

Abstract
Mutations in the adenomatous polyposis coli (APC) tumor suppressor gene initiate a majority of colorectal cancers. Acquisition of chromosomal instability is an early event in these tumors. We provide evidence that the loss of APC leads to a partial loss of interkinetochore tension at metaphase and alters mitotic progression. Furthermore, we show that inhibition of APC in U2OS cells compromises the mitotic spindle checkpoint. This is accompanied by a decrease in the association of the checkpoint proteins Bub1 and BubR1 with kinetochores. Additionally, APC depletion reduced apoptosis. As expected from this combination of defects, tetraploidy and polyploidy are consequences of APC inhibition in vitro and in vivo. The removal of APC produced the same defects in HCT116 cells that have constitutively active β-catenin. These data show that the loss of APC immediately induces chromosomal instability as a result of a combination of mitotic and apoptotic defects. We suggest that these defects amplify each other to increase the incidence of tetra- and polyploidy in early stages of tumorigenesis.
Introduction
The loss of functional adenomatous polyposis coli (APC) is an early event in the development of most polyploid colorectal cancers (Kinzler and Vogelstein, 1996; Polakis, 1997). Mutated APC has been linked to genetic instability, which is an early hallmark of colorectal tumors and a factor that greatly promotes tumor development (Fodde et al., 2001; Kaplan et al., 2001; Shih et al., 2001). Exactly how APC is involved in maintaining chromosomal stability remains unknown. APC performs multiple roles, including negative regulation of the Wnt signaling pathway (Bienz and Clevers, 2000; Polakis, 2000), organization of the cytoskeleton, and regulation of cell migration (Dikovskaya et al., 2001; Zumbrunn et al., 2001; Hanson and Miller, 2005; Nathke, 2005). This functional diversity places APC in an important position in maintaining gut epithelia, but it is not clear how it relates to the function of APC in safeguarding a normal karyotype.
The formation of a protein complex between APC and the spindle assembly checkpoint proteins Bub1 and BubR1 and the ability of these kinases to phosphorylate APC in vitro (Kaplan et al., 2001) raise the intriguing possibility that APC “talks” directly to the mitotic checkpoint machinery. Insufficient microtubule plus end attachment resulting from the expression of dominant, truncated fragments of APC inhibits chromosome congression at metaphase and results in abnormal chromosome segregation (Green and Kaplan, 2003; Green et al., 2005). Consequently, the overexpression of N-terminal APC fragments (like those commonly found in tumors) in cells with wild-type APC can lead to premature exit from mitosis and aneuploidy (Tighe et al., 2004).
The loss of heterozygosity in the APC locus, which initiates most colorectal tumors, has two direct consequences: the loss of normal functional APC and the expression of a truncated N-terminal APC fragment. It is likely that both the absence of functional APC and the presence of N-terminal APC fragments contribute in separate but interactive ways to the phenotype of APC-deficient cancers. The majority of previous work examined the dominant effects of N-terminal APC fragments commonly found in tumors (Green and Kaplan, 2003; Tighe et al., 2004; Green et al., 2005). In most cases, the effects of N-terminal fragments were assessed in cells that, unlike tumors, also express wild-type, full-length APC. The interpretation of these data requires an understanding of the effects produced by the absence of APC in order to distinguish the effects of N-terminal APC fragments from the effects that result from loss of APC itself. Therefore, we specifically addressed the role of the full-length APC molecule by depleting full-length APC in several different systems.
Using Xenopus laevis egg extracts, we previously showed that lack of APC causes mitotic spindles to have several defects, including a disorganized microtubule network with reduced total microtubule mass, particularly in the midspindle area (Dikovskaya et al., 2004). In the present study, we show that in addition to spindle defects, the mitotic spindle assembly checkpoint is not functioning properly in cells lacking APC. Consequently, we find that the loss of APC leads to the accumulation of tetraploid cells. Additionally, we observed decreased apoptosis in APC-deficient cells. We propose that this combination of defects can aid in the longevity of cells with abnormal DNA content to promote polyploidy. Importantly, we provide direct evidence for the appearance of tetra- and polyploid cells extremely early after APC is inactivated not only in cultured cells but also in gut tissue. Additionally, we eliminate a major role for β-catenin signaling in the observed effects by showing identical defects in HCT116 cells, which carry an activating mutation in β-catenin.
Results
Lack of APC leads to reduced tension at kinetochores metaphase
To address how the lack of APC affects mitotic spindle function in cells, we removed APC from human osteosarcoma U2OS cells using RNAi. Transfection of 5 nM siRNA targeting APC reduced the level of APC protein to 14.7 ± 8.6% of that in cells transfected with nontargeting control siRNA (Fig. 1 A). The increase in β-catenin in cells treated with anti-APC siRNA further confirmed the reduction of functional APC in such cells (Fig. 1 A).
Figure 1.

Mitotic defects in APC-deficient cells. (A) A reduction in the amount of APC protein in U2OS cells using RNAi. Lysates from U2OS cells transfected with short RNA duplexes directed against APC or with control RNA duplexes were collected 72 h after transfection. Proteins were separated by PAGE and probed for APC, β-catenin, and actin (loading control). (B) Tension between kinetochores of metaphase sister chromatids is reduced in APC-deficient U2OS cells. Interkinetochore distances in U2OS cells transfected with nontargeting (iC) or APC-targeting (iA) siRNA as in A were measured as the distances between CREST-stained kinetochores in prophase or metaphase chromosomes as indicated. Mean values are shown as red bars. (C) APC inhibition in U2OS cells decreases the time from mitotic entry to anaphase onset. U2OS cells transfected with control (iC) or APC-targeting (iA) siRNA together with H2B-RFP were presynchronized by a single thymidine block and released for 6 h before imaging their mitotic progression for 6 h. Time from initiation of mitosis to anaphase onset was measured for 14 control and 32 APC-deficient cells and is shown as box plots using a five-point summary. Pairs with a statistically significant difference (P < 0.005) in two-tailed t tests are indicated by red hashes (#). (D and E) Reducing APC causes a mitotic checkpoint defect. U2OS cells transfected for 52 h with control or APC-directed siRNA duplexes were incubated with increasing amounts of nocodazole (A) or taxol (B) for a further 20 h before staining with propidium iodide and antiphosphohistone H3 antibodies. Mitotic index was determined by flow cytometry and is shown as the percentage of live cells that were phosphohistone H3 positive and had high (∼4n) DNA content. Data are represented as the mean ± SD (error bars).
Mitotic spindles in cells lacking APC were not grossly perturbed. However, the inability of such spindles to create full tension at kinetochores showed compromised spindle function (Fig. 1 B). The distance between sister kinetochores in prophase, when there is no bipolar attachment and therefore no tension, was almost identical in APC-inhibited and control cells (0.74 ± 0.03 μm and 0.70 ± 0.03 μm [mean ± SEM], respectively). In control cells, the distance between sister kinetochores increased to 1.38 ± 0.02 μm in metaphase, indicating the development of tension. Notably, interkinetochore distance in metaphase chromosomes of APC-deficient cells only reached 1.15 ± 0.02 μm, which is significantly different (P < 0.001; Mann-Whitney rank sum test and t test) from the control metaphase dataset. This demonstrates that metaphase kinetochores in APC-deficient spindles were under reduced tension.
In mouse fibroblasts in which APC expression can be conditionally inactivated (Sansom et al., 2004), APC removal produced the same effect (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1). Thus, the loss of APC leads to defects in the mitotic spindle that are translated into reduced tension on metaphase kinetochores.
Loss of APC correlates with reduced Bub1 at kinetochores
Insufficient interkinetochore tension induced by APC loss should lead to the accumulation of mitotic checkpoint proteins like Bub1 and BubR1 on kinetochores. Therefore, we quantitated the amount of kinetochore-associated Bub1 and BubR1 in APC-deficient and control mitotic cells by measuring the intensities of Bub1 and BubR1 immunofluorescence at kinetochores, which were defined by costaining with CREST (Fig. 2 A; also see Materials and methods; Schiffmann et al., 2006). Bub1 and BubR1 accumulate at kinetochores at prophase and gradually disappear throughout mitosis, with only minimal kinetochore localization at metaphase and anaphase (Jablonski et al., 1998; Howell et al., 2004). Our measurements confirmed these dynamics of Bub1 and BubR1 at kinetochores (Fig. 2, B and C). However, despite insufficient tension at kinetochores, unsynchronized U2OS cells transfected with APC-targeting siRNA accumulated 1.84-fold less Bub1 per cell (mean reduced by 45.6%, which is significant; P < 0.001 in a two-tailed t test) and 1.66-fold less BubR1 (mean reduced by 39.7%, which is significant; P < 0.01 in a two-tailed t test) at kinetochores during prometaphase than cells transfected with control siRNA (Fig. 2, B and C). The difference was still detectable at the prometaphase→metaphase transition and in metaphase cells; however, it was not statistically significant at these stages.
Figure 2.

Reduction in kinetochore-associated Bub1 and BubR1 in APC-deficient cells. (A) Examples of cells stained with antibodies against the kinetochore marker CREST, Bub1, and BubR1, which were used to measure kinetochore-associated Bub1 and BubR1. U2OS cells were fixed and stained with DAPI, anti-CREST, anti-Bub1, and anti-BubR1 antibodies as indicated. Each image represents a projection of several 0.2-μm sections. Insets show magnified regions of the image (indicated in the CREST image; arrow); a circle in the insets indicates the region identified by CREST staining that was then used as the kinetochore domain to calculate the amount of Bub1 or BubR1 intensity at this site. Bars, 5 μm. (B and C) The relative amount of Bub1 (B) and BubR1 (C) at mitotic kinetochores of U2OS cells decreases after APC depletion. Cells treated with control (white) or APC-targeted (gray) siRNA as in Fig. 1 A were fixed and stained as in A (this figure). The total intensity of Bub1 and BubR1 at kinetochores was measured in 3D images of individual cells using the OME system optimized for high throughput analysis as described in Materials and methods. Box plots show the amount of kinetochore-associated Bub1 and BubR1 per cell normalized to the amount of CREST in this region using a five-point summary. For each box plot, the number of cells on which it was based is indicated below the x axis. (D) Immunoblot of a constitutive APC mutant (ΔAPC) and wild-type (wt) counterpart mouse fibroblasts with anti-APC antibodies reveals the reduced level of APC in mutant fibroblasts. Actin was used as a loading control. (E and F) APC-deficient (ΔAPC; gray) and wild-type (wt; white) fibroblasts were prepared as in A, and the amount of Bub1 (E) or BubR1 (F) at kinetochores was quantitated as in B and C, with the difference that Bub1- and BubR1-stained cells were collected in separate datasets rather than from the same cells. APC-deficient cells accumulate relatively less Bub1 and BubR1 at their kinetochores during mitosis, particularly during prometaphase. Pairs with a statistically significant difference in two-tailed t tests are indicated by red hashes (#). B, P < 0.001; C, P < 0.01; E, P < 0.05; F, P < 0.001. Mild outliers are indicated by circles, and extreme outliers are shown by asterisks.
We also used fibroblasts from mice with constitutively reduced APC protein (Kaplan et al., 2001) and compared them with fibroblasts isolated from wild-type littermates (Fig. 2 D). Again, we found that APC-deficient cells had less Bub1 and BubR1 at kinetochores than control cells (Fig. 2, E and F). In prometaphase, the mean kinetochore-associated Bub1 intensity per cell in APC-deficient fibroblasts was reduced 1.65-fold (39.3% of that in wild-type cells, which is significant; P < 0.05 in a two-tailed t test; mean kinetochore-associated BubR1 per cell was reduced 2.37-fold [58.3% of wild-type counterpart], which is significant; P < 0.001 in a two-tailed t test). Thus, two independent APC-deficient cell systems showed a decrease in the amount of both Bub1 and BubR1 at kinetochores.
Effect of APC inhibition on mitotic progression
Monitoring the mitotic progression of logarithmically growing APC-inhibited and control U2OS cells revealed that inhibiting APC resulted in a decrease in the time from entry into mitosis to the onset of anaphase (27.9 ± 1.1 min in control and 23.3 ± 0.6 min in APC-inhibited cells), which was statistically significant (P < 0.005 by a Mann-Whitney rank sum test; Fig. 1 C).
Thus, APC inhibition resulted in the loss of both checkpoint proteins at kinetochores despite decreased interkinetochore tension. This correlated with a decrease in the time to anaphase onset in APC-negative cells. Together, these data indicate that in addition to spindle damage, the mitotic checkpoint might be compromised by APC removal.
APC inhibition leads to a defective mitotic spindle checkpoint
To assess mitotic checkpoint function, we treated cells with microtubule poisons that induce mitotic arrest if the mitotic spindle checkpoint is intact and counted phosphohistone H3–positive cells. Histone H3 is specifically phosphorylated in mitosis (Hendzel et al., 1997), and this event is commonly used as a marker for mitotic cells.
We observed that in U2OS cells, both nocodazole and taxol treatment led to a dose-dependent accumulation of cells in mitosis (Fig. 1, D and E; iContr). Inhibition of APC reproducibly resulted in a reduction in the number of mitotically arrested cells after 20 h of treatment with a broad range of concentrations of both taxol and nocodazole compared with cells transfected with control nontargeting siRNA (Fig. 1, D and E). This was not caused by a G2 block or delay, which would prevent cells from entering mitosis, as G2 progression in presynchronized cells was not altered by APC removal (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1). Thus, APC inhibition does not completely obliterate but substantially compromises the mitotic checkpoint in U2OS cells.
Inhibiting APC increases mitotic slippage
One of the consequences of a damaged mitotic checkpoint is the inappropriate exit of cells from mitosis into G1 to produce tetraploid cells. To distinguish cells that had exited mitosis with a 4n DNA content from the rest of the 4n DNA population, representing a mixture of cells in G2 and M, we costained cells with 7AAD (for DNA profile) and FITC-labeled anti– cyclin B1 antibody (Fig. 3 A). The cyclin B1 level is lowest in G1, gradually accumulates throughout S and G2, and peaks in mitosis (Hwang et al., 1995). Thus, the 4n DNA/cyclin B1–negative population (Fig. 3 A, red) represents cells that inappropriately exited mitosis with double chromosome content. This population was clearly increased in APC-deficient cultures even in the absence of mitotic poisons (Fig. 3, A and B). Mitotic arrest induced by taxol or nocodazole increased the number of 4n DNA/cyclin B1–negative cells. Importantly, such tetraplolid G1 cells were 1.5–2 times more abundant after APC depletion (Fig. 3 B).
Figure 3.

Inhibiting APC causes mitotic slippage and tetraploidy. (A) Flow cytometry profiles of cells 3 d after transfection with control (iContr) or APC-directed (iAPC) siRNA stained with FITC-labeled anti–cyclin B1 antibodies and 7AAD to detect DNA content. The 4n DNA cyclin B1–negative population representing tetraploid cells in G1 is shown in red; the polyploid population (>4n DNA) falls into the area shaded in blue. (B) APC inhibition leads to an increase in the number of tetraploid cells. The proportion of cells with a 4n DNA content that were cyclin B1 negative (tetraploid G1) in the total live cell population of U2OS cells was measured in cells 3 d after transfection with control or APC-targeting siRNA and, where indicated, after 20 h of treatment with 1.25 μg/ml taxol or nocodazole before harvesting and staining as in A. (C) Premature cyclin B degradation in mitotically arrested APC-deficient cells. Control (iContr) and APC-deficient (iAPC) U2OS cells were synchronized by double thymidine block and released into 1.25 μg/ml taxol (as outlined in Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1). Cells were collected at the indicated times after the addition of taxol and were stained as in A. The loss of cyclin B1 in APC-deficient cells 25.5 h after arrest is indicated by an arrow. (B and C) Data are represented as the mean ± SD (error bars).
Comparing cyclin B1 profiles for control and APC-negative cells arrested by taxol after synchronization (see Fig. S2 A for protocol) revealed a massive degradation of cyclin B1 in APC-negative cells when the level of cyclin B1 in control cells was only slightly decreased (Fig. 3 C). Thus, premature cyclin B1 degradation is likely to be involved in the increased mitotic slippage of APC-deficient cells.
APC removal alters exit from drug-induced mitosis
To determine the fate of APC-deficient cells that exit mitosis prematurely, we imaged U2OS cells expressing H2B–red fluorescent protein (RFP). H2B-RFP localizes to chromatin at all times, making it possible to observe chromatin structures in vivo. Observation of control (n = 175) and APC-inhibited (n = 172) cells arrested in taxol for 20 h revealed several differences between these cells. First, we found that as expected, APC inhibition shortened the time that cells remained arrested in mitosis (Fig. 4 C). The number of cells arrested for >10 h was 25% lower when APC was inhibited (88% of all mitoses in control and 66% in APC-deficient cells), and the number of cells arrested in mitosis for >15 h was halved by APC inhibition (52% in control and 27% in APC-deficient cells). This confirmed that the loss of APC induces spindle checkpoint defects.
Figure 4.

Visualization of mitotic slippage in APC-deficient cells arrested with taxol. (A) Representative images of control siRNA-treated U2OS cells showing mitotic arrest followed by cell death. 2 d after cotransfection with H2B-RFP and siRNA, cells were treated with 1.25 μg/ml taxol. Mitotic arrest was followed by the imaging of fluorescent chromatin-associated H2B (right) or brightfield (left) for 19 h starting 1 h after taxol addition. Both entry and exit from mitotic arrest are depicted as well as images of cells at the end of the recording (end). Note the membrane blebbing, chromatin tightening, and fragmentation as well as the change in the cell shape at the end of mitotic exit. (B) Representative image of a U2OS cell treated with APC-targeting siRNA that spreads after mitotic arrest. Cells were treated and imaged as in A. Images of control and APC-deficient cells were collected in parallel. Mitotic entry and mitotic exit of an APC-inhibited cell are depicted using both brightfield (left) and fluorescent chromatin (right) images at the indicated times as well as fluorescent images of final chromatin structure at the end of the video. Note the chromatin decondensation (most apparent at 2 h and 0 min), cell spreading, and irregular chromatin structure at the end of mitotic exit. (C) Taxol-induced mitotic arrest is shortened by APC inhibition. U2OS cells were treated and imaged as in A, and the times of mitotic entry (as judged by rounding up or/and chromatin condensation) and exit (as judged by membrane blebbing or/and chromatin decondensation) were recorded for each individual cell. The graph shows the percentage of control (blue) and APC-inhibited (pink) cells that remained in mitosis for longer than the indicated times. 175 control and 172 APC-deficient cells were analyzed. (D) An example of chromatin structure in interphase APC-deficient U2OS cells. 3 d after APC inhibition by siRNA, U2OS cells were fixed and stained with DAPI. The image represents a projection of several 0.2-μm sections. Bar, 15 μm. (E) Proportion of different types of mitotic exit in APC-deficient and control U2OS cells. Cells were treated and imaged as in A–C, and the number of cells that spread after exiting mitosis (purple; as in B) and that did not spread after exiting mitosis (pink; as in A) were counted and plotted to show the proportion of each type of mitotic exit. (F) An example of a taxol-arrested APC-deficient cell that attempts to form a cleavage furrow (black arrow in brightfield image; left) and in which the chromatin is stretched (white arrow; right), as if the cell is attempting to divide. After this event, the chromatin decondensed, indicating mitotic exit.
Second, careful monitoring of cells revealed two types of mitotic exit. One type (Fig. 4 A) was accompanied by prolonged membrane blebbing that coincided with mild chromosomal decondensation and was followed by the tightening and fragmentation of chromatin, which are typical signs of apoptosis. Eventually, such a cell acquired an irregular shape, lost all movements, and floated (Fig. 4 A, bottom). The second typeof mitotic exit (Fig. 4 B) displayed mild, if any, membrane blebbing but slight and short waves of cell reshaping. This coincided with chromatin decondensation and was followed by cell spreading. After cell spreading, decondensed chromatin remained partially fragmented for some time and contained holes that were gradually sealed (Fig. 4 B, bottom). This phenotype suggested that cells exited mitosis with a 4n DNA content and remained alive. Note that even in the absence of mitotic poison, we frequently observed misshapen interphase chromatin in APC-deficient cultures that could be the result of such alternative mitotic exit (Fig. 4 D). Quantitating these types of mitotic exit in control and APC-deficient cells revealed striking differences (Fig. 4 E). APC-deficient cells exited mitosis by the second route (spreading) twice as often as control cells, with 85% of APC-negative cells exiting in this manner, whereas only 41% of control cells spread after mitotic exit. Additionally, in a few cases, APC-deficient cells that spread after exiting mitosis initiated furrow formation and chromosome stretching (Fig. 4 F) despite the presence of taxol, which prevented spindle function.
Inhibition of APC decreases apoptosis
The prevalence of cell spreading as a route of mitotic exit in APC-negative cells suggested that these cells survived taxol treatment better than control cells. Therefore, we asked whether APC inhibition altered apoptotic response to microtubule poisons. Consistent with this idea, we found that inhibiting APC invariably decreased the relative size of the sub-G1 DNA fraction, which is an indicator of apoptosis (Fig. 5, A and B). This was true for both untreated cells and cells treated with taxol at various concentrations and for different amounts of time (10 h in Fig. 5 A and 20 h in Fig. 5 B).
Figure 5.

The loss of APC inhibits apoptosis. (A) U2OS cells transfected with nontargeting (iCONTR) or APC-targeting (iAPC) siRNA were treated with no or increasing amounts of taxol for 10 h. They were ethanol fixed and stained with propidium iodide to measure their DNA profiles by flow cytometry. The fraction of cells with sub-G1 DNA content, which is an indicator of apoptosis, is shown as a percentage of the total number of cells. (B) U2OS cells transfected with nontargeting (iContr) or APC-targeting (iAPC) siRNA were treated with 1.25 μg/ml taxol for 20 h, and the sub-G1 fraction was measured as in A. (C) APC-deficient cells can apoptose. Mouse fibroblasts constitutively lacking APC (ΔAPC fibroblasts; see Fig. 2 D for Western blot) or U2OS cells treated with APC-targeting siRNA (U2OS + siAPC) were treated for 12 h with 0.1 μM staurosporine (right) or were left untreated (left). Cells were fixed and stained with PE-labeled antibody against active (cleaved) caspase 3 and analyzed by flow cytometry. Cells positive for active caspase 3 fall into polygon R2. The percentage of active caspase 3–positive (apoptotic) cells is indicated in the right top corner of each dot blot. Note a dramatic increase in apoptosis after staurosporine treatment in all APC-deficient cells. (D) U2OS cells transfected with nontargeting (iContr) or APC-targeting (iAPC) siRNA were treated with 1.25 μg/ml nocodazole (noc) or taxol (tax) for 20 h or were left untreated (no), were stained with PE-labeled antiactive caspase 3 antibody, and were analyzed as in C. Bars show the percentage of apoptotic (active caspase 3 positive) cells. (E) Constitutively APC-deficient mouse fibroblasts (ΔAPC) and their wild-type counterpart (wt) were stained with PE-labeled antiactive caspase 3 antibody and were analyzed as in C. Bars show the percentage of apoptotic cells. (B and E) Data show a representative example of one out at least three independent experiments; for each experiment, duplicate samples were analyzed by flow cytometry. (B, D, and E) Data are represented as the mean ± SD (error bars).
To measure apoptosis directly in APC-negative cells, we stained cells with fluorescently labeled antibody directed against the cleaved version of caspase 3 that is specific for cells in apoptosis and counted the number of cells positive for active caspase 3 using flow cytometry (Fig. 5 C, R2 area). All APC-negative cells described here, constitutively APC-deficient mouse fibroblasts (Fig. 5 C, top), anti-APC siRNA-treated U2OS cells (Fig. 5 C, bottom), and mouse fibroblasts with conditionally inactivated APC (Fig. S1) responded to staurosporine with a dramatic increase in the number of apoptotic cells. These data validated the antiactive caspase 3 antibody as a useful tool to detect apoptosis in these cells and indicated that the apoptotic machinery is generally functional in the absence of APC. Using this method, we found that inhibiting APC reduced apoptosis in untreated and nocodazole- or taxol-treated U2OS cells (Fig. 5 D). In addition, the baseline level of apoptosis in mouse fibroblasts that are constitutively deficient for APC (ΔAPC) was reduced in comparison with that in the wild-type control cells (Fig. 5 E). Decreased apoptotic response to virus treatment was also detected in fibroblasts in which APC expression was conditionally inactivated (Fig. S1). Together, these results show that inhibiting APC results in a decrease in apoptosis.
Inhibiting APC leads to polyploidy
Tetraploid cells are often eliminated by apoptosis (Blajeski et al., 2001; Rieder and Maiato, 2004), and failure to do so would allow such cells to progress to polyploidy. The relative number of polyploid cells (>4n DNA; Fig. 3 A, blue shaded area) was up to three times higher in APC-deficient than in control cultures, which is consistent with the predicted consequence of an increased tetraploid G1 pool and decreased apoptosis. This was true for untreated or drug-treated cells (Fig. 6 A). Notably, deleting APC caused an overproportional increase in the number of polyploid cells relative to the increase in the tetraploid G1 cells (Fig. 6 B; compare Figs. 3 B with 6 A), suggesting that relatively more tetraploid cells proceeded to cycle. This is consistent with an apoptotic deficiency in these cells and confirms that lack of APC promotes polyploidy.
Figure 6.

Loss of APC leads to polyploidy. (A) Control or APC-targeting siRNA-transfected U2OS cells were treated with 1.25 μg/ml taxol or nocodazole for 20 h or were left untreated before harvesting and staining with FITC-labeled anti–cyclin B1 antibody and 7AAD for DNA content as in Fig. 3. The proportion of polyploid cells (>4n DNA; indicated as blue shaded area in Fig 3 A) in APC-positive (iContr) and APC-deficient (iAPC) cultures was determined. Graph represents the percentage of polyploid cells as the mean ± SD (error bars). (B) Accumulation of polyploid (>4n DNA) cells is accelerated in the absence of APC. U2OS cells were transfected with APC or control targeted siRNA, arrested, and stained as in A. The ratio of the polyploid population measured as in A to the tetraploid G1 population measured as in Fig. 3 was calculated. The graph represents the mean of polyploid/tetraploid G1 ratios ± SD for APC-deficient (iAPC) and control (iContr) U2OS cells, which were either unsynchronized or treated with mitotic poisons as indicated. (A and B) Data show a representative example of one out at least three independent experiments; for each experiment, duplicate samples were collected for flow cytometry analysis.
Effect of APC inhibition in cells insensitive to β-catenin activation
The multifunctional nature of APC raises the question of the mechanism for APC control of mitotic progression. To discriminate between the effects of an increase in β-catenin signaling resulting from APC inhibition and possible direct contributions of the APC molecule to the mitotic checkpoint and apoptotic machinery, we analyzed the consequences of APC inhibition in HCT116 colon cancer cells. HCT116 cells have constitutively transcriptionally active β-catenin as a result of a mutation in one of their β-catenin alleles. Removing APC in these cells allowed us to assess the direct role of APC independently of its β-catenin–mediated targets.
Using a β-catenin/T cell factor (TCF)–responsive promoter element fused to luciferase (Morin et al., 1997) to measure the effect of APC inhibition on the activation of β-catenin signaling, we confirmed that in HCT116 cells, APC inhibition produced a minute effect (3.8× activation) on β-catenin–activated transcription compared with the massive (1,903×) activation in U2OS cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1). If β-catenin/TCF target genes make a major contribution to controlling mitotic progression, the effect of APC inhibition in HCT116 cells should be minimal.
However, APC inhibition in HCT116 cells resulted in quantitatively nearly identical phenotypical changes to those observed in U2OS cells. Mitotic index in both taxol- and nocodazole-arrested HCT116 cells was decreased by APC inhibition (Fig. 7 B), indicating a mitotic spindle checkpoint defect. The relative size of both the tetraploid G1 and polyploid populations were increased in mitotically arrested and unsynchronized HCT116 cells upon APC removal (Fig. 7, C and D). Additionally, similar to results in U2OS cells, the sub-G1 fraction was decreased by APC depletion in unsynchronized and taxol/nocodazole-treated HCT116 (Fig. 7 E), indicating apoptotic deficiency. Thus, the effects of APC on the mitotic checkpoint, apoptosis, and, consequently, on the loss of euploidy can be exerted independently of downstream targets of β-catenin.
Figure 7.

Depleting APC affects the mitotic checkpoint and apoptosis and leads to tetra- and polyploidy in HCT116 cells. (A) HCT116 cells transfected with control (first lane) or APC-targeting (second lane) siRNA for 36 h were lysed, separated by PAGE, and blotted for APC and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; loading control). (B) APC inhibition results in mitotic spindle checkpoint defects in HCT116 cells. HCT116 cells were transfected with control (purple) or APC-targeting (pink) siRNA for 36 h and were either arrested with 10 ng/ml taxol or 100 ng/ml nocodazole for 12 h or were left untreated. Cells were harvested, fixed, and stained with phosphohistone H3 antibody to detect mitotic cells. (C) The G1 tetraploid population is increased in APC-deficient HCT116. HCT116 cells transfected and treated as in B were harvested and stained for cyclin B1 and DNA content as in Fig. 3. (D) The polyploid population is increased in APC-deficient HCT116 cells. The graph represents the polyploid population in control (purple) and APC-deficient HCT116 cells treated and stained as in C. (E) Apoptosis is decreased in APC-deficient HCT116 cells. The sub-G1 fractions in control (purple) and APC-deficient HCT116 cells treated and stained as in C are shown. (B–E) Values are given as the mean ± SD (error bars).
Loss of APC induces tetraploidy in vivo
To test whether the loss of euploidy demonstrated by increased tetra- and polyploidy in APC-deficient cells in vitro was also detectable in vivo, we determined whether a similar phenomena occurred when APC was deleted from intestinal cells using Cre-lox technology. Mice bearing a lox/P-flanked APC and an inducible form of Cre-recombinase (AhCre+Apcfl/fl) lose APC expression specifically in the gut (Sansom et al., 2004) upon the induction of Cre-recombinase expression. At day 8 after Cre induction, we observed a subset of cells within the AhCre+Apcfl/fl intestine with up-regulated p21, a marker for the loss of euploidy (Fig. 8, A–D). This was apparent as early as 3 d after induction (Fig. 8 E). The average nuclear area of these cells at day 3 (Fig. 8 F) or 8 (Fig. 8 G) showed an approximately twofold increase (P = 0.01; Kolmorov-Smirnov test), implying polyploidy. To confirm ploidy changes in these cells, the DNA content of intestinal epithelium from induced Cre+Apcwt/wt and Cre+Apcfl/fl mice was examined at day 6 by flow cytometry (Fig. 8 H). In Cre+Apcwt/wt epithelium, only 0.63% of cells had a DNA content >4n, whereas 9.1% of cells extracted from Cre+Apcfl/fl mice had a >4n DNA content. Thus, the loss of euploidy is also an early consequence of APC depletion in the intestinal epithelium.
Figure 8.

Loss of APC leads to tetraploidy in vivo. (A–E) Immunohistochemistry on intestinal tissue sections from mice with inducible Cre-recombinase (AhCre+) and wild-type APC (wt; A and C) and from AhCre+ mice carrying floxed APC (APCfl/fl; APC mut; B, D, and E) 8 (A–D) or 3 d (E) after Cre induction to inactivate APC in APCfl/fl but not APCwt/wt mice. Sections were stained for p21 and counterstained with haematoxylin to outline nuclei. C and D are magnifications of areas in A and B, respectively. Note that only tissue from AhCre+APCfl/fl, when APC was lost, had up-regulated p21 in the epithelial cells in crypts. (F and G) The nuclear area of epithelial cells in Cre-induced AhCre+APCwt/wt (wt/wt) or AhCre+APCfl/fl tissue that were p21 negative (fl/fl) or p21 positive (fl/fl p21 positive) were measured 3 (F) or 8 d (G) after Cre induction. The median nuclear area in p21-positive cells is twice that of p21-negative cells in APCfl/fl mice, suggesting that the former cells are tetraploid. (H) Flow cytometric analysis of intestinal samples 6 d after the initiation of Cre-mediated excision in AhCre+APCwt/wt (wt; top) and AhCre+ APCfl/fl (APC mut) mice shows an increase in the number of tetra- and polyploid cells in tissue without APC. (I) A tumor from the intestine of an APCmin mouse. Sections are counterstained with β-catenin, which is up-regulated in tumors. The arrow and arrowhead indicate the position of adenomas with high β-catenin levels. Note that nuclei showing a strong signal for β-catenin are larger than those in the surrounding tissue (inset shows a magnified view of the boxed area).
Together, our data provide evidence that the loss of APC in several systems rapidly and directly leads to tetra- and polyploidy both in vitro and in vivo. A combination of defects is likely to be responsible for this: first, abnormal spindles; second, a compromised spindle assembly checkpoint; and, third, a decrease in apoptosis that allows inappropriate survival of defective cells. We propose that incapacitating one single molecule, APC, creates this unique combination of defects that leads to the simultaneous disruption of several layers of cellular control (Fig. 9) and, thus, provides a selective advantage for APC-negative tumor cells.
Figure 9.
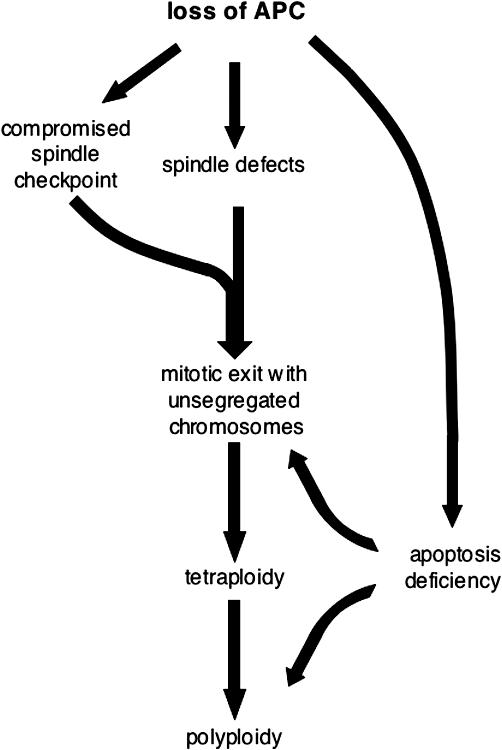
Consequences of APC loss. See Discussion for details.
Discussion
Tetraploidy has been correlated with poor cancer prognosis (Ladenstein et al., 2001; Sudbo et al., 2001; Melegh et al., 2005). Recent evidence suggests that it is not just a harmless side effect of cancer-promoting chromosomal instability but could be tumorigenic in its own right (Fujiwara et al., 2005). Thus, it is important to understand the mechanisms involved in producing and maintaining tetraploidy in cancer and the mechanisms that normally prevent cells from becoming tetraploid.
In this study, we show that depletion of the APC tumor suppressor is sufficient to produce tetraploidy. Our data are consistent with the idea that this occurs, at least in part, via mitotic slippage with nonseparated chromosomes. When cells divide, the spindle checkpoint machinery ensures that all kinetochores are attached to spindle microtubules and are under tension. When this process is completed, the anaphase-promoting complex is activated to initiate a proteolytic cascade that, on one hand, results in chromosome separation because of the release of separase and, on the other hand, initiates mitotic exit that begins with inactivation of the CDK1–cyclin B complex and leads to cytokinesis (Cleveland et al., 2003; Castro et al., 2005). If the mitotic checkpoint cannot be satisfied because of spindle damage, cells arrest at this stage for a limited length of time that depends on many factors and varies between cell types (Rieder and Maiato, 2004). At this point, cells are either cleared by apoptosis or escape mitotic arrest to reenter G1 with a 4n DNA content. Our live imaging data (Fig. 4) suggest that in the absence of APC, the second route for mitotic exit dominates. In U2OS cells depleted of APC by RNAi, 85% of cells that exited a taxol-induced arrest spread again and remained alive. Although U2OS cells have a relatively high spontaneous slippage rate, this number was more than doubled when APC was removed.
At this time, we do not understand the mechanism of mitotic slippage well enough to clearly dissect the role of APC in this process. However, there are several clues. First, we show that the mitotic spindle checkpoint is compromised after APC removal. Mitotic progression of logarithmically grown cells was faster despite spindle damage induced by APC loss (Fig. 1), and mitotic checkpoint proteins Bub1 and BubR1 were not localized efficiently to the kinetochores. Importantly, the accelerated exit from drug-induced mitotic arrest in APC-deficient cells reflects a defect in this checkpoint (Figs. 1 and 4). The reduced accumulation of Bub1 and BubR1 at kinetochores precedes and, thus, might be the cause of premature mitotic exit. Indeed, we found that the depletion of Bub1 by RNAi from U2OS cells caused a similar decrease in mitotic arrest as the depletion of APC (unpublished data). However, it is also formally possible that these events occur independently of each other. Experiments from another laboratory did not reveal a mitotic checkpoint defect in APC-depleted HeLa cells (Draviam et al., 2006). This discrepancy is likely caused by differences in the experimental approach used to measure such defects. Our experimental setup was able to detect a compromised mitotic checkpoint, whereas that used by Draviam et al. (2006) was designed to only detect the complete absence of a mitotic checkpoint.
Second, we observed extensive degradation of cyclin B1 in these cells after prolonged mitotic arrest. Of course, this could be a consequence of the mitotic checkpoint defect, as its abrogation would induce the cyclosome-driven degradation of cyclin B as cells exit mitosis. On the other hand, the destruction of cyclin B was previously reported to accompany mitotic slippage in mammalian cells with an active mitotic checkpoint (Jablonski et al., 1998). At this point, we cannot distinguish whether increased cyclin B degradation in APC-deficient cells is a cause or a consequence of premature mitotic exit.
Third, the failure of APC-deficient cells to die after they had escaped mitotic arrest could be related to the decreased apoptosis we detected in all APC-negative cells (Fig. 5). Bax-dependent apoptosis can be induced by mitotic slippage after prolonged mitotic arrest; however, this requires an intact mitotic checkpoint (Tao et al., 2005). Thus, apoptosis deficiency of APC-negative cells could be partially related to the mitotic checkpoint defect in these cells. However, this can only be part of the story because we detect a decreased rate of apoptosis in the absence of APC even in cells that were not exposed to mitotic poisons. An alternative explanation places both apoptotic and mitotic checkpoint defects downstream of one event, namely Bub1/BubR1 insufficiency. The proapoptotic function of the mitotic checkpoint proteins BubR1 and Bub1 (kinetochore-associated pools of both of them are decreased in APC-deficient cells) can trigger apoptosis specifically in polyploid cells (Shin et al., 2003). It is possible that a defect in this function of Bub1 and BubR1 in APC-depleted cells is responsible for their inability to undergo apoptosis efficiently. Consistent with this idea, the relative number of polyploid cells is overproportionally increased in APC-negative cultures (Fig. 6 B). Importantly, overexpressing recombinant Bub1 reduced polyploidy that was induced by APC loss and rendered the mitotic spindle checkpoint less sensitive to APC inhibition (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1).
In this context, it is important to note that we found decreased apoptosis in all of the APC-deficient systems we tested, including fibroblasts with a floxed APC allele that allows the conditional inactivation of APC (Fig. S1). These particular cells lack a functional mitotic checkpoint; both floxed fibroblasts and their wild-type counterparts did not maintain a mitotic arrest when treated with mitotic poisons (unpublished data). Interestingly, in these cells, only Bub1 but not BubR1 was reduced at the kinetochores when the APC level was reduced. Thus, the apoptotic defect in these cells could not be mediated by changes in BubR1. Importantly, data from these cells suggest that the downregulation of Bub1 at kinetochores is a primary consequence of APC loss and that the lack of BubR1 in the two other cell types may be secondary to the Bub1 deficiency. Indeed, BubR1 was shown to require Bub1 to be recruited to kinetochores (Johnson et al., 2004).
APC is a known negative regulator of the Wnt pathway. To determine whether this function contributes to the effect of APC on ploidy, we used HCT116 cells as a model. These cells have constitutively active β-catenin that is insensitive to APC depletion. Using these cells, we showed that APC is likely to affect the mitotic checkpoint and apoptosis independently of β-catenin targets (Fig. 7). This is in contrast to recent data that implicate the upregulation of conductin downstream of activated β-catenin in establishing genomic instability in the absence of APC (Hadjihannas et al., 2006). However, activation of Wnt signaling by APC inhibition in HCT116 cells used in this study was not well described, making a direct comparison difficult.
Our current data are consistent with the idea that the effect of APC on the spindle checkpoint and possibly on apoptosis involves Bub1 and, in some cases, BubR1. Recently, caspases were implicated in the removal of Bub1 and BubR1 from kinetochores during prolonged mitotic arrest and the resulting mitotic slippage (Baek et al., 2005). Because APC is found at kinetochores (Fodde et al., 2001; Kaplan et al., 2001), where many spindle checkpoint components accumulate, we considered the possibility that APC could protect checkpoint proteins from degradation by caspases at these sites. However, the defect in mitotic arrest induced by APC depletion was not altered by caspase inhibitors (unpublished data), suggesting that this is not the case. Furthermore, the removal of APC did not alter the phosphorylation of Bub1 and BubR1 in mitotically arrested cells (unpublished data). Therefore, the molecular mechanism of Bub1 and/or BubR1 regulation by APC remains unresolved.
The combination of spindle, spindle checkpoint, and apoptosis defects allows APC-negative cells in culture to become polyploid. Importantly, our data are supported by in vivo evidence showing that the inhibition of APC in intestinal mouse epithelia also leads to the loss of euploidy 3 d after APC loss is induced. It is intriguing that in this case, both prominent tetraploidy and a decrease in the number of apoptotic bodies in intestinal crypts (relative to increased apoptosis in the rest of the tissue; unpublished data) were restricted to the same area of the epithelium. Furthermore, a section of intestine from an APCmin mouse that produces β-catenin–positive adenomas (Fig. 8 I) after the loss of wild-type APC also revealed a striking difference in the nuclear size between cells in small β-catenin–positive lesions and the surrounding APC heterozygous (APCwt/min) normal tissue. Consistent with the idea that the mitotic checkpoint is compromised in the absence of APC, APC-Min mouse adenomas were arrested in mitosis by vinorelbine treatment less efficiently than normal tissues in these mice (our preliminary data).
In summary, we show that the loss of APC has immediate consequences: spindle defects together with a compromised mitotic checkpoint can produce tetraploid cells; combined with decreased apoptosis, this promotes the expansion of the polyploid population (Fig. 9). Furthermore, we demonstrate the loss of euploidy in the mouse gut after the inactivation of APC and in adenomas from Min mice, suggesting that tetraploidy is indeed a common feature of APC-deficient cancers.
Lack of APC induces a spectrum of cell cycle defects that can amplify each other to promote genetic instability. Furthermore, in the context of deregulated β-catenin, which maintains cells in a proliferative state inappropriately, the resulting defects in genetic stability could be particularly effective in producing tumors.
Materials and methods
Cell culture and treatment
Human colon carcinoma HCT116 cells and mouse embryonic fibroblasts constitutively deficient or wild type for APC (a gift from R.A. Weinberg, Whitehead Institute, Cambridge, MA) were cultured in DME supplemented with 10% FCS and 1% penicillin-streptavidin stock solution (MP Biomedicals). For human osteosarcoma U2OS cells, nonessential amino acids (1:100; Sigma-Aldrich) and 1% l-glutamine were added.
For siRNA-mediated inhibition, U2OS cells were transfected using siPORT-NeoFX reagent (Ambion) according to the manufacturer's instructions with 5 nM of either siRNA-targeting human APC (SmartPool reagent; Dharmacon) or nontargeting siCONTROL siRNA (Dharmacon) and were grown for 2–3 d. Alternatively, transfection with 16.7 nM siRNA using OligofectAMINE transfection reagent (Invitrogen) gave a similar level and timing of APC inhibition. For time-lapse imaging, U2OS cells were cotransfected with 0.2 μg H2B-RFP (a gift from M. Posch, Dundee University, Dundee, UK) together with 12.5 nM of appropriate siRNA (Dharmacon) using LipofectAMINE 2000 reagent (Invitrogen) according to the manufacturer's instructions. In HCT116 cells, APC was inhibited by transfection for 2 d consecutively with 50 nM of control or APC-targeting siRNA (Dharmacon) using OligofectAMINE and were allowed to grow for another day.
For mitotic arrest, U2OS cells were treated with the indicated amount of nocodazole or taxol for 20 h (unless stated otherwise), and HCT116 cells were treated with 10 ng/ml taxol or 100 ng/ml nocodazole for 12 h. For synchronization, U2OS cells received two rounds of 2 mM thymidine for 22 h with a 10-h interval. When needed, 1.25 μg/ml taxol or nocodazole was added at the time of release from the second thymidine block. For time-lapse imaging of mitotic progression (Fig. 1), U2OS cells were presynchronized with a single 18-h thymidine treatment and were released 6 h before taking images to enrich for cells dividing at the time of filming. For apoptosis induction, cells were treated with 0.1 μM staurosporine (Calbiochem) for 12 h before analysis.
Immunofluorescence
Cells were fixed in warm 1.85% PFA in PHEM buffer (60 mM Pipes, 4 mM MgSO4, 25 mM Hepes, and 10 mM K-EGTA, pH 6.9) for 15 min, washed in PBS, and blocked in blocking buffer containing 0.1% Triton X-100, 2% BSA, 5% donkey serum, and 0.02% NaN3 in PBS supplemented with 50 mM NH4Cl for at least 15 min. Alternatively, cells were fixed in ice-cold methanol for 5 min, rehydrated in PBS, and blocked in the aforementioned blocking buffer for at least 30 min. Primary antibodies were diluted in blocking buffer as follows: anti-Bub1 and -BubR1 (gift from S. Taylor, University of Manchester, Manchester, UK) were used at 1:130 for mouse cell lines and at 1:1,000 for human cell lines, anti-Bub1 mAb (Chemicon) was used at 1:500, and CREST was used at 1:100 for mouse cell lines and at 1:300 for human cell lines (gift from B. McStay [Biomedical Research Centre, Ninewells Hospital, Dundee, Dundee, UK] and W. Earnshaw [University of Edinburgh, Edinburgh, UK]). Secondary antibodies raised in donkey (Jackson ImmunoResearch Laboratories) conjugated with either FITC, Texas red, or Cy5 were used at a 1:250 dilution. Cells were counterstained with DAPI at 1 μg/ml for 2 min.
Microscopy
High resolution images were collected with an imaging system (DeltaVision Restoration; Applied Precision) built on an inverted microscope or stand (Eclipse TE200; Nikon; or 1X70; Olympus) using a 100× NA 1.4 objective lens. Images were acquired at 0.2-μm intervals in the z dimension and were deconvolved, and, where required, projections of multiple sections were built using SoftWoRx software (Applied Precision). Interkinetochore distances were measured in 3D images using SoftWoRx. H2B fluorescence/brightfield time lapses were acquired on a DeltaVision Restoration imaging system that was built on a stand (1X70; Olympus) equipped with a 37°C chamber using a 40× NA 1.4 dry objective lens. We collected five fluorescent sections at 2-μm intervals in the z dimension and one brightfield reference image in the middle of the stack every 10 min for 19 h for mitotically arrested U2OS cells or every 3 min for 6 h for the mitotic progression of logarithmically grown U2OS cells. Fluorescent H2B projections were built from the sections containing in-focus images using SoftWoRx software.
Analysis of images to measure the amount of Bub1 and BubR1 at kinetochores
Deconvolved 3D images of cells stained for CREST (kinetochore marker) and either Bub1, BubR1, or both were cropped around the nuclear area using SoftWoRx Explorer (Applied Precision) and analyzed in an open source microscopy image management system (Open Microscopy Environment [OME]; Swedlow et al., 2003; Goldberg et al., 2005; Hogan, 2005). The FindSpots algorithm available within OME was used to automatically analyze entire datasets of images. This algorithm identified as kinetochores any 3D objects with a volume of >30 voxels and intensities of CREST staining above the threshold, which was set up as μ + nσ, where μ is the mean, σ is the standard deviation of all voxels in the image stack, and n is either three or four but is constant throughout each experiment. The total Bub1 or BubR1 kinetochore intensity in each cell was determined by adding the integrated intensity of Bub1 or BubR1 signal for all 3D objects defined by the thresholded CREST signal in the image. After subtracting the background, which was calculated as Vk × μBub, where μBub is the mean voxel intensity in the Bub1 or BubR1 channel and Vk is the total CREST-positive (kinetochore) volume of that image, this figure was standardized using the total kinetochore volume to yield a background-corrected kinetochore-specific intensity: K*bub = (KBub − Vk × μBub)/Vk.
This analysis was achieved through mostly automated data processing, which facilitated the analysis of a large number of images (Schiffmann et al., 2006). The data was summarized visually using box and whisker plots. Each shows the normalized per cell kinetochore levels of Bub1 or BubR1 through a five-point summary: the median (thick middle line), lower quartile (bottom boundary of box), upper quartile (top boundary of box), and the lower and upper extents of the data (bottom and top whiskers drawn from the box, respectively). This occurred after excluding outliers as defined by the standard 1.5 interquartile range (IQR) rule: mild outliers were defined as those points lying >1.5 times the IQR away from the lower or upper quartiles and are indicated by small circles; extreme outliers (lying more than three times the IQR) are shown by asterisks (Fig. 2). Box plots were generated using SPSS software version 11 (SPSS, Inc.). Two-tailed t tests were performed using the Analysis ToolPak in Excel 2004 (Microsoft) to determine whether there was a statistically significant difference in the mean of the kinetochore intensities after removing the outliers identified by SPSS. Alternatively, t tests and Mann-Whitney rank sum tests were performed using the SigmaSTAT program (Systat Software, Inc.).
Western blots
Cells were lysed in MEBC buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM EGTA, 5 mM EDTA, 0.5% NP-40, and 40 mM β-glycerol phosphate) supplemented with 10 μg/ml each of leuptin, pepstatin A, and chymotrypsin. Soluble fractions were run on 4–12% gradient SDS gels (Invitrogen) using MOPS running buffer and transferred to nitrocellulose membrane (Protran) with a 0.1-μm pore size (Schleicher & Schuell). For APC detection, Ali mouse monoclonal antibody (against N terminus; Cancer Research UK; Efstathiou et al., 1998) or crude serum of rabbit polyclonal antibody raised against the middle portion of APC (anti-APCII; Nathke et al., 1996) were used at 1:1,000 in blocking solution (TBS containing 5% nonfat milk, 1% donkey serum, and 0.02% Triton X-100). Anti–α-tubulin mouse monoclonal antibody DM1a (Sigma-Aldrich) was used at a dilution of 1:2,000, anti-actin (C4 clone; MP Biomedicals) was used at 1:2,000, anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Abcam) was used at 1:5,000, anti-Bub1 and -BubR1 sheep polyclonal antibodies (gifts from S. Taylor) were diluted 1:500, and anti–β-catenin polyclonal antibody against C terminus (Hinck et al., 1994) was diluted 1:1,000. Secondary anti–rabbit, –mouse or –sheep HRP-labeled antibodies (Scottish Antibody Production Unit) or IRDye 800/700–conjugated anti–sheep and –mouse secondary antibodies (either Invitrogen or Rockland) were used at 1:5,000 dilutions.
Flow cytometry
For mitotic index measurements, cells were collected and fixed in 1% PFA in PBS at 37°C for 20 min, washed in PBS, and postfixed and permeabilized in 70% ethanol at −20°C for >30 min before staining with phosphohistone H3 antibodies (Upstate Biotechnology) diluted to 10 μg/ml in PBS with 1% BSA followed by AlexaFluor488-labeled anti–rabbit secondary antibody (Invitrogen) at a 1:150 dilution in PBS with 1% BSA. This was followed by 50 μg/ml propidium iodide in PBS containing 50 μg/ml RNase A and 0.1% Triton X-100. Alternatively, cells were stained with FITC-labeled antiphosphohistone H3 antibody (Upstate Biotechnology) followed by staining with 20 μg/ml 7AAD in PBS for the DNA profile. To measure G2/M or sub-G1 fractions, cells were collected, washed in 1% BSA in PBS, and fixed/permeabilized in 70% ethanol at −20°C for >30 min before staining with 50 μg/ml propidium iodide in PBS containing 50 μg/ml RNase A and 0.1% Triton X-100. For identification of early tetraploid cells, cells were fixed in 70% EtOH at −20°C, permeabilized on ice for 5 min in PBS containing 1% BSA and 0.25% Triton X-100, and stained with FITC-conjugated anti–cyclin B1 antibody or FITC-conjugated isotype control antibody (BD Biosciences) followed by staining with 20 μg/ml 7AAD in PBS. The cell profile was analyzed on a flow cytometer (FACSCalibur; Becton Dickinson) using CellQuest Pro software (BD Biosciences). Alternative data analysis was performed using Flowjo software (Tree Star, Inc.). In any kind of DNA profile analysis, clusters of two or more cells were excluded from analysis by gating. The cyclin B1–negative population was defined using the profile obtained with a FITC-labeled isotype control antibody combined with 7AAD. For active caspase 3 staining, cells were fixed and stained using the active caspase 3 phycoerythrin (PE) monoclonal antibody apoptosis kit (BD Biosciences) according to the manufacturer's instructions.
To determine DNA content in the mouse intestine, cells were extracted from the top 10 cm of the mouse small intestine. Intestines were opened, and the epithelium was removed with a scalpel blade. Cells were suspended in 1 ml of detergent solution (0.1 mol/ml citric acid and 0.5% vol/vol Tween 20), gently shaken for 20 min at room temperature, and passed through a 100-μm sieve. DRAQ5 (deep red anthraquinone; Biostatus) was added to a concentration of 20 μM before the detection of DNA content on a flow cytometry system (FACSVantage; Becton Dickinson).
Induction of APC loss in mice, immunocytochemistry, and determination of nuclear area
To induce recombination, AchCre+APCfl/fl and AchCre+APCwt/wt mice (Sansom et al., 2004) were given either single or multiple intraperitoneal injections of 80 mg/kg β-napthoflavone. At each time point, mice were killed, and the small intestine was removed and flushed with water. The proximal 10 cm of intestines was divided into 1-cm lengths, bundled using surgical tape, and fixed in 4% formaldehyde at 4°C for no more than 24 h before processing.
Immunohistochemistry of mouse intestines was performed using two different antibodies for p21, which gave the same pattern of expression (1:5, Neomarkers; and 1:25, Santa Cruz Biotechnology, Inc.). Nuclear area was assessed using standard protocols (Nunez et al., 2000) after image capture with analySIS software (Soft Imaging Systems). Immunohistochemistry with β-catenin antibody (dilution of 1:50; Becton Dickinson) on a small intestine of an APCmin mouse (Moser et al., 1990) was performed as described previously (Sansom et al., 2004).
Online supplemental material
Fig. S1 shows analysis of mitotic and apoptotic defects caused by APC reduction in mouse fibroblasts with conditional APC expression. Fig. S2 presents a comparison of cell cycle and G2 progression of presynchronized APC-negative and control U2OS. Fig. S3 shows comparative activation of the β-catenin/TCF-responsive promoter by APC depletion in U2OS and HCT116 cells. Fig. S4 shows the rescue effects of recombinant Bub1 overexpression in U2OS cells on mitotic checkpoint damage and polyploidy induced by APC depletion. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200610099/DC1.
Acknowledgments
We thank Stephen Taylor, Bill Earnshaw, Brian McStay, and others who contributed reagents as indicated in the manuscript. We thank John Rouse, Paul Clark, and all members of the Näthke laboratory for helpful discussions. We are also grateful for support and help with image analysis from Sam Swift, the OME team in Dundee, and Ilya Goldberg (National Institutes of Health, Bethesda, MD).
This study was funded by a project and program grant from Cancer Research UK. I.S. Näthke is a Cancer Research UK Senior Research Fellow.
Abbreviations used in this paper: APC, adenomatous polyposis coli; IQR, interquartile range; PE, phycoerythrin; RFP, red fluorescent protein; TCF, T cell factor.
References
- Baek, K.H., H.J. Shin, S.J. Jeong, J.W. Park, F. McKeon, C.W. Lee, and C.M. Kim. 2005. Caspases-dependent cleavage of mitotic checkpoint proteins in response to microtubule inhibitor. Oncol. Res. 15:161–168. [DOI] [PubMed] [Google Scholar]
- Bienz, M., and H. Clevers. 2000. Linking colorectal cancer to Wnt signaling. Cell. 103:311–320. [DOI] [PubMed] [Google Scholar]
- Blajeski, A.L., T.J. Kottke, and S.H. Kaufmann. 2001. A multistep model for paclitaxel-induced apoptosis in human breast cancer cell lines. Exp. Cell Res. 270:277–288. [DOI] [PubMed] [Google Scholar]
- Castro, A., C. Bernis, S. Vigneron, J.C. Labbe, and T. Lorca. 2005. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 24:314–325. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W., Y. Mao, and K.F. Sullivan. 2003. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 112:407–421. [DOI] [PubMed] [Google Scholar]
- Dikovskaya, D., J. Zumbrunn, G.A. Penman, and I.S. Näthke. 2001. The adenomatous polyposis coli protein: in the limelight out at the edge. Trends Cell Biol. 11:378–384. [DOI] [PubMed] [Google Scholar]
- Dikovskaya, D., I.P. Newton, and I.S. Näthke. 2004. The adenomatous polyposis coli protein is required for the formation of robust spindles formed in CSF Xenopus extracts. Mol. Biol. Cell. 15:2978–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draviam, V.M., I. Shapiro, B. Aldridge, and P.K. Sorger. 2006. Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J. 25:2814–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou, J.A., M. Noda, A. Rowan, C. Dixon, R. Chinery, A. Jawhari, T. Hattori, N.A. Wright, W.F. Bodmer, and M. Pignatelli. 1998. Intestinal trefoil factor controls the expression of the adenomatous polyposis coli-catenin and the E-cadherin-catenin complexes in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA. 95:3122–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde, R., J. Kuipers, C. Rosenberg, R. Smits, M. Kielman, C. Gaspar, J.H. van Es, C. Breukel, J. Wiegant, R.H. Giles, and H. Clevers. 2001. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat. Cell Biol. 3:433–438. [DOI] [PubMed] [Google Scholar]
- Fujiwara, T., M. Bandi, M. Nitta, E.V. Ivanova, R.T. Bronson, and D. Pellman. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 437:1043–1047. [DOI] [PubMed] [Google Scholar]
- Goldberg, I.G., C. Allan, J.M. Burel, D. Creager, A. Falconi, H. Hochheiser, J. Johnston, J. Mellen, P.K. Sorger, and J.R. Swedlow. 2005. The Open Microscopy Environment (OME) data model and XML file: open tools for informatics and quantitative analysis in biological imaging. Genome Biol. 6:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, R.A., and K.B. Kaplan. 2003. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J. Cell Biol. 163:949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, R.A., R. Wollman, and K.B. Kaplan. 2005. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol. Biol. Cell. 16:4609–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjihannas, M.V., M. Bruckner, B. Jerchow, W. Birchmeier, W. Dietmaier, and J. Behrens. 2006. Aberrant Wnt/beta-catenin signaling can induce chromosomal instability in colon cancer. Proc. Natl. Acad. Sci. USA. 103:10747–10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, C.A., and J.R. Miller. 2005. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene. 361:1–12. [DOI] [PubMed] [Google Scholar]
- Hendzel, M.J., Y. Wei, M.A. Mancini, A. Van Hooser, T. Ranalli, B.R. Brinkley, D.P. Bazett-Jones, and C.D. Allis. 1997. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 106:348–360. [DOI] [PubMed] [Google Scholar]
- Hinck, L., I.S. Näthke, J. Papkoff, and W.J. Nelson. 1994. Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J. Cell Biol. 125:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan, H. 2005. Where's that picture? Biophotonics International. 12:32–36. [Google Scholar]
- Howell, B.J., B. Moree, E.M. Farrar, S. Stewart, G. Fang, and E.D. Salmon. 2004. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14:953–964. [DOI] [PubMed] [Google Scholar]
- Hwang, A., A. Maity, W.G. McKenna, and R.J. Muschel. 1995. Cell cycle-dependent regulation of the cyclin B1 promoter. J. Biol. Chem. 270:28419–28424. [DOI] [PubMed] [Google Scholar]
- Jablonski, S.A., G.K. Chan, C.A. Cooke, W.C. Earnshaw, and T.J. Yen. 1998. The hBUB1 and hBUBR1 kinases sequentially assemble onto kinetochores during prophase with hBUBR1 concentrating at the kinetochore plates in mitosis. Chromosoma. 107:386–396. [DOI] [PubMed] [Google Scholar]
- Johnson, V.L., M.I. Scott, S.V. Holt, D. Hussein, and S.S. Taylor. 2004. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J. Cell Sci. 117:1577–1589. [DOI] [PubMed] [Google Scholar]
- Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Näthke. 2001. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat. Cell Biol. 3:429–432. [DOI] [PubMed] [Google Scholar]
- Kinzler, K.W., and B. Vogelstein. 1996. Lessons from hereditary colorectal cancer. Cell. 87:159–170. [DOI] [PubMed] [Google Scholar]
- Ladenstein, R., I.M. Ambros, U. Potschger, G. Amann, C. Urban, F.M. Fink, K. Schmitt, R. Jones, M. Slociak, F. Schilling, et al. 2001. Prognostic significance of DNA di-tetraploidy in neuroblastoma. Med. Pediatr. Oncol. 36:83–92. [DOI] [PubMed] [Google Scholar]
- Melegh, Z., E. Csernak, E. Toth, Z. Veleczki, E. Magyarosy, K. Nagy, and Z. Szentirmay. 2005. DNA content heterogeneity in neuroblastoma analyzed by means of image cytometry and its potential significance. Virchows Arch. 446:517–524. [DOI] [PubMed] [Google Scholar]
- Morin, P.J., A.B. Sparks, V. Korinek, N. Barker, H. Clevers, B. Vogelstein, and K.W. Kinzler. 1997. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 275:1787–1790. [DOI] [PubMed] [Google Scholar]
- Moser, A.R., H.C. Pitot, and W.F. Dove. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 247:322–324. [DOI] [PubMed] [Google Scholar]
- Näthke, I. 2005. Relationship between the role of the adenomatous polyposis coli protein in colon cancer and its contribution to cytoskeletal regulation. Biochem. Soc. Trans. 33:694–697. [DOI] [PubMed] [Google Scholar]
- Näthke, I.S., C.L. Adams, P. Polakis, J.H. Sellin, and W.J. Nelson. 1996. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J. Cell Biol. 134:165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez, F., M.D. Chipchase, A.R. Clarke, and D.W. Melton. 2000. Nucleotide excision repair gene (ERCC1) deficiency causes G(2) arrest in hepatocytes and a reduction in liver binucleation: the role of p53 and p21. FASEB J. 14:1073–1082. [DOI] [PubMed] [Google Scholar]
- Polakis, P. 1997. The adenomatous polyposis coli (APC) tumor suppressor. Biochim. Biophys. Acta. 1332:F127–F147. [DOI] [PubMed] [Google Scholar]
- Polakis, P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837–1851. [PubMed] [Google Scholar]
- Rieder, C.L., and H. Maiato. 2004. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell. 7:637–651. [DOI] [PubMed] [Google Scholar]
- Sansom, O.J., K.R. Reed, A.J. Hayes, H. Ireland, H. Brinkmann, I.P. Newton, E. Batlle, P. Simon-Assmann, H. Clevers, I.S. Näthke, et al. 2004. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 18:1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann, D.A., D. Dikovskaya, P.L. Appleton, I.P. Newton, D.A. Creager, C. Allan, I.S. Näthke, and I.G. Goldberg. 2006. Open microscopy environment and findspots: integrating image informatics with quantitative multidimensional image analysis. Biotechniques. 41:199–208. [DOI] [PubMed] [Google Scholar]
- Shih, I.M., W. Zhou, S.N. Goodman, C. Lengauer, K.W. Kinzler, and B. Vogelstein. 2001. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 61:818–822. [PubMed] [Google Scholar]
- Shin, H.J., K.H. Baek, A.H. Jeon, M.T. Park, S.J. Lee, C.M. Kang, H.S. Lee, S.H. Yoo, D.H. Chung, Y.C. Sung, et al. 2003. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 4:483–497. [DOI] [PubMed] [Google Scholar]
- Sudbo, J., M. Bryne, A.C. Johannessen, W. Kildal, H.E. Danielsen, and A. Reith. 2001. Comparison of histological grading and large-scale genomic status (DNA ploidy) as prognostic tools in oral dysplasia. J. Pathol. 194:303–310. [DOI] [PubMed] [Google Scholar]
- Swedlow, J.R., I. Goldberg, E. Brauner, and P.K. Sorger. 2003. Informatics and quantitative analysis in biological imaging. Science. 300:100–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, W., V.J. South, Y. Zhang, J.P. Davide, L. Farrell, N.E. Kohl, L. Sepp-Lorenzino, and R.B. Lobell. 2005. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 8:49–59. [DOI] [PubMed] [Google Scholar]
- Tighe, A., V.L. Johnson, and S.S. Taylor. 2004. Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability. J. Cell Sci. 117:6339–6353. [DOI] [PubMed] [Google Scholar]
- Zumbrunn, J., K. Kinoshita, A.A. Hyman, and I.S. Näthke. 2001. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr. Biol. 11:44–49. [DOI] [PubMed] [Google Scholar]