

Abstract
Radial organization of nuclei with peripheral gene-poor chromosomes and central gene-rich chromosomes is common and could depend on the nuclear boundary as a scaffold or position marker. To test this, we studied the role of the ubiquitous nuclear envelope (NE) component lamin B1 in NE stability, chromosome territory position, and gene expression. The stability of the lamin B1 lamina is dependent on lamin endoproteolysis (by Rce1) but not carboxymethylation (by Icmt), whereas lamin C lamina stability is not affected by the loss of full-length lamin B1 or its processing. Comparison of wild-type murine fibroblasts with fibroblasts lacking full-length lamin B1, or defective in CAAX processing, identified genes that depend on a stable processed lamin B1 lamina for normal expression. We also demonstrate that the position of mouse chromosome 18 but not 19 is dependent on such a stable nuclear lamina. The results implicate processed lamin B1 in the control of gene expression as well as chromosome position.
Introduction
Chromosomes occupy discrete, mutually exclusive volumes in interphase nuclei (Cremer and Cremer, 2001). These chromosome territories do not intermingle, except at their boundaries (Zink et al., 1998; Edelmann et al., 2001; Branco and Pombo, 2006), and their size is approximately related to DNA content (Gilbert et al., 2005). Although most proteins diffuse rapidly throughout nuclei (Misteli, 2001), the diffusion of individual chromosome territories is constrained (Manders et al., 2003; Walter et al., 2003). Territories are not randomly distributed, and the rules governing positioning are incompletely understood and vary between cell types (Parada et al., 2004) and growth state (Parada et al., 2002). The most constant finding is of a conserved radial organization; gene-poor chromosomes (e.g., human chromosome 18) are found at the periphery, and gene-rich chromosomes (e.g., human chromosome 19) are found more centrally (Croft et al., 1999; Cremer and Cremer, 2001; Tanabe et al., 2005). A similar although weaker radial organization with gene-poor chromosomes more peripheral has recently been reported for the mouse, based on position analysis of chromosomes 1, 2, 9, 11, 14, and X in a range of cell types (Mayer et al., 2005). No specific nuclear envelope (NE) component has been implicated in these large-scale localizations, and the inner nuclear membrane protein emerin and lamina protein lamin A have been excluded (Boyle et al., 2001; Meaburn et al., 2005).
The significance of the nonrandom distribution of chromosome territories within the interphase nucleus remains poorly understood. The nonrandom organization of territories may reflect functional associations between specific chromatin regions and other nuclear structures, the nucleolus being one obvious example. The inner face of the nuclear periphery, consisting of an inner nuclear membrane decorated with a nuclear lamina pierced by nuclear pore complexes (NPCs) represents another large domain for potential chromatin interaction. Experimental evidence from several groups has shown that associations between certain gene loci and the nuclear periphery may play important roles in the transcriptional regulation of those genes. The association has been shown to act at the level of chromosomal subregions and generally has a repressive or silencing role, with activation involving movement away from the periphery into the nuclear interior (Hewitt et al., 2004; Zink et al., 2004; Chuang et al., 2006).
However, peripheral association does not always result in repression; in yeast, transcription-dependent association with the NPC couples transcriptional activity with message export (Casolari et al., 2004; Taddei et al., 2006). Significantly, engineered enhancement or weakening of the association of the hexokinase I test locus with the NPC modulated the gene expression changes seen in response to physiological triggers, suggesting a positional effect operating in tandem with regulation because of transcription factor binding (Taddei et al., 2006). Using live-cell imaging of the yeast GAL1 gene marked by adjacent Tet operator sequences detected with GFP-tagged Tet repressor protein, Cabal et al. (2006) demonstrated that transcription is necessary but not sufficient for perinuclear confinement of active loci, which requires, in addition, direct interactions between components of the histone acetyltransferase complex and NPC components. Intriguingly, in this study, the subnuclear localization of the activated test locus demonstrated a peripheral confinement that was not static but, rather, involved a 2D sliding motion along the nuclear periphery, suggestive of molecular interactions continually formed and released.
In parallel with the emerging picture of active gene association with NPCs in yeast, association of chromatin with the nuclear lamina is increasingly recognized in mammalian cells. Known chromatin–lamina protein interactions (Taniura et al., 1995; Goldberg et al., 1999), together with the observed genetic defects in the human laminopathies (Mounkes et al., 2003; Broers et al., 2004; Worman and Courvalin, 2005), also implicate this structure in additional levels of nuclear organization and regulation (Gruenbaum et al., 2005). Recently, a lamina-dependent chromatin position effect has been observed during analysis of the localization of the human 4q35.2 region implicated in fascioscapulohumeral muscular dystrophy (FSHD; Masny et al., 2004). In this case, the tightly peripheral position of the subtelomeric FSHD locus requires a lamina containing functional lamin A. The overall nuclear position of the chromosome 4 territory, however, is unchanged between wild-type (WT) and lamin A–null fibroblasts, suggesting that the 4q35.2 region migrates within the chromosome territory when lamin A–dependent peripheral association is lost.
The nuclear lamina is a protein network that underlies the inner nuclear membrane, where it maintains nuclear shape and plays roles in attaching heterochromatin (Goldman et al., 2002). The mammalian nuclear lamina contains lamins A and C (alternatively spliced products of a single gene, LMNA), together with lamins B1 and B2, products of two additional genes, LMNB1 and LMNB2. B-type lamins are expressed in all cells, whereas A-type lamins are developmentally regulated. Lamin A contains a C-terminal CAAX motif (cysteine, aliphatic, aliphatic, any of several residues) that undergoes a ras-like processing comprising farnesylation, endoproteolysis, and carboxymethylation, but this modified C terminus is then removed by a Zmpste24-dependent maturation cleavage, which removes the C-terminal 15 amino acids. Mature lamin A therefore lacks the farnesylated and carboxymethylated C-terminal anchor. A point mutation that alters splicing to generate lamin A lacking the maturation cleavage site produces “progerin,” a lamin A with the CAAX attachment site intact. The resulting severe multisystem premature aging phenotype (Hutchinson-Gilford progeria syndrome) suggests that modified C-terminal anchoring of the nuclear lamina is important for its function (Eriksson et al., 2003). The dominant toxic effect of progerin may be the result of competition for limited binding sites on the inner nuclear membrane, and it has recently been shown that reducing the amount of progerin within the nucleus using farnesyl transferase inhibitors (FTIs) restores nuclear shape (Glynn and Glover, 2005; Mallampalli et al., 2005; Meaburn et al., 2005; Yang et al., 2005) and may be of potential use in the treatment of progeria (Fong et al., 2006).
Lamin B1 also undergoes CAAX processing; the mature protein retains the hydrophobic processed CAAX anchor and remains stably associated with the lamina and with the inner nuclear membrane. CAAX processing is important for functional lamin B1 expression at the nuclear periphery (Kitten and Nigg, 1991). Processing has three stages; farnesylation of the cysteine at −4 by a unique farnesyl transferase, endoproteolysis to remove the last three residues by the Ras converting enzyme 1 (Rce1), and carboxymethylation of the newly terminal cysteine by isoprenylcysteine carboxyl methyltransferase (Icmt). Endoproteolysis only occurs after farnesylation. The first two steps are essential for stable association of lamin B1 with the nuclear periphery, whereas the final carboxymethylation step is only important in the context of the isolated C terminus lacking the coiled-coil domains (Maske et al., 2003).
In the current study, we hypothesize that the stability of associations of the nuclear lamina with chromatin are important for gene expression. We first use live-cell imaging to show that the stability of the lamin B1 network of the nuclear lamina is dependent on lamin B1 processing by the endoprotease Rce1. Using three MIAME (minimum information for annotation of microarray experiments)–compliant microarray datasets, we show that either absence of full-length lamin B1 or lack of C-terminal processing affects gene expression and that some of the dysregulated genes form clusters on certain chromosomes. We identify a significant cluster of three dysregulated genes within an ∼4–mega bp (Mbp) region on chromosome 18 and use this chromosome as a model for how loss of interaction with the nuclear lamina affects chromosome position and, hence, gene expression. This is, to our knowledge, the first report linking a defect in the NE, an altered chromosome position, and changes in gene expression and supports the view that peripheral nuclear architecture is important for aspects of genome organization that play a role in the regulation of gene expression.
Results
Nuclear lamina stability is dependent on lamin B1 processing
We previously demonstrated the importance of lamin B1 processing for the integrity of the nuclear lamina and its association with the NE using sequential extraction of nuclear proteins (Maske et al., 2003). Here, we first looked at the effects of defects in lamin B1 processing on the stability of the NE using fluorescence loss in photobleaching (FLIP) of GFP-tagged lamin B1 expressed in cells lacking either of the CAAX processing enzymes, Rce1 or Icmt (Fig. 1). FLIP shows that lamin B1 endoproteolysis by Rce1 is important for its stability within the nuclear lamina (Fig. 1 b), whereas loss of carboxymethylation by Icmt has little effect on the GFP–lamin B1 stability (Fig. 1 c). Restoring tagged full-length lamin B1 expression to Lmnb1−/− cells gives a stability indistinguishable from WT, confirming that secondary changes due to selection on the transgenic cells are not responsible for altered stability. Lamin C dynamics are not altered in the absence of lamin B1 or lack of its processing, as FLIP of YFP–lamin C in all the knockout cells used in the current study shows no significant difference from that in WT cells (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200607054/DC1). These results suggest that farnesylation and endoproteolysis are essential for integrity of lamin B1 in the NE and, hence, for those aspects of nuclear architecture that depend on it. Given the association between chromosome position and gene expression, we set out to investigate the effect of the lack of lamin B1 or defects in its processing on gene expression.
Figure 1.

The stability of lamin B1 interactions is dependent on posttranslational modification by Rce1. (a) FLIP in WT mouse embryonic fibroblasts, Rce−/−, and Icmt−/− cells expressing GFP–lamin B1. A specific ROI was photobleached at full laser power for 250 s. The postbleach image shows the extent of fluorescence loss in the nonbleached area, which is indicative of the stability of GFP–lamin B1 at the nuclear periphery. Bar, 10 μm. (b) Quantitative FLIP. An ROI outside the photobleached area shown in panel a was used to measure fluorescence loss after photobleaching (mean values ± SD; n = 5). The background intensity was subtracted and the values normalized by dividing by the intensities of an ROI in an adjacent control cell to account for photobleaching of the 5% laser power used for scanning the nonbleached areas. GFP–lamin B1 interactions at the nuclear periphery are more stable in WT and Icmt−/− (c) than in Rce−/− (b), indicating that carboxymethylation has little or no effect on the stability of the nuclear lamina. (d) FLIP of GFP–lamin B1 in the Lmnb1−/− cells used in the current study does not show any significant difference from its behavior in WT cells, indicating that the difference observed in the Rce1−/− cells is due to a lack of processing and not another cell-specific factor.
Effect of lamin B1 processing and NE stability on gene expression
To study the effect of lamin B1 and its processing on gene expression, we performed 18 genome-wide microarray experiments to compare the gene expression profiles of Lmnb1−/−, Rce1−/−, and Icmt−/− cells with their WT backgrounds. We reasoned that genes with altered expression in both Lmnb1−/− and Rce1−/− cells are dependent on endoproteolyzed lamin B1, whereas those dysregulated in Lmnb1−/−, Rce1−/−, and Icmt−/− cells are dependent on carboxymethylated lamin B1. In contrast, genes dysregulated in either Rce1−/− or Icmt−/− cells but not in Lmnb1−/− cells would be dependent on Rce1 and/or Icmt processing of proteins other than lamin B1. The microarray datasets were MIAME complaint and included six biological replicates of RNA preparations of each cell type, with duplicate comparisons on each slide. Dye swaps were also included to eliminate any dye-specific effects on the microarray hybridizations. All of the raw data may be downloaded from Array Express (www.ebi.ac.uk/arrayexpress/). The microarray datasets were also validated by quantitative real-time PCR (qRT-PCR) for samples of genes up-regulated, down-regulated, or unchanged for each cell type (Table I; qRT-PCR values are shown in parentheses). All of the data discussed in this paper were statistically significant at P < 0.05 for a 1.5-fold change in expression between the test and WT samples. We used a 1.5-fold change as a cutoff for biological significance; by this criterion, there were significantly up- and down-regulated genes for each knockout type. For example, in the Lmnb1−/− cells, 834 genes were down-regulated, in contrast to only 129 genes that were up-regulated. For Rce1−/− cells, the corresponding values were 42 genes down-regulated compared with 422 up-regulated.
Table I.
Examples of differentially expressed genes in the three transgenic cell populations and their RT-qPCR confirmation
|
Ratio (KO/WT)
|
||||
|---|---|---|---|---|
| Ref Seq ID | Gene | Lmnb1 | Rce1 | Icmt |
| NM_009964 | Crystallin, α B (Cryab) | 3.0 (5.8) | 2.7 (2.7) | 1.8 (4.4) |
| NM_007486 | Rho, GDP dissociation inhibitor β (Arhgdib) | 2.5 | 3.2 | 1.7 |
| NM_010415 | Heparin-binding EGF-like growth factor (Hegfl) | 3.6 (6.1) | 3.0 (2.6) | 1.6 (4.9) |
| NM_019760 | Tumor differentially expressed 2 (Tde2) | 1.6 | 2.2 | 1.6 |
| NM_007874 | Deleted in polyposis 1 (Dp1) | 1.9 | 4.0 | 1.2 |
| NM_016774 | ATP synthase, H+ transporting mitochondrial F1 complex, β subunit (Atp5b) |
1.44 | 1.7 | 1.1 |
| NM_010288 | Gap junction membrane channel protein α1 (Gja1) | 5.5 | 8.3 (8.4) | 0.47 |
| XM_203363 | Slit homologue 3 (Slit3) | 0.39 | 0.52 | 0.83 |
| NM_011333 | Chemokine (C-C motif) ligand 2 (Ccl2) | 0.41 | 0.33 | 0.66 |
| NM_013496 | Cellular retinoic acid binding protein 1 (Crabp1) | 38 (50) | 0.07 | 0.3 (0.5) |
| NM_019390 | Lamin A (Lmna) | 2.8 (2.3) | 1.6 (1.3) | (0.3) |
| NM_010722 | Lamin B2 (Lmnb2) | (0.8) | (1.9) | (0.8) |
Values in parentheses represent fold changes determined using qRT-PCR. KO, knockout.
Three-way comparisons of expression changes were then made to find genes affected in more than one knockout cell type. First, genes with altered expression in only one of the knockout cell types were identified. We found 614 genes with altered expression (predominantly down-regulation) only in Lmnb1−/− cells. Because the normal expression of these genes depends on lamin B1 expression but is not altered when the CAAX processing machinery is defective, we conclude that their expression is influenced by an unmodified lamin B1 pool. Similarly, we found 249 genes with altered expression (predominantly up-regulation) only in Rce1−/− cells. Because the normal expression of these genes requires cellular CAAX processing but is not altered when lamin B1 is absent, we conclude that their expression is modulated by CAAX processed proteins other than lamin B1; ras is an obvious candidate.
Fig. 2 a shows part of a tree view from a clustering analysis that takes account of all three knockout cell types. The section shown includes genes with altered expression (in either direction) in any two of the cell types. Using the 1.5-fold change as a cutoff, we identified a group of 16 genes that were coordinately dysregulated in the Rce1−/− and Lmnb1−/− cells, 11 up-regulated and 5 down-regulated (Fig. 2 b). To confirm the similarity in the pattern of expression change in the two cell types, we performed correlation analysis on the fold changes for these 16 genes in Lmnb−/− and Rce1−/− cells. The resulting Pearson product–moment correlation coefficient was 0.897, with a value of 1.0 representing perfect correlation; this confirms that this group of genes is dysregulated in a similar way in the two transgenic cell types. Because these genes were coordinately either up- or down-regulated in each of the knockout cells, we conclude that their normal expression requires both lamin B1 expression and an intact CAAX farnesylation and endoproteolysis machinery, indicating that their expression depends on processed lamin B1.
Figure 2.

A summary of the gene expression changes in cells lacking full-length lamin B1, Rce1, or Icmt. (a) A tree view diagram showing the fold change of genes that are differentially expressed in Lmnb1−/−, Rce1−/−, and Icmt−/− cells. (b) A detailed view of the 16 genes with coordinately altered expression. The top box shows genes up-regulated in both Lmnb1−/− and Rce1−/−, and the bottom box shows genes down-regulated in both these knockout (KO) cells. Fold changes are shown as KO/WT. (c) A Venn diagram showing the proportion of genes that are regulated by 1.5-fold because of abnormalities in lamin B1 processing. The percentages represent the proportion of up-regulated genes in relation to all dysregulated genes within a specific category. (d) Summary of changes in gene expression in Lmnb−/− and Rce1−/− cells, showing changes unique to one knockout cell type.
To confirm that this dysregulation is indeed a consequence of interfering with lamin B1 and its processing, we performed additional experiments on WT fibroblasts in which CAAX processing was abolished by treatment of the cells with an inhibitor of the farnesyl transferase enzyme. qRT-PCR on several of these genes in FTI-treated WT cells confirmed that they showed similar changes as a result of interfering with the first step of lamin B1 processing as they do in the Rce1−/− cells (Table II). FTI treatment is expected to alter the gene expression of several genes, but the subset of genes selected here are also dysregulated in the Lmnb1−/− cells, which means that we are examining genes that are dysregulated as a result of interfering with the farnesylation of lamin B1.
Table II.
Fold changes of gene expression following FTI treatment of WT cells, showing that interfering with the farnesylation of lamin B1 has an effect that is similar to lack of endoproteolysis
| Ratio (KO/WT or treated/untreated)
|
||||
|---|---|---|---|---|
| Ref Seq ID | Gene | Lmnb1 | Rce1 | FTI treated |
| NM_009964 | Crystallin, α B (Cryab) | 3.0 | 2.7 | 6.9 |
| NM_010415 | Heparin-binding EGF-like growth factor (Hegfl) | 3.6 | 3.0 | 7.0 |
| NM_010288 | Gap junction membrane channel protein α1 (Gja1) | 5.6 | 9.0 | 1.7 |
| NM_019390 | Lamin A (Lmna) | 2.8 | 1.6 | 1.14 |
| NM_010722 | Lamin B2 (Lmnb2) | 0.8 | 1.9 | 1.21 |
Bold indicates up-regulation; all others were not significantly changed. KO, knockout.
After identifying this group of 16 coordinately dysregulated genes (Fig. 2 b), we sought common features or characteristics between them. First, we tried to cluster these dysregulated genes according to function using the Gene Ontology database but were unable to find a common pattern. To exclude any functional clustering more completely, we took all genes showing significant dysregulation in either direction in one or more of the knockout cell types (a total of 4,144 genes) and repeated the Gene Ontology clustering. The entire dataset is given in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200607054/DC1), but a small representative area is shown in Fig. 3. This analysis should find clusters of dysregulated genes associated with specific functions or pathways even if expression levels are changing in opposite directions in different knockout cells. We could find no evidence for clustering of dysregulated genes by function even by this more relaxed criterion.
Figure 3.

Genes that are dysregulated in the absence of full-length lamin B1 or Rce1 do not show any functional clustering. A tree view of genes that are changed by 1.5-fold or more in Rce−/− and Lmnb1−/− cells (red indicates up-regulated genes, and green indicates down-regulated genes), illustrating that there is no functional clustering based on the following GO entries: Calcium ion binding (0005509), transcription (0003700), cell adhesion (0007155), zinc ion binding (0008270), heparin binding (0008201), development (0007275), growth factor activity (0008083), and transferase activity (0016740). Note that some genes are represented more than once because they have multiple entries in the GO database.
To continue the search for shared characteristics in this small group of genes with expression dependent on processed lamin B1, the position of each gene was mapped onto the mouse karyotype. Inspection of the result suggested that the distribution, especially of the up-regulated genes, was not random but bunched (Fig. 4). In particular, a group of three up-regulated genes were found within a short 4-Mbp region on chromosome 18. To test whether this apparent clustering was statistically significant, we used a bootstrap sampling method to repeatedly draw 16 genes at random from the set of genes common to both lamin B1 and Rce1 knockout experiments. This method takes account of the genes present in both datasets from the microarray experiment and their nonuniform distribution within the karyotype. Analysis of clustering of the results of 10,000 trials confirmed that the experimental cluster found on chromosome 18 is significant (P < 0.02). We then considered this significant cluster of up-regulated genes on chromosome 18 and reasoned that its presence might be a consequence of movement away from the periphery as a result of the loss of fully processed lamin B1.
Figure 4.

The distribution of genes that are dysregulated in the absence of full-length lamin B1 or Rce1 shows some positional clustering. Genes that are differentially expressed by at least 1.5-fold (red indicates up-regulated genes, and blue indicates down-regulated genes) in both Lmnb1−/− and Rce1−/− cells were mapped to their specific chromosomal positions using Ensembl KaryoView. Note that the cluster on chromosome 18 contains three up-regulated genes not resolved at this scale.
Chromosome 18 position in WT versus knockout cells
There have been no previous reports of the radial position of the gene-poor mouse chromosome 18. We therefore studied its position using FISH. The method used to determine its radial position involved dividing the nucleus into five concentric shells and measuring the distribution of the chromosome 18 signal within these shells in 50 nuclei. We found that in three independent WT mouse embryonic fibroblast populations, chromosome 18 occupies a strongly peripheral location (Fig. 5, a and b, WT), possibly in association with the nuclear lamina. Indeed, when Con A was used as a marker for high-mannose glycoproteins in the intermembrane space of the NE in further FISH experiments, we found no detectable separation between the NE and the outermost border of the chromosome 18 territory (Fig. 5 c). As the same result was obtained in three independent WT fibroblast populations, the peripheral location of mouse chromosome 18 is unlikely to be an artifact because of inadvertent clonal selection during culture.
Figure 5.

Endoproteolyzed but not carboxymethylated lamin B1 is required for the peripheral localization of mouse chromosome 18. (a) Adherent cells were fixed in methanol acetic acid and hybridized with probes to mouse chromosomes 18 (red) and 19 (green). Nuclei were then counterstained with DAPI (blue). (b) 50 nuclei of normal morphology from each cell type were used to quantitate the distribution of chromosomes 18 and 19 in five concentric shells eroded from the nuclear periphery (left) to the center (right). The y axis shows the proportion of FISH signal in each of five zones; error bars show SEM. (c) Con A labeling (blue) alongside chromosome 18 FISH (red) shows that chromosome 18 is in close contact with the nuclear periphery.
We also performed FISH for chromosome 19, a very gene-rich chromosome that would not be expected to be associated with the nuclear periphery. We found that chromosome 19 has a more central location in all the studied cell types (Fig. 5, a and b), consistent with the observation that no chromosome 19 genes are dysregulated as a result of the lack of processed lamin B1 (Fig. 4).
Once the peripheral position of chromosome 18 in normal primary fibroblasts was established, we repeated the chromosome 18 FISH in Lmnb1−/− and Rce1−/− cells, to test whether detectable chromosome territory movement had occurred. Vergnes et al. (2004) reported that 38–39% of the Lmnb1−/− cells they examined had misshapen nuclei, compared with 2–8% of the WT cells. We also observed many misshapen nuclei in the Lmnb−/− cells. To exclude any secondary effects of gross morphological abnormalities on measured chromosome distribution, we only considered nuclei that exhibited normal morphology in all of our FISH analyses. Strikingly, in both Rce1−/− and Lmnb1−/− knockout cell types, chromosome 18 was no longer found at the periphery (Fig. 5, a and b, Rce1−/− and Lmnb1−/−). In contrast, however, Icmt−/− cells showed a peripheral distribution for chromosome 18 that was very similar to WT (Fig. 5, a and b, Icmt−/−), suggesting that just as carboxymethylation is not essential for the stability of lamin B1 in the lamina (Fig. 1), it is also not important in the maintenance of chromosome 18 at the nuclear periphery.
To confirm this striking result, we sought an additional experimental test. In particular, we were concerned with excluding the possibility that the observed new position of chromosome 18 was the result of selection of an unusual subclone during culture, although this would have had to occur independently for both Rce1−/− and Lmnb1−/− cell types. We reasoned that a demonstration of chromosome 18 relocation as a result of an acute perturbation of WT cells would offer strong evidence that the altered position of chromosome 18 in both transgenic knockout cells was not the result of inadvertent simultaneous selection of both Lmnb1−/− and Rce1−/− subclones with altered chromosomal position. Accordingly, we used a selective inhibitor of farnesylation, the first and mandatory step of CAAX processing, in WT cells and repeated the FISH experiment for chromosomes 18 and 19. As shown in Fig. 6, the relocation of chromosome 18 was also observed in FTI-treated WT cells, confirming that it is indeed processed lamin B1 that is essential for the localization of chromosome 18 at the nuclear periphery.
Figure 6.

Farnesylated lamin B1 is required for the peripheral localization of mouse chromosome 18. (a) FISH analysis of chromosome 18 position after FTI treatment, which disrupts the first step of lamin B1 processing, shows that loss of farnesylated lamin B1 has an effect that is similar to that of loss of endoproteolysis. Bar, 10 μm. (b) Quantitative analysis of chromosome 18 position as described above after FTI treatment shows loss of peripheral localization. Error bars indicate SEM.
Discussion
Previous sequential extraction studies of lamin B1 association with the nuclear lamina after inhibition of CAAX processing demonstrated the importance of these posttranslational modifications for stable lamina formation (Maske et al., 2003). Quantitative live-cell microscopy using GFP-tagged lamin B1 in FLIP experiments in WT, Lmnb1−/−, Rce1−/−, and Icmt−/− cells confirms and extends this biochemical data. Using FLIP analysis, we show that the stability of full-length lamin B1 lacking only carboxymethylation is almost identical to that of fully processed lamin B1 (Fig. 1 c). In contrast, the stability of nonendoproteolyzed lamin B1 in Rce1−/− is lower, reflected by the faster loss of fluorescence in FLIP experiments (Fig. 1 b). These results suggest that lamin B1 is much less able to contribute to a stable lamina when CAAX endoproteolysis does not occur, with consequences for any aspect of nuclear organization or function that depends on a stable lamin B1 scaffold. This effect is specific for the lamin B1 component, as defects in the lamin B1 processing machinery do not affect the stability of lamin C (Fig. S1), which does not undergo the same series of posttranslational modifications.
We hypothesized that cells with defects in lamin B1 expression or in the CAAX processing machinery would exhibit altered gene expression levels, partly as a result of the absence of processed lamin B1 in the lamina. We examined this proposal using microarray analyses to compare gene expression profiles in WT murine fibroblasts with those in cells deficient in full-length lamin B1 (Lmnb1−/−) or components of the processing pathway. Although Rce1−/− cells deficient in the ras-converting enzyme endoprotease have defective CAAX processing and exhibit the gene expression consequences of loss of processed Ras, Lmnb1−/− cells deficient in lamin B1 exhibit normal CAAX processing and normal Ras function. The Icmt−/− cells, which are deficient in the carboxymethyl transferase, occupy an intermediate position because the final carboxymethylation step is important for Ras targeting and function (Svensson et al., 2005), whereas the FLIP analysis (Fig. 1 c) suggests that this step is less critical for proper deployment of lamin B1 to a stable NE. Thus, we reasoned that genes showing coordinate dysregulation in both the lamin B1 and Rce1 knockouts would likely represent genes whose expression is dependent on an NE containing correctly processed lamin B1.
On this basis, we identified 16 genes that showed a 1.5-fold or greater change in expression in the same direction in both Lmnb1−/− and Rce1−/− cells; of these, five also showed altered expression in Icmt−/− cells. In most cases, the change represented up-regulation in the knockout cells (Fig. 2 b). Taking a more stringent twofold cutoff, no genes were either up- or down-regulated in all three cell types, suggesting that few, if any, of the genes examined were affected by a failure of carboxymethylation of lamin B1. In contrast, at this stringent twofold cut-off, seven genes were up-regulated and two were down-regulated in both Lmnb1−/− and Rce1−/− cells (Table I), suggesting that their expression depends on the presence of farnesylated, proteolyzed lamin B1, irrespective of carboxymethylation. When the cutoff for the p-values associated with the fold changes is raised from 0.05 to 0.1, we find that 51 genes are coordinately dysregulated in Lmnb1−/− and Rce1−/− cells, 32 of which are up-regulated and 19 of which are down-regulated.
Conversely, genes with altered expression only in the lamin B1 hypomorph cells are presumably responding to a deficit other than the lack of processed lamin B1 in the NE; otherwise, they would also be dysregulated in Rce1−/− cells that expressed normal lamin B1, but which were unable to process it to the mature form. The discovery of 498 genes with at least 1.5-fold change in expression that are unique to the Lmnb1−/− cells (Fig. 2 a) is consistent with a model in which unprocessed full-length lamin B1 in the nuclear interior plays an important role in regulating gene expression. The mechanism by which nucleoplasmic lamin B1 might play a role in regulated gene expression remains unknown, although observations in other systems provide some clues (Hutchison and Worman, 2004). Intranuclear lamin B1 might associate with the machinery of transcription and RNA processing in a way similar to that observed for lamin A. A possible direct interaction with a polymerase complex is suggested by the discovery that the germline-specific lamin of Xenopus laevis oocytes (Liii) associates with RNA polymerase II, and that Pol II activity is inhibited by dominant-negative lamin mutants (Spann et al., 2002). A more indirect effect via specific transcription factors is suggested by reports of lamin B1 binding to the repressor protein, Oct1, although in this case the presence of elevated levels of Oct1 at the nuclear periphery may implicate processed lamin B1 in the lamina, rather than an unprocessed nucleoplasmic pool (Imai et al., 1997).
We concentrated next on the small group of 16 genes showing a consistent expression dependence on processed lamin B1 (listed in Fig. 2). First, we attempted to cluster them by function using the Gene Ontology database. No clustering into any functional grouping could be detected (Fig. 3 and Table S1). Second, the genes were mapped to identify their chromosomal positions; clustering at specific chromosomal locations was observed (Fig. 4). In particular, a cluster of three genes within ∼4 Mbp coordinately up-regulated in both Lmnb1−/− and Rce1−/− cells was found on chromosome 18, a chromosome that we found to have a strongly peripheral localization in WT cells (Fig. 5). This result is consistent with an envelope-dependent suppression of gene expression that is lost when processed lamin B1 is unavailable.
In contrast to this up-regulated cluster on chromosome 18, none of the genes on chromosome 19 showed such up-regulation. We have shown that chromosomes 18 and 19 have peripheral and central locations, respectively. The presence of the up-regulated cluster on chromosome 18 might therefore be due to disruption of peripheral localization because of the absence of proteolyzed lamin B1. We therefore analyzed the localization patterns of chromosomes 18 and 19 in the three knockout cell types and their WT background cells by two-color FISH (Fig. 5). Because mouse chromosome 19 has one of the highest gene densities in the genome (14.1 genes/Mbp) and gene-dense chromosomes are usually more central, and no coordinate dysregulation of its genes was detected, its localization would not be expected to be altered in the knockout cells; therefore, it serves as an internal control. Mouse chromosome 18 has the lowest gene density of any mouse chromosome (7.5 genes/Mbp), suggesting that a peripheral localization was likely. We found that chromosome 18 is indeed located at the nuclear periphery in three independent WT mouse embryo fibroblast populations. This peripheral localization was preserved in Icmt−/− cells, but in both Lmnb1−/− and Rce1−/− cells (Fig. 5), it became centrally located, like chromosome 19. This suggests that farnesylated and proteolyzed lamin B1 anchors chromosome 18 to the periphery and that this positioning in turn plays a role in the expression of a group of genes on this chromosome. Chromosome 19, on the other hand, showed a central location in the WT cells and in all the knockouts, and none of its genes were coordinately up-regulated in both Lmnb1−/− and Rce1−/− cells. This is the behavior expected of a chromosome that is indifferent to the presence or absence of peripheral processed lamin B1. Mayer et al. (2005) recently reported that the distribution of chromosome territories is cell type specific. Although they observed that the gene-rich mouse chromosome 11 is generally more centrally located, they reported that it also had contact points with the nuclear periphery. Our current observation of two dysregulated genes on chromosome 11 in the absence of processed lamin B1 from the periphery supports this observation because chromosome 11 (121.7 Mbp), which is larger than chromosome 18 (90.7 Mbp), may be anchored at the nuclear periphery at specific points but still exhibit an internal location.
The published gene expression changes in the absence of lamin A or lack of its processing by Zmpste24 (Varela et al., 2005) are distinct from genes that are coordinately dysregulated in Lmnb1−/− and Rce1−/− cells. Only 12 genes are coordinately dysregulated in Lmnb1−/−, Lmna−/−, and Zmpste24−/− cells, with 8 up-regulated and 4 down-regulated. Therefore, the genes that we report to be dysregulated in the absence of processed lamin B1 are specific to that defect and are not due to a general abnormality in the nuclear lamina. Furthermore, the change in chromosome position as a result of the absence of lamin B1 or its processing is specific, as cells lacking the inner nuclear membrane protein emerin do not show altered chromosome organization (Boyle et al., 2001). It has more recently also been reported that cells from patients with emerin and lamin A mutations do not show a significant change in chromosome locations (Meaburn et al., 2005).
Although lamin B1 deficits have not to date been associated with any human disease, such defects are certainly not inconsequential. Lmnb1 mutant mice, from which the Lmnb1−/− embryonic fibroblasts used in the current study were obtained, die shortly after birth, with lung and bone abnormalities (Vergnes et al., 2004). Although the groups of up-regulated genes on chromosome 18 do not seem to have a common function, heparin-binding EGF-like growth factor (Hegfl), plays important roles in development (Arkonac et al., 1998; Iwamoto and Mekada, 2006), which might explain some of the phenotypic aspects of the lamin B1 mutant mice. Furthermore, although no functional clustering is observed when the genes that are coordinately dysregulated in two or more of the transgenic cells used in the current study are considered, genes that are dysregulated only in the absence of full-length lamin B1 do show some functional clustering. A group of down-regulated genes—Bmp4, Cutl1, Fgf18, Fgf2, Fgfr4, Foxa1, Foxa2, Gli3, Hsd11b1, Nfib, Sim2, and Wnt5a—are involved in lung development (Gene Ontology accession no. 0030324), which is consistent with the Lmnb1−/− mice failing to survive after birth as a result of respiratory failure and having reduced numbers of alveoli and thickened mesenchymal tissue.
It was recently reported that overexpression of lamin B1 because of a genomic duplication covering the 5q31 region containing the lamin B1 gene causes autosomal dominant leukodystrophy (ADLD; Padiath et al., 2006). The increased gene dosage results in increased lamin B1 message and protein in brain tissue of affected individuals. The clinical result is a late (adult) onset progressive, symmetrical demyelinating disease that resembles multiple sclerosis except that oligodendroglia are preserved in lesions and there is no astrogliosis. This result suggests that altered lamin B1 expression can be associated with severe human disease and that ADLD should be added to the list of laminopathies.
Alongside lamin B1, lamin A and B2 are major components of the nuclear lamina of most cells. We therefore speculated that a compensatory mechanism involving the up-regulation of their corresponding genes might be taking place to maintain the structure of the nuclear lamina. qRT-PCR revealed about a threefold up-regulation in the Lmna transcript in Lmnb1−/− cells, suggesting that this may indeed be an attempt to compensate for the loss of functional lamin B1. This, however, does not reverse the effect of the lamin B1 defect, as observed by abnormalities in gene expression and chromosome localization. This indicates that there are functions that are quite unique to lamin B1 that cannot be compensated for by an excess of lamin A. In contrast, transcription of Lmnb2 remains almost unchanged, suggesting, perhaps surprisingly, that lamin B2 is subject to entirely independent regulation.
Some investigators have observed an activation of some genes while still at the nuclear periphery but by changing location within that vicinity (Heun et al., 2001; Ralph et al., 2005). Therefore, the nuclear periphery probably contains regions of effective suppression and other regions where additional factors may be contributing to the regulation of gene expression. This might explain why not all the genes that we analyzed on chromosome 18 are up-regulated although the whole chromosome moves away from the periphery in the knockout cells. Such a conclusion would be supported by recent high-resolution studies of a short, 4-Mbp chromosome segment showing that zigzagging of the chromatin can bring discontiguous genes together, while intervening genes are looped out (Shopland et al., 2006). A similar arrangement of the genes within the 4-Mbp cluster on chromosome 18 would permit lamina-dependent regulation of three genes that are not contiguous, although they are clustered in a small region.
In summary, our results represent the first report of a role for a nuclear lamina component, specifically, farnesylated and endoproteolyzed lamin B1, in the positional organization of chromosomes in the interphase nucleus. We demonstrate that processed lamin B1 is required to anchor chromosome 18 at the nuclear periphery and that disruption of this interaction (directly or indirectly) results in dispersion of this chromosome from the nuclear periphery together with an up-regulation of certain genes on the chromosome, consistent with a context-dependent gene-silencing role for the NE on these genes. Alterations to the global organization of chromosomes in the nucleus may lead to the severe consequences observed in laminopathies and may also provide insights into the normal process of aging.
Materials and methods
Cell culture
Mouse embryonic fibroblasts Rce1+/+, Rce1−/− (Kim et al., 1999), Lmnb1+/+, Lmnb1−/− (an insertional mutation lacking 6 exons of the lamin B1 gene encompassing the C-terminal 273 amino acid residues, including the chromatin interaction and CAAX domains; Vergnes et al., 2004), Icmt+/+, and Icmt−/− (Bergo et al., 2001) were cultured in DME supplemented with 10% FCS, l-glutamine, and nonessential amino acids at 37°C in a humidified atmosphere. The GFP-tagged full-length lamin B1 construct (GFP–lamin B1) has been described before (Maske et al., 2003). Cells were grown in 25-cm2 flasks for RNA isolation, on glass-bottomed 35-mm dishes (MatTek) for photobleaching experiments, and on glass coverslips for FISH. For photobleaching experiments, cells were transfected with GFP–lamin B1 using Lipofectamine 2000 (Invitrogen), and experiments were performed 48 h after transfection. For microarray analyses, early passage cells were seeded at 50% confluency, and RNA was extracted when cells reached ∼90% confluency. FTI-treated cells were incubated with 100 μM FPT inhibitor III (Calbiochem) for 48 h.
Photobleaching experiments
FLIP was performed using a confocal laser-scanning system (Radiance 2000 MP; Bio-Rad Laboratories) on an inverted microscope (Eclipse TE300; Nikon) at 37°C using the 488-nm line of a Kr/Ar laser with a 60× 1.4 NA objective. Some cells with distorted nuclear morphology were observed in transgenic knockout populations (Vergnes et al., 2004); only cells with normal nuclear morphology were selected for FLIP analysis. A region of interest (ROI) was photobleached at full laser power while scanning at 5% laser power elsewhere with 1-s intervals between scans over a period of 250 s. Image acquisition was controlled by Lasersharp (Bio-Rad Laboratories), and images were analyzed using MetaMorph (Universal Imaging Corp.). For quantitative analysis, background intensity was subtracted and intensities of a specific ROI outside the photobleached area were measured over time and normalized using intensities of an ROI in a transfected but nonbleached cell.
RNA isolation and data extraction
RNA was extracted from six biological replicates of early passage cultures for each cell type, using TRIZOL reagent (Invitrogen), further purified with RNeasy mini columns (QIAGEN), and quantitated on a Nanodrop spectrophotometer. RNA integrity was confirmed before labeling using Nanochips on an 2100 Bioanalyzer (Agilent Technologies), according to the manufacturers' instructions. Two protocols for microarray labeling and processing were used during these experiments, an indirect amino-allyl dUTP Alexa Fluor labeling system and, subsequently, a 3DNA dendrimer-based system. For the indirect amino-allyl dUTP-based method, RNA (5 μg of total RNA) was labeled and hybridized using the HiSpot RT kit (Genetix) according to the manufacturer's instructions, with the exception that Superscript III was used in place of the supplied reverse transcriptase, followed by labeling with the ARES kit (Invitrogen) with Alexa Fluor 555 and 647 dyes, according to the manufacturer's instructions. For the 3DNA dendrimer-based system, RNA (1 μg of total RNA) was labeled using the 3DNA Array 900 kit (Genisphere), using Superscript III reverse transcriptase (Invitrogen) in the first strand cDNA synthesis. The hybridization and detection steps were performed using a two-step hybridization procedure on a SlideBooster (Advalytix), each with a power setting of 25 and a pulse ratio of 3:7 at 55°C. The first hybridization was for 16 h using hybridization buffer EB, and the second hybridization was for 4 h using SDS buffer. Microarrays containing probes for 6,482 mouse genes were fabricated using the Mouse Known Gene SGC Oligo set, printed in duplicate, designed by Compugen, synthesized by Sigma-Genesys, and printed and supplied by the MRC Human Genome Mapping Project Resource Centre. After the hybridization and washing steps, microarray slides were scanned using the ScanArray ExpressHT system (PerkinElmer), and images for analysis were obtained using autocalibration with 100% laser power, a variable PMT, and a target saturation of 90%. Spot features were identified, poor quality spots were manually flagged, and intensity values were extracted using BlueFuse for microarrays version 2 (BlueGnome). Full details of the slide layout, culture conditions, detailed protocols, and primary extracted data files have been submitted to, and are publicly available in a MIAME-compliant form from, ArrayExpress (www.ebi.ac.uk/arrayexpress/; experiment references E-MEXP-538 DJVaux_MEF_Lmnb1, E-MEXP-539 DJVaux_MEF_Rce1, and E-MEXP-540 DJVaux_MEF_Icmt).
Microarray data analysis
Intensity values, extracted using BlueFuse, were analyzed using BASE (Saal et al., 2002). Only median fold ratio values with P < 0.05 using t test were used for subsequent analysis. Cluster and TreeView (rana.lbl.gov/ EisenSoftware.htm) were used to generate the tree diagrams. For the functional cluster analysis, Gene Ontology IDs associated with genes that are differentially expressed in the three cell types were obtained using Ensembl MartView (www.ensembl.org). Ensembl KaryoView was used for mapping gene positions.
Statistical testing
The ordering of fold change ratios in selected groups of dysregulated genes was compared for different cell types and tested for significance using the Pearson product–moment correlation coefficient. To assess the significance of groups of n genes identified as the intersection of independent experimental datasets, we drew n genes at random from each dataset and counted the number of individual genes that appeared in both lists. This sampling was repeated 100,000 times and enabled us to assign a probability for the observed intersection subsets occurring by chance. The significance of the distribution of selected groups of genes was tested using a bootstrap method in which 16 genes were drawn at random from the list of genes meeting specific criteria and mapped onto the genome. The sampling was repeated 10,000 times, and the distribution of each of these 16-mer gene sets was assessed using predetermined test criteria. To assess apparent clustering of genes, this approach was used with the test criteria of three genes (out of the 16 selected in each trial) mapping within a 5-Mbp region as the definition of a cluster.
RT-PCR validation
Quantitative PCR was performed using a Rotor-Gene 3000 (Corbett Research) using the Platinum two-step qRT-PCR kit with SYBR green according to the manufacturer's instructions (Invitrogen). Primers were designed using the OligoPerfect designer (Invitrogen) and tested for single product generation in control end-stage PCR before qRT-PCR. The housekeeping gene β-actin (Actb) was used as an internal standard for the qPCR verification of Cryab, Hegfl, Mgst1, Crabp1, Lmna, and Lmnb2. Relative gene expression values were determined using the 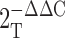 method (Livak and Schmittgen, 2001).
method (Livak and Schmittgen, 2001).
Two-color FISH and image analysis
Cells were fixed in 3:1 methanol acetic acid as described previously (Bickmore and Carothers, 1995; Croft et al., 1999; Boyle et al., 2001) with the modification that the cells were grown and fixed as adherent monolayers rather than in suspension. Denaturation was performed for 4 min at 65°C in 70% deionized formamide/2× SSC. Mouse chromosome 18 and 19 paints labeled with Cy3 and FITC, respectively, were denatured according to the manufacturer's instructions (Cambio) followed by hybridization to coverslips for 16 h at 37°C in a humidified box. Coverslips were washed three times for 5 min at 45°C in 50% deionized formamide/2× SSC, washed two times for 5 min at 50°C in 1× SSC, and mounted in Mowiol supplemented with DAPI. Cells stained with Con A–Alexa Fluor 633 (Con A 633; Invitrogen) were incubated with 100 μg/ml of the Con A conjugate for 1 h at room temperature after the 1× SSC washing step and washed three times for 5 min in PBS before mounting. Cells were examined using a fluorescence microscope (Axioplan 2e; Carl Zeiss MicroImaging, Inc.) or a Radiance 2000 MP confocal laser-scanning microscope. The Con A and DAPI stains were used to identify cells with distorted nuclear morphology, and these cells were excluded from the analysis. Images were viewed in MetaMorph or ImageJ 1.33u. For chromosome position analysis, images were successively partitioned into five shells. For each partition, a length, r, was determined such that all pixels with a distance less than r away from the immediate outer partition (or the boundary if they constitute the outermost partition) are grouped as one partition, and such that the resulting partition has ∼20% of the overall area of the nucleus. The intensity associated with each shell is the sum of the “bright” pixels within that shell. Because of the discrete nature of the image, shells may not have exactly 20% of the total area; hence, the intensity corresponding to each shell is renormalized by the actual area of the shell. Unlike some conventional analyses, this method does not depend on prior definition of a centroid for the cell.
Online supplemental material
Fig. S1 shows FLIP of YFP–lamin C expressed in WT, Lmnb1−/−, Rce1−/−, and Icmt−/− cells. The figure shows that there is no significant difference between lamin C dynamics in the three different cell types. This indicates that the loss of lamin B1 or any of its processing steps does not affect the stability of lamin C interactions or the lamin C lamina. Table S1 provides a summary of the gene expression data (expressed as fold ratios) for Lmnb1−/−, Rce1−/−, and Icmt−/− cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200607054/DC1.
Acknowledgments
The authors are grateful to Peter Cook and Nick Proudfoot for critical comments on the manuscript. We wish to acknowledge the BioImaging Facility and the Computational Biology Research Group, Medical Sciences Division, Oxford, for use of their services in this project.
This work was funded by grants from the EP Abraham Trust and Tim and Kit Kemp. A. Malhas is a Kemp Postdoctoral Fellow of Lincoln College, Oxford.
The authors declare no competing commercial interests.
R. Sanders's current address is Department of Clinical Biochemistry, University of Cambridge, Cambridge CB2 2QR, England, UK.
Abbreviations used in this paper: FLIP, fluorescence loss in photobleaching; FTI, farnesyl transferase inhibitor; Mbp, Mega bp; MIAME, minimum information for annotation of microarray experiments; NE, nuclear envelope; NPC, nuclear pore complex; qRT-PCR, quantitative real-time PCR; ROI, region of interest; WT, wild-type.
References
- Arkonac, B.M., L.C. Foster, N.E. Sibinga, C. Patterson, K. Lai, J.C. Tsai, M.E. Lee, M.A. Perrella, and E. Haber. 1998. Vascular endothelial growth factor induces heparin-binding epidermal growth factor-like growth factor in vascular endothelial cells. J. Biol. Chem. 273:4400–4405. [DOI] [PubMed] [Google Scholar]
- Bergo, M.O., G.K. Leung, P. Ambroziak, J.C. Otto, P.J. Casey, A.Q. Gomes, M.C. Seabra, and S.G. Young. 2001. Isoprenylcysteine carboxyl methyltransferase deficiency in mice. J. Biol. Chem. 276:5841–5845. [DOI] [PubMed] [Google Scholar]
- Bickmore, W.A., and A.D. Carothers. 1995. Factors affecting the timing and imprinting of replication on a mammalian chromosome. J. Cell Sci. 108:2801–2809. [DOI] [PubMed] [Google Scholar]
- Boyle, S., S. Gilchrist, J.M. Bridger, N.L. Mahy, J.A. Ellis, and W.A. Bickmore. 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10:211–219. [DOI] [PubMed] [Google Scholar]
- Branco, M.R., and A. Pombo. 2006. Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol. 4:e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broers, J.L., C.J. Hutchison, and F.C. Ramaekers. 2004. Laminopathies. J. Pathol. 204:478–488. [DOI] [PubMed] [Google Scholar]
- Cabal, G.G., A. Genovesio, S. Rodriguez-Navarro, C. Zimmer, O. Gadal, A. Lesne, H. Buc, F. Feuerbach-Fournier, J.C. Olivo-Marin, E.C. Hurt, and U. Nehrbass. 2006. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 441:770–773. [DOI] [PubMed] [Google Scholar]
- Casolari, J.M., C.R. Brown, S. Komili, J. West, H. Hieronymus, and P.A. Silver. 2004. Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell. 117:427–439. [DOI] [PubMed] [Google Scholar]
- Chuang, C.H., A.E. Carpenter, B. Fuchsova, T. Johnson, P. de Lanerolle, and A.S. Belmont. 2006. Long-range directional movement of an interphase chromosome site. Curr. Biol. 16:825–831. [DOI] [PubMed] [Google Scholar]
- Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301. [DOI] [PubMed] [Google Scholar]
- Croft, J.A., J.M. Bridger, S. Boyle, P. Perry, P. Teague, and W.A. Bickmore. 1999. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145:1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann, P., H. Bornfleth, D. Zink, T. Cremer, and C. Cremer. 2001. Morphology and dynamics of chromosome territories in living cells. Biochim. Biophys. Acta. 1551:M29–M39. [DOI] [PubMed] [Google Scholar]
- Eriksson, M., W.T. Brown, L.B. Gordon, M.W. Glynn, J. Singer, L. Scott, M.R. Erdos, C.M. Robbins, T.Y. Moses, P. Berglund, et al. 2003. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 423:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, L.G., J.K. Ng, J. Lammerding, T.A. Vickers, M. Meta, N. Cote, B. Gavino, X. Qiao, S.Y. Chang, S.R. Young, et al. 2006. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest. 116:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, N., S. Gilchrist, and W.A. Bickmore. 2005. Chromatin organization in the mammalian nucleus. Int. Rev. Cytol. 242:283–336. [DOI] [PubMed] [Google Scholar]
- Glynn, M.W., and T.W. Glover. 2005. Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum. Mol. Genet. 14:2959–2969. [DOI] [PubMed] [Google Scholar]
- Goldberg, M., A. Harel, M. Brandeis, T. Rechsteiner, T.J. Richmond, A.M. Weiss, and Y. Gruenbaum. 1999. The tail domain of lamin Dm0 binds histones H2A and H2B. Proc. Natl. Acad. Sci. USA. 96:2852–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, R.D., Y. Gruenbaum, R.D. Moir, D.K. Shumaker, and T.P. Spann. 2002. Nuclear lamins: building blocks of nuclear architecture. Genes Dev. 16:533–547. [DOI] [PubMed] [Google Scholar]
- Gruenbaum, Y., A. Margalit, R.D. Goldman, D.K. Shumaker, and K.L. Wilson. 2005. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 6:21–31. [DOI] [PubMed] [Google Scholar]
- Heun, P., T. Laroche, K. Shimada, P. Furrer, and S.M. Gasser. 2001. Chromosome dynamics in the yeast interphase nucleus. Science. 294:2181–2186. [DOI] [PubMed] [Google Scholar]
- Hewitt, S.L., F.A. High, S.L. Reiner, A.G. Fisher, and M. Merkenschlager. 2004. Nuclear repositioning marks the selective exclusion of lineage-inappropriate transcription factor loci during T helper cell differentiation. Eur. J. Immunol. 34:3604–3613. [DOI] [PubMed] [Google Scholar]
- Hutchison, C.J., and H.J. Worman. 2004. A-type lamins: guardians of the soma? Nat. Cell Biol. 6:1062–1067. [DOI] [PubMed] [Google Scholar]
- Imai, S., S. Nishibayashi, K. Takao, M. Tomifuji, T. Fujino, M. Hasegawa, and T. Takano. 1997. Dissociation of Oct1 from the nuclear peripheral structures induces the cellular aging associated collagenase gene expression. Mol. Biol. Cell. 8:2407–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto, R., and E. Mekada. 2006. ErbB and HB-EGF signaling in heart development and funciton. Cell Struct. Funct. 31:1–14. [DOI] [PubMed] [Google Scholar]
- Kim, E., P. Ambroziak, J.C. Otto, B. Taylor, M. Ashby, K. Shannon, P.J. Casey, and S.G. Young. 1999. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J. Biol. Chem. 274:8383–8390. [DOI] [PubMed] [Google Scholar]
- Kitten, G.T., and E.A. Nigg. 1991. The CaaX motif is required for isoprenylation, carboxyl methylation, and nuclear membrane association of lamin B2. J. Cell Biol. 113:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
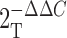 method. Methods.
25:402–408.
[DOI] [PubMed] [Google Scholar]
method. Methods.
25:402–408.
[DOI] [PubMed] [Google Scholar] - Mallampalli, M.P., G. Huyer, P. Bendale, M.H. Gelb, and S. Michaelis. 2005. Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 102:14416–14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders, E.M., A.E. Visser, A. Koppen, W.C. de Leeuw, R. van Liere, G.J. Brakenhoff, and R. van Driel. 2003. Four-dimensional imaging of chromatin dynamics during the assembly of the interphase nucleus. Chromosome Res. 11:537–547. [DOI] [PubMed] [Google Scholar]
- Maske, C.P., M.S. Hollinshead, N.C. Higbee, M.O. Bergo, S.G. Young, and D.J. Vaux. 2003. A carboxyl-terminal interaction of lamin B1 is dependent on the CAAX endoprotease Rce1 and carboxymethylation. J. Cell Biol. 162:1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masny, P.S., U. Bengtsson, S. Chung, J.H. Martin, B. van Engelen, S.M. van der Maarel, and S.T. Winokur. 2004. Localization of 4q35.2 to the nuclear periphery: is FSHD a nuclear envelope disease? Hum. Mol. Genet. 13:1857–1871. [DOI] [PubMed] [Google Scholar]
- Mayer, R., A. Brero, J. von Hase, T. Schroeder, T. Cremer, and S. Dietzel. 2005. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaburn, K.J., N. Levy, D. Toniolo, and J.M. Bridger. 2005. Chromosome positioning is largely unaffected in lymphoblastoid cell lines containing emerin or A-type lamin mutations. Biochem. Soc. Trans. 33:1438–1440. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science. 291:843–847. [DOI] [PubMed] [Google Scholar]
- Mounkes, L., S. Kozlov, B. Burke, and C.L. Stewart. 2003. The laminopathies: nuclear structure meets disease. Curr. Opin. Genet. Dev. 13:223–230. [DOI] [PubMed] [Google Scholar]
- Padiath, Q.S., S. Kazumasa, R. Schiffman, H. Asahara, T. Yamada, A. Koeppen, K. Hogan, L.J. Ptacek, and Y.-H. Fu. 2006. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 38:1114–1123. [DOI] [PubMed] [Google Scholar]
- Parada, L.A., P.G. McQueen, P.J. Munson, and T. Misteli. 2002. Conservation of relative chromosome positioning in normal and cancer cells. Curr. Biol. 12:1692–1697. [DOI] [PubMed] [Google Scholar]
- Parada, L.A., P.G. McQueen, and T. Misteli. 2004. Tissue-specific spatial organization of genomes. Genome Biol. 5:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph, S.A., C. Scheidig-Benatar, and A. Scherf. 2005. Antigenic variation in Plasmodium falciparum is associated with movement of var loci between subnuclear locations. Proc. Natl. Acad. Sci. USA. 102:5414–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal, L.H., C. Troein, J. Vallon-Christersson, S. Gruvberger, A. Borg, and C. Peterson. 2002. BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol. 3:software0003.1–software0003.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland, L.S., C.R. Lynch, K.A. Peterson, K. Thornton, N. Kepper, J.V. Hase, S. Stein, S. Vincent, K.R. Molloy, G. Kreth, et al. 2006. Folding and organization of a contiguous chromosome region according to the gene distribution pattern in primary genomic sequence. J. Cell Biol. 174:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spann, T.P., A.E. Goldman, C. Wang, S. Huang, and R.D. Goldman. 2002. Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J. Cell Biol. 156:603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson, A.W., P.J. Casey, S.G. Young, and M.O. Bergo. 2005. Genetic and pharmacologic analyses of the role of Icmt in ras membrane association and function. Methods Enzymol. 407:144–159. [DOI] [PubMed] [Google Scholar]
- Taddei, A., G. Van Houwe, F. Hediger, V. Kalck, F. Cubizolles, H. Schober, and S.M. Gasser. 2006. Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature. 441:774–778. [DOI] [PubMed] [Google Scholar]
- Tanabe, H., K. Kupper, T. Ishida, M. Neusser, and H. Mizusawa. 2005. Inter- and intra-specific gene-density-correlated radial chromosome territory arrangements are conserved in Old World monkeys. Cytogenet. Genome Res. 108:255–261. [DOI] [PubMed] [Google Scholar]
- Taniura, H., C. Glass, and L. Gerace. 1995. A chromatin binding site in the tail domain of nuclear lamins that interacts with core histones. J. Cell Biol. 131:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela, I., J. Cadinanos, A.M. Pendas, A. Gutierrez-Fernandez, A.R. Folgueras, L.M. Sanchez, Z. Zhou, F.J. Rodriguez, C.L. Stewart, J.A. Vega, et al. 2005. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 437:564–568. [DOI] [PubMed] [Google Scholar]
- Vergnes, L., M. Peterfy, M.O. Bergo, S.G. Young, and K. Reue. 2004. Lamin B1 is required for mouse development and nuclear integrity. Proc. Natl. Acad. Sci. USA. 101:10428–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, J., L. Schermelleh, M. Cremer, S. Tashiro, and T. Cremer. 2003. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman, H.J., and J.C. Courvalin. 2005. Nuclear envelope, nuclear lamina, and inherited disease. Int. Rev. Cytol. 246:231–279. [DOI] [PubMed] [Google Scholar]
- Yang, S.H., M.O. Bergo, J.I. Toth, X. Qiao, Y. Hu, S. Sandoval, M. Meta, P. Bendale, M.H. Gelb, S.G. Young, and L.G. Fong. 2005. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA. 102:10291–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink, D., T. Cremer, R. Saffrich, R. Fischer, M.F. Trendelenburg, W. Ansorge, and E.H. Stelzer. 1998. Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 102:241–251. [DOI] [PubMed] [Google Scholar]
- Zink, D., M.D. Amaral, A. Englmann, S. Lang, L.A. Clarke, C. Rudolph, F. Alt, K. Luther, C. Braz, N. Sadoni, et al. 2004. Transcription-dependent spatial arrangements of CFTR and adjacent genes in human cell nuclei. J. Cell Biol. 166:815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]