig. 8). It will be of interest to learn if other, yet to be identified, key effectors will share this characteristic. In contrast, p40 shows a stronger preference for Rab9-GTP (Diaz et al., 1997) and may also interact with PIK-FYVE kinase on earlier endosomal compartments (Ikonomov et al., 2003). That interaction may explain why p40 failed to relocalize Rab5/9 to late endosomes. We cannot fully rule out the alternative possibility that p40 failed to relocalize the hybrid Rab proteins because it did not bind the chimeras as well as TIP47.
If TIP47 is essential for Rab9 localization on late endosomes, TIP47 depletion would be expected to lead to Rab9 mislocalization. In previous work, we could only deplete cells of 80–90% of total cellular TIP47; under these conditions, the fraction of total, remaining TIP47 that was membrane associated increased compared with control cells. Thus, we could never completely deplete cells of membrane-associated TIP47 without killing them; indeed, TIP47 siRNA treatment led to a cellular growth arrest (Ganley et al., 2004). Even if we could deplete all TIP47, it is likely that other effector interactions could contribute to Rab9 localization; Rabs may have more than one key interaction that can provide their membrane-domain entry and stabilization.
Once located within either the secretory or endocytic pathways, our data suggest that effector-mediated incorporation and stabilization within a membrane microdomain underlies Rab localization at steady state in eukaryotic cells. Localization of a newly delivered Rab requires either the preexistence of a microdomain already containing that Rab (into which the new Rab becomes incorporated), or de novo formation of a microdomain that forms when multiple constituents coalesce and assemble (Zerial and McBride, 2001; Pfeffer, 2003). Indeed, effectors also need Rabs for their membrane association. For example, active Rabs recruit p115 (Moyer et al., 2001) onto the Golgi. Rab9 recruits TIP47 onto endosomes (Carroll et al., 2001; Ganley et al., 2004). Other effectors, such as GM130 and GRASP65, associate with the Golgi in a Rab-independent manner (Short et al., 2005). Together, these data reveal a complex interdependence and potential for cooperativity between Rabs and effectors for membrane localization and domain stabilization. A role for key effectors can easily explain all of the previously reported localization data for chimeric Rab GTPases.
Materials and methods
Recombinant protein expression and purification
NH2-terminal His6-tagged p40 (Diaz, et al., 1997), His6-tagged TIP47 (152–434; Carroll et al., 2001), and Rab9-CLLL (Shapiro et al., 1993) were purified. Human Rab5a, Rab1a, and Rab9/5, Rab5/9, Rab9/1, and Rab1/9 were cloned into pET14b (Novagen), for expression in E. coli as unprenylated Rabs, or into pGEX-4T1 (GE Healthcare). Rab9-CLLL was also cloned into pGEX-4T1. Untagged or GST-tagged Rab1a, Rab5, Rab1/9, Rab9/1, Rab5/9, Rab9/5, and Rab9-CLLL were expressed in E. coli BL21(DE3) RIL (Stratagene) or Rosetta (Invitrogen) cells and induced with 0.5 mM IPTG for 3 h at 37°C (Rab5 and Rab9/1) and 16 h at 25°C (Rab1a, Rab1/9, and Rab5/9, GST-Rab9CLLL, GST-Rab1a, GST-Rab1/9, and GST-Rab9/1), and with 1 mM IPTG for 4 h at 30°C (Rab9/5). Cells (6 L) were resuspended in 40 ml of 50 mM Tris-HCl, pH 8.0, 8 mM MgCl2, 2 mM EDTA, 0.5 mM DTT, 10 μM GDP, and protease inhibitor cocktail (Roche; Rab1a, Rab5, Rab9/1, Rab1/9, GST-Rab1a, GST-Rab1/9, GST-Rab9CLLL, and GST-Rab9/1), or 50 mM MES, pH 6.5, 8 mM MgCl2, 2 mM EDTA, 0.5 mM DTT, 10 μM GDP, protease inhibitor cocktail (Rab9/5 and Rab5/9). Cells were lysed using a French press, and lysates were centrifuged at 15,000 rpm for 20 min (JA 20 rotor; Beckman Coulter). Homogenates were centrifuged at 55,000 rpm for 30 min (70Ti rotor; Beckman Coulter). Supernatants were diluted 10-fold with lysis buffer and loaded onto a 25-ml Q–Sepharose Fast Flow column (GE Healthcare; Rab1a, Rab5, Rab1/9, Rab9/1, Rab9/5, GST-Rab1a, GST-Rab1/9, GST-Rab9CLLL, and GST-Rab9/1), or a SP–Sepharose Fast Flow column (GE Healthcare; Rab5/9). With Rab5, Rab9/5, Rab5/9, Rab1/9, and Rab9/1, proteins were eluted with 0–400 mM NaCl in lysis buffer. With Rab5, Rab1/9, and Rab9/5, fractions were precipitated with 60% (Rab5) or 40% (Rab1/9, Rab9/5, GST-Rab1a, GST-Rab1/9, GST-Rab9CLLL, and GST-Rab9/1) (NH4)2SO4, resuspended in buffer A (20 mM Hepes, pH 7.4, 200 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, and 10 μM GDP) and loaded onto a Superdex 200 column (GE Healthcare). Pools were brought to 10% (vol/vol) glycerol and stored at −80°C. With Rab9/1 and Rab5/9, fractions were pooled and 4 ml loaded onto a Superdex 200 column. With Rab1a, the Q–Sepharose flow-through was subjected to 60% (NH4)2SO4 precipitation, and the precipitate was resuspended in buffer A. GST-Rab5, GST-Rab5/9, and GST-Rab9/5 were expressed and purified on glutathione–Sepharose beads (GE Healthcare), as recommended by the manufacturer. Rab nucleotide-binding activity was performed as previously described (Shapiro et al., 1993), except for nucleotide exchange; Rabs (200 nM) were incubated in 50 mM Hepes, pH 7.5, 150 mM KCl, 2 mM EDTA, 1 mM MgCl2, 0.1% BSA, and 2 μM [35S]GTPγS (4 nCi/μl) for 45 min at 25°C in 50 μl of extract.
TIP47 binding to wild-type and chimeric Rab GTPases
Rab proteins (2.6 μM) were preloaded with 3 μM [35S]GTPγS or [3H]GDP (4.5 nCi/μl) in 2.2 ml of extract containing 50 mM Hepes, pH 7.4, 150 mM KCl, 15 mM imidazole, 4.5 mM EDTA, 5 mM MgCl2, 0.5 mM DTT, and 100 μg/ml BSA. Rab-GTPγS was separated from free nucleotide by gel filtration (G-25). TIP47-binding reactions (450 μl) were in 50 mM Hepes, pH 7.4, 150 mM KCl, 15 mM imidazole, 5 mM MgCl2, and 100 μg/ml BSA. Rab–[35S]GTPγS complex (571 nM) was incubated with TIP47 (2.5 μM) at room temperature for 1.5 h. TIP47-bound Rabs were recovered on Ni-NTA resin (QIAGEN; 50 μl of a 50% slurry) and counted.
p40 binding to Rab GTPases
Rab–[35S]GTPγS complexes (571 nM) were incubated with 2.5 μM p40 at room temperature for 1.5 h in 50 mM Hepes, pH 7.4, 150 mM KCl, 25 mM imidazole, 5 mM MgCl2, and 100 μg/ml BSA. p40-bound GTPases were recovered on Ni-NTA resin (QIAGEN; 50 μl of a 50% slurry) and counted.
Rab5 effector binding to Rab GTPases
GST-Rab fusion proteins (3.0 μM) were incubated in binding buffer (20 mM Hepes, pH 7.5, 100 mM NaCl, 4.5 mM MgCl2, 5 mM EDTA, 300 μM GTPγS, and 0.2 mM DTT) for 1.5 h at room temperature in 200 μl of extract. Bovine brain cytosol (40% (NH4)2SO4 precipitate; 200 μl of ∼15 mg/ml) in binding buffer (+1 mM PMSF, 5 mM MgCl2 and protease inhibitor cocktail, but no EDTA) was added at 20°C for 1 h. Glutathione–Sepharose (GE Healthcare; 10% slurry in binding buffer) was added for 20 min on a rotator. Beads were washed three times with 1 ml of wash buffer (20 mM Hepes, pH 7.5, 100 mM NaCl, 5 mM MgCl2, and 0.2 mM DTT). Proteins were eluted with SDS-PAGE loading buffer and analyzed by immunoblot with anti-EEA1 and anti–rabaptin-5.
Rab1 effector binding to Rab GTPases
Binding of Rab1 effectors from rat liver Golgi extracts to GST-Rabs was measured as previously described (Satoh et al., 2003), with minor modifications. In brief, 10 nmol (∼0.5 mg) of each GST-Rab was loaded on glutathione–Sepharose. Bead-bound GST-Rab1 and GST-Rab1/9 were loaded with GTPγS (Christoforidis et al., 1999), whereas GST-Rab9CLLL and GST-Rab9/1 were loaded with GTPγS in 20 mM Hepes, pH 7.4, 150 mM KCl, 5 mM EDTA, 4.5 mM MgCl2, 1 mM DTT, and 100 μM GTPγS for 1.5 h at room temperature, followed by a 30-min incubation in 20 mM Hepes, pH 7.4, 100 mM NaCl, 5 mM MgCl2, and 100 μM GTPγS at room temperature. Golgi extracts were made by diluting rat liver Golgi membranes to 0.5 mg/ml with 20 mM Hepes, pH 7.4, 100 mM KCl, and 5 mM MgCl2, and adding 1% Triton X-100, 1 mM DTT, protease inhibitor cocktail (Roche), 1 mg/ml soybean trypsin inhibitor, and MgCl2 to a final concentration of 8 mM. After incubation on ice for 30 min, extracts were spun at 16,000 g for 20 min. 1 mM GTPγS was added to the supernatant. Extract (200 μl) was incubated with GST-Rab–loaded beads for 1 h at 4°C. Beads were washed, eluted, and analyzed by SDS-PAGE and Western blot, as previously described (Satoh et al., 2003).
Chimeric Rab localizations
cDNAs for canine Rab9a and human Rab5a, Rab1a, and Rab9/5, Rab5/9, Rab9/1, and Rab1/9 were cloned into pECFP-C1, pEYFP-C1, and pEGFP-C3 (CLONTECH Laboratories, Inc.) for expression. For fixed cell indirect immunofluorescence, HeLa cells were grown and transfected on glass coverslips. Cells were transfected (Fugene 6; Roche); 16–24 h after transfection, cells were fixed, permeabilized, and stained (Ganley, et al., 2004), and then mounted in ProFade mounting medium (Invitrogen). Micrographs in Figs. 6 and 7 and Figs. S1–S4 were acquired using a deconvolution microscopy system (Spectris; Applied Precision, LLC) with an inverted epifluorescence microscope (IX70; Olympus), a PlanApo 60×, 1.40 NA, oil immersion objective (Olympus), a charge-coupled device (CCD) camera (CoolSNAP HQ; Roper Scientific), and acquisition and deconvolution software (DeltaVision; Applied Precision, LLC). Micrographs in Fig. 5 were acquired using a microscope (Axioplan2; Carl Zeiss MicroImaging, Inc.) fitted with 63×, 1.30 NA, and 100×, 1.3 NA, Plan Neofluar objective lenses, a CCD camera (AxioCamHRc; Carl Zeiss MicroImaging, Inc.), and controlled by Axiovision 4.2 software (Carl Zeiss MicroImaging, Inc.). Pictures were analyzed using ImageJ (National Institutes of Health) and Photoshop (Adobe) software. For late endosome labeling, cells were incubated with 30 μg/ml of mouse anti–CI-MPR (2G11) IgG with a 20-min pulse and a 40-min chase in complete medium. Endogenous organelle markers used were as follows: anti-p115 (Golgi; gift from G. Waters, Merck Research Laboratories, Rahway, NJ), anti-TGN46 (TGN; Serotec), anti-EEA1 and anti-rabaptin-5 (early endosomes; BD Biosciences), and CI-MPR (primarily late endosomes; Diaz and Pfeffer, 1998). Rabbit anti-GFP and secondary antibodies derivatized with Alexa Fluor dyes (Invitrogen) were used. The extent of colocalization between endosomal Rabs and organelle markers was quantitated by generating contour maps of each image by overlaying the Rab and marker channels. The total number of vesicles and the number showing overlap between both markers were tallied. All fixed cell microscopy was performed at room temperature. For live cell microscopy, BS-C-1 cells on glass coverslips were observed 16–20 h after transfection at 37°C on a temperature-controlled stage of an inverted microscope (Diaphot-300; Nikon) equipped with a cooled CCD camera (NDE/CCD; Princeton Instruments), Plan Apo 60×, 1.40 NA, and 100×, 1.40 NA, oil objectives, and using MetaMorph software (Molecular Devices); images were analyzed using Photoshop (Adobe). Dual CFP/YFP filters were obtained from Chroma Technology Corp. CFP- or YFP-tagged wild-type Rabs or CFP-Golgi (CLONTECH Laboratories, Inc.) were used as organelle markers.
Analysis of Rab chimera membrane association
HeLa cells on 100-mm dishes were transfected for expression of Rab fusion proteins with GFP, CFP, or YFP. After 23 h, cells were washed three times with 10 ml of phosphate buffered saline, and then two times with 10 ml ice-cold HM buffer (25 mM Hepes, pH 7.4, and 2.5 mM Mg(OAc)2). Cells were swollen on ice for 5 min, washed gently with 5 ml SEAT buffer (10 mM ethanolamine, 10 mM acetic acid, 1 mM EDTA, and 0.25 M sucrose) leaving ∼0.5 ml of buffer, and then scraped off of the plate with a rubber policeman and transferred to microfuge tubes. Protease inhibitor cocktail (Complete EDTA-Free; Roche) and 0.1 M PMSF were added to each sample. Lysates were passed through a 27-gauge needle 20 times and spun for 20 min at 341,000 g and 4°C in a rotor (TLA-100.1; Beckman Coulter). Pellets and supernatants were analyzed by SDS-PAGE and an anti-GFP Western blot.
Measurement of Rab activity
Rab nucleotide–binding activity was performed according to Shapiro et al. (1993), with minor changes. Rabs (200 nM) were incubated in 50 mM Hepes, pH 7.5, 150 mM KCl, 2 mM EDTA, 1 mM MgCl2, 0.1% BSA, and 2 μM [35S]GTPγS (4 nCi/μl) for 45 min at 25°C in 50 μl of extract. Reactions were in triplicate; 40-μl samples were removed, diluted with 3 ml of ice-cold wash buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, and 25 mM MgCl2) and passed through 24-mm HA filters. Filters were washed three times with 3 ml of wash buffer, dried, and counted in scintillation fluid.
Online supplemental material
Fig. S1 shows deconvolution microscopic localizations of Rab9/5 and Rab9/1 in fixed HeLa cells. Fig. S2 shows deconvolution microscopic localizations of Rab5/9 and Rab5 in fixed HeLa cells. Fig. S3 shows deconvolution microscopic localizations of Rab1/9 and Rab1 in fixed HeLa cells. Fig. S4 shows p40 is not a key effector for Rab9 localization. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200510010/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Pierre Barbero for initiating this project, Collin Melton for generating several of the chimeras, and Drs. Ayano Satoh and Graham Warren for help with Rab1 effector binding.
This work was supported by grants from the American Heart Association and the National Institutes of Health National Institute of Diabetes and Digestive Kidney Diseases to S. Pfeffer and a postdoctoral fellowship from the Leukemia and Lymphoma Society to D. Aivazian.
Abbreviations used in this paper: CCD, charge-coupled device; CI, cation-independent; EEA, early endosome antigen; GDF, GDI displacement factor; GDI, GDP dissociation inhibitor; GEF, guanine nucleotide exchange factor; MPR, mannose 6-phosphate receptor; TIP47, tail-interacting protein of 47 kD.
References
- Ali, B.R., C. Wasmeier, L. Lamoreux, M. Strom, and M.C. Seabra. 2004. Multiple regions contribute to membrane targeting of Rab GTPases. J. Cell Sci. 117:6401–6412. [DOI] [PubMed] [Google Scholar]
- Allan, B.B., B.D. Moyer, and W.E. Balch. 2000. Rab1 recruitment of p115 into a cis-SNARE complex: programming budding COPII vesicles for fusion. Science. 289:444–448. [DOI] [PubMed] [Google Scholar]
- Brennwald, P., and P. Novick. 1993. Interactions of three domains distinguishing the Ras-related GTP-binding proteins Ypt1 and Sec4. Nature. 362:560–563. [DOI] [PubMed] [Google Scholar]
- Burguete, A.S., P.B. Harbury, and S.R. Pfeffer. 2004. In vitro selection and successful prediction of TIP47 protein-interaction interfaces. Nat. Methods. 1:55–60. [DOI] [PubMed] [Google Scholar]
- Carroll, K.S., J. Hanna, I. Simon, J. Krise, P. Barbero, and S.R. Pfeffer. 2001. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science. 292:1373–1376. [DOI] [PubMed] [Google Scholar]
- Chavrier, P., M. Vingron, C. Sander, K. Simons, and M. Zerial. 1990. Molecular cloning of YPT1/SEC4-related cDNAs from an epithelial cell line. Mol. Cell. Biol. 10:6578–6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier, P., J.-P. Gorvel, E. Stelzer, K. Simons, J. Gruenberg, and M. Zerial. 1991. Hypervariable C-terminal domain of rab proteins acts as a targeting signal. Nature. 353:769–772. [DOI] [PubMed] [Google Scholar]
- Chen, L., E. DiGiammarino, X.E. Zhou, Y. Wang, D. Toh, T.W. Hodge, and E.J. Meehan. 2004. High resolution crystal structure of human Rab9 GTPase: a novel antiviral drug target. J. Biol. Chem. 279:40204–40208. [DOI] [PubMed] [Google Scholar]
- Christoforidis, S., H.M. McBride, R.D. Burgoyne, and M. Zerial. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 397:621–625. [DOI] [PubMed] [Google Scholar]
- Colicelli, J. 2004. 2004. Human RAS superfamily proteins and related GTPases. Sci. STKE. 250:RE13. [DOI] [PMC free article] [PubMed]
- Diaz, E., and S.R. Pfeffer. 1998. TIP47: a cargo selection device for mannose 6-phosphate receptor trafficking. Cell. 93:433–443. [DOI] [PubMed] [Google Scholar]
- Diaz, E., F. Schimmöller, and S.R. Pfeffer. 1997. A novel Rab9 effector required for endosome-to-TGN transport. J. Cell Biol. 138:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley, I.G., K. Carroll, L. Bittova, and S.R. Pfeffer. 2004. Rab9 GTPase regulates late endosome size and requires effector interaction for its stability. Mol. Biol. Cell. 15:5420–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna, J., K. Carroll, and S.R. Pfeffer. 2002. Identification of residues in TIP47 essential for Rab9 binding. Proc. Natl. Acad. Sci. USA. 99:7450–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomov, O.C., D. Sbrissa, K. Mlak, R. Deeb, J. Fligger, A. Soans, R.L. Finley Jr., and A. Shisheva. 2003. Active PIKfyve associates with and promotes the membrane attachment of the late endosome-to-trans-Golgi network transport factor Rab9 effector p40. J. Biol. Chem. 278:50863–50871. [DOI] [PubMed] [Google Scholar]
- Krise, J.P., P.M. Sincock, J.G. Orsel, and S.R. Pfeffer. 2000. Quantitative analysis of TIP47-receptor cytoplasmic domain interactions: implications for endosome-to-trans Golgi network trafficking. J. Biol. Chem. 275:25188–25193. [DOI] [PubMed] [Google Scholar]
- Merithew, E., S. Hatherly, J.J. Dumas, D.C. Lawe, R. Heller-Harrison, and D.G. Lambright. 2001. Structural plasticity of an invariant hydrophobic triad in the switch regions of Rab GTPases is a determinant of effector recognition. J. Biol. Chem. 276:13982–13988. [DOI] [PubMed] [Google Scholar]
- Merithew, E., C. Stone, S. Eathiraj, and D.G. Lambright. 2003. Determinants of Rab5 interaction with the N terminus of early endosome antigen 1. J. Biol. Chem. 278:8494–8500. [DOI] [PubMed] [Google Scholar]
- Moyer, B.D., B.B. Allan, and W.E. Balch. 2001. Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis-Golgi tethering. Traffic. 2:268–276. [DOI] [PubMed] [Google Scholar]
- Nair, P., B.E. Schaub, and J. Rohrer. 2003. Characterization of the endosomal sorting signal of the cation-dependent mannose 6-phosphate receptor. J. Biol. Chem. 278:24753–24758. [DOI] [PubMed] [Google Scholar]
- Orsel, J.G., P.M. Sincock, J.P. Krise, and S.R. Pfeffer. 2000. Recognition of the 300K mannose 6-phosphate receptor cytoplasmic domain by TIP47. Proc. Natl. Acad. Sci. USA. 97:9047–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier, C., and A.T. Brunger. 1999. Structural basis of Rab effector specificity: crystal structure of the small G protein Rab3A complexed with the effector domain of rabphilin–3A. Cell. 96:363–374. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal, J.B., and M.C. Seabra. 2000. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J. Mol. Biol. 301:1077–1087. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal, J.B., and M.C. Seabra. 2001. Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 313:889–901. [DOI] [PubMed] [Google Scholar]
- Pettersen, E.F., T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, and T.E. Ferrin. 2004. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Pfeffer, S. 2003. Membrane domains in the secretory and endocytic pathways. Cell. 112:507–517. [DOI] [PubMed] [Google Scholar]
- Pfeffer, S., and D. Aivazian. 2004. Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 5:886–896. [DOI] [PubMed] [Google Scholar]
- Preisinger, C., R. Korner, M. Wind, W.D. Lehmann, R. Kopajtich, and F.A. Barr. 2005. Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 24:753–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh, A., Y. Wang, J. Malsam, M.B. Beard, and G. Warren. 2003. Golgin-84 is a rab1 binding partner involved in Golgi structure. Traffic. 4:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, A.D., M.A. Riederer, and S.R. Pfeffer. 1993. Biochemical analysis of rab9, a ras-like GTPase involved in protein transport from late endosomes to the trans Golgi network. J. Biol. Chem. 268:6925–6931. [PubMed] [Google Scholar]
- Short, B., A. Haas, and F.A. Barr. 2005. Golgins and GTPases, giving identity and structure to the Golgi apparatus. Biochim. Biophys. Acta. 1744:383–395. [DOI] [PubMed] [Google Scholar]
- Simonsen, A., R. Lippe, S. Christoforidis, J.-M. Gaullier, A. Brech, J. Callaghan, B.-H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 394:494–498. [DOI] [PubMed] [Google Scholar]
- Sincock, P.M., I.G. Ganley, J. Krise, S. Diederichs, U. Sivars, B. O'Connor, L. Ding, and S.R. Pfeffer. 2003. Self-assembly is important for TIP47 function in mannose 6-phosphate receptor transport. Traffic. 4:18–25. [DOI] [PubMed] [Google Scholar]
- Sivars, U., D. Aivazian, and S.R. Pfeffer. 2003. Yip3 catalyses the dissociation of endosomal Rab-GDI complexes. Nature. 425:856–859. [DOI] [PubMed] [Google Scholar]
- Soldati, T., A.D. Shapiro, A.B. Svejstrup, and S.R. Pfeffer Sr. 1994. Membrane targeting of the small GTPase Rab9 is accompanied by nucleotide exchange. Nature. 369:76–78. [DOI] [PubMed] [Google Scholar]
- Stenmark, H., A. Valencia, O. Martinez, O. Ullrich, B. Goud, and M. Zerial. 1994. Distinct structural elements of rab5 define its functional specificity. EMBO J. 13:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark, H., G. Vitale, O. Ullrich, and M. Zerial. 1995. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 83:423–432. [DOI] [PubMed] [Google Scholar]
- Ullrich, O., H. Horiuchi, C. Bucci, and M. Zerial. 1994. Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange. Nature. 368:157–160. [DOI] [PubMed] [Google Scholar]
- Waters, M.G., D.O. Clary, and J.E. Rothman. 1992. A novel 115-kD peripheral membrane protein is required for intercisternal transport in the Golgi stack. J. Cell Biol. 118:1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M., W. Tuanlao, E. Loh, W. Hong, and H. Song. 2005. Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J. 24:1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
- Zhu, G., P. Zhai, J. Liu, S. Terzyan, G. Li, and X.C. Zhang. 2004. Structural basis of Rab5-Rabaptin5 interaction in endocytosis. Nat. Struct. Mol. Biol. 11:975–983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}