

Abstract
Epithelial–mesenchymal transition (EMT) occurs during embryogenesis, carcinoma invasiveness, and metastasis and can be elicited by transforming growth factor-β (TGF-β) signaling via intracellular Smad transducers. The molecular mechanisms that control the onset of EMT remain largely unexplored. Transcriptomic analysis revealed that the high mobility group A2 (HMGA2) gene is induced by the Smad pathway during EMT. Endogenous HMGA2 mediates EMT by TGF-β, whereas ectopic HMGA2 causes irreversible EMT characterized by severe E-cadherin suppression. HMGA2 provides transcriptional input for the expression control of four known regulators of EMT, the zinc-finger proteins Snail and Slug, the basic helix-loop-helix protein Twist, and inhibitor of differentiation 2. We delineate a pathway that links TGF-β signaling to the control of epithelial differentiation via HMGA2 and a cohort of major regulators of tumor invasiveness and metastasis. This network of signaling/transcription factors that work sequentially to establish EMT suggests that combinatorial detection of these proteins could serve as a new tool for EMT analysis in cancer patients.
Introduction
Epithelial–mesenchymal transition (EMT) converts polarized epithelial cells to motile mesenchymal cells (Hay, 2005). EMT operates during embryonic cell layer movements and tumor cell invasiveness (Huber et al., 2005). During EMT, the epithelial proteins E-cadherin and zonula occludens 1 (ZO-1) are down-regulated, and the mesenchymal proteins vimentin, α-smooth muscle actin, and fibronectin are up-regulated.
Receptor tyrosine kinase, Wnt, Notch, and TGF-β pathways trigger EMT (Huber et al., 2005). TGF-β binds to receptor serine/threonine kinases, which activate intracellular Smad and other signaling pathways that regulate gene expression (Feng and Derynck, 2005). TGF-β inhibits epithelial cell growth, acting as a tumor suppressor, but it also promotes carcinoma progression and metastasis (Roberts and Wakefield, 2003). The tumor-promoting effects of TGF-β are based on its ability to induce (a) EMT, matrix invasiveness, and blood vessel intravasation by carcinoma cells (Huber et al., 2005); (b) cytostatic effects on surveilling immune cells (Li et al., 2006); and (c) proangiogenic effects (Bhowmick and Moses, 2005). TGF-β elicits EMT and in vivo metastasis via Smads and complementary non-Smad effectors, such as Rho GTPases and p38 MAPK (Moustakas and Heldin, 2005). TGF-β represses inhibitor of differentiation (Id) 2 and 3 expression and induces expression of the Notch ligand Jagged-1, which are critical events during EMT (Kondo et al., 2004; Kowanetz et al., 2004; Zavadil et al., 2004).
Here, we describe the role of high mobility group (HMG) A2, also known as HMGI-C, as an effector of TGF-β that causes EMT. HMGA2 and -1 constitute a family of nuclear factors that bind AT-rich DNA sequences (Reeves, 2001; Sgarra et al., 2004). HMGA factors contribute to transcriptional regulation by organizing nucleoprotein complexes such as enhanceosomes (Merika and Thanos, 2001). HMGA2 is expressed during embryogenesis and becomes silent in adult tissues (Sgarra et al., 2004). However, HMGA2 is abundantly expressed by transformed cells or tumors of mesenchymal and epithelial origin (for reviews see Reeves, 2001; Sgarra et al., 2004). In contrast, depletion of HMGA2 by antisense cDNA in thyroid cells eliminates their transformation by myeloproliferative and Kirsten murine sarcoma viruses (Berlingieri et al., 1995). Here, we show that HMGA2 regulates the transcription factors Snail, Slug, Twist, and Id2, thus linking TGF-β signaling to regulators of tumor invasiveness and metastasis.
Results and discussion
TGF-β/Smad signaling induces Hmga2 transcription
Transcriptomic analysis of TGF-β–induced EMT in mammary epithelial NMuMG cells identified Hmga2 as a prominent TGF-β target (Valcourt et al., 2005). Hmga2 mRNA increased after 2 and 8 h and returned to basal levels after 36 h of TGF-β stimulation (Fig. 1 A). In contrast, Hmga1 mRNA was not regulated and dropped significantly upon cell confluence at 36 h (Fig. 1 A). HMGA2 protein increased after 8 h and peaked at 12 h of TGF-β stimulation (Fig. 1 B). A TGF-β type I receptor kinase inhibitor (LY580276; Peng et al., 2005) did not affect basal HMGA2 levels, demonstrating the absence of autocrine TGF-β (Fig. 1 B). Hmga2 mRNA induction by TGF-β was not impaired by the protein synthesis inhibitor cycloheximide while it was blocked by the RNA polymerase inhibitor II actinomycin D (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200512110/DC1). A constitutively active form of the TGF-β type I receptor increased Hmga2 expression more efficiently than TGF-β itself, whereas a kinase-dead mutant of this receptor inhibited it (Fig. S1 B). When the constitutively active type I receptor was expressed at higher levels, it often failed to induce Hmga2 at higher levels than TGF-β (Valcourt et al., 2005). This reflects mechanisms of pathway desensitization, as TGF-β signaling is controlled in a timed fashion by activation and inactivation of receptor and Smads. The results suggest that Hmga2 is a direct TGF-β target.
Figure 1.

TGF-β/Smad signaling induces Hmga2 transcription. (A) RT-PCR analysis of Hmga2 and Hmga1 expression in NMuMG cells stimulated with 5 ng/ml TGF-β for the indicated times. (B) Immunoblot analysis of endogenous Hmga2 in NMuMG cells treated with vehicle (0), TGF-β type I receptor inhibitor LY580276 (2.5 μM; LY) for 4 h, or stimulated with TGF-β for the indicated periods of time. Histone H1 serves as a loading control. Molecular mass markers are in bp (A) and kD (B). (C) Hmga2 promoter assays of the indicated deletion constructs in HepG2 cells stimulated (gray bars) or not (white bars) with 5 ng/ml TGF-β for 24 h. The black box in the Hmga2 promoter corresponds to a TCC repeat-rich sequence. (D) Quantitative RT-PCR analysis of Hmga2 expression in NMuMG clones expressing dominant-negative Smad2 (S2 SA) or empty vector (mock) induced or not with 10 μM CdCl2 for 24 h, before stimulation with 5 ng/ml TGF-β for 4 h. (E) Promoter assays of the Hmga2 BaP construct in HepG2 cells transfected with Smad2 SA and stimulated (gray bars) or not (white bars) with 5 ng/ml TGF-β for 24 h. (F and G) Quantitative RT-PCR analysis of Hmga2 expression in NMuMG (F) and MDA-MB-231 (G) clones expressing short hairpin vectors (sh-Smad) directed against Smad2, -3, and -4, or the empty vector and treated with 5 ng/ml TGF-β for 6 h. Asterisks indicate statistically significant gene expression or promoter activity differences between TGF-β–stimulated and nonstimulated conditions (P < 0.05). (H) ChIP assays in NMuMG cells treated or not with 5 ng/ml TGF-β for 2 h using a Smad4 antibody or a preimmune serum (Ctrl) and amplification of Hmga2 promoter fragments.
Mouse Hmga2 promoter analysis showed that basal promoter activity varied according to the deletion construct used, and TGF-β stimulation led to a 2.5–3-fold induction (Fig. 1 C). Basal promoter activity and induction by TGF-β were lost when the proximal region containing TCC repeats was deleted. Sequence inspection of 4 kbp upstream from the transcription initiation site showed few noncanonical Smad binding elements between −700 and −100 bp (unpublished data). We now examine the role of these elements on Hmga2 transcriptional induction by TGF-β.
Hmga2 mRNA induction and promoter activation by TGF-β was blocked in cells expressing dominant-negative Smad2 (Smad2 SA; Fig. 1, D and E); Smad2 SA cannot be phosphorylated by the TGF-β type I receptor and blocks TGF-β– induced EMT (Valcourt et al., 2005). Knockdown of Smad2 by 80% or Smad3 by 65% after RNAi had no effect on Hmga2 induction by TGF-β or on the EMT response (Fig. 1 F and Fig. S1, C and D). However, knockdown of the common partner of Smad2 and -3, Smad4, by 95% effectively blocked Hmga2 induction by TGF-β and the EMT response (Fig. 1 F and Fig. S1, C and D; Deckers et al., 2006). The lack of effect by knockdown of Smad2 or -3 may indicate that the protein depletion achieved was insufficient. Alternatively, both Smad2 and -3 may be involved in EMT, as we previously proposed (Valcourt et al., 2005), and for effective block of EMT, both Smad2 and -3 need to be depleted. Experiments are under way to test this possibility. In another cell line, metastatic breast cancer MDA-MB-231 cells, TGF-β weakly induced HMGA2 expression, and knockdown of Smad3 or -4 blocked this response, whereas knockdown of Smad2 did not (Fig. 1 G and Fig. S1, E and F). Based on these data, it appears that single Smad3 or -4 knockdown is sufficient in blocking TGF-β–induced HMGA2 expression (Fig. 1 G and Fig. S1 F). A more robust knockdown of Smad2 is needed to reach final conclusions about the role of this Smad isoform in HMGA2 regulation and EMT.
Finally, immunoprecipitation of chromatin bound Smad4 from NMuMG cells confirmed a TGF-β–inducible association of Smad4 in the proximal (−195/+5) and upstream (−495/+245) but not in the distal (−3420/−3320) promoter region of the Hmga2 gene (Fig. 1 H). The −500 to +5 Hmga2 promoter region where Smad4 binds overlapped with putative Smad binding elements. The results establish that Smad signaling is involved in Hmga2 induction by TGF-β, with Smad4 clearly being implicated. However, we cannot conclude whether Smad4 cooperates with Smad2, Smad3, or both during Hmga2 regulation.
Ectopic HMGA2 weakly inhibits epithelial cell proliferation
To address the functional role of HMGA2, we established stable NMuMG clones inducibly expressing human HMGA2. Ectopic HMGA2 was expressed in the absence of an inducer, and induction increased its expression further (Fig. 2 A). Ectopic HMGA2 localized in the nucleus as expected, and its localization was not affected by TGF-β (Fig. 2 B). In the absence of TGF-β, HMGA2 clones grew slower than mock cells (Fig. 2 C). TGF-β inhibited growth of mock and HMGA2-expressing cells (Fig. 2 D). Thus, TGF-β induces growth arrest despite ectopic HMGA2 expression.
Figure 2.

HMGA2 inhibits cell proliferation. (A) Analysis of ectopic HMGA2 expression in NMuMG clones (mock, 5, and 13) transfected with empty or HMGA2 vector. Cells were stimulated with 5 ng/ml TGF-β for 36 h, 24 h after induction with 10 μM CdCl2. Immunoblots were incubated with anti-HA antibody. β-Tubulin is loading control. Molecular mass markers are in kD. (B) Immunostaining with anti-HA antibody of mock NMuMG and HMGA2 clone 5 stimulated with 5 ng/ml TGF-β for 36 h. Bar, 10 μm. (C) Cell proliferation assays with mock (diamonds) and HMGA2 clones (5, squares; 13, triangles). (D) Cell proliferation assays with mock and HMGA2 clones 5 and 13 stimulated (gray bars) or not (white bars) with TGF-β for 4 d.
HMGA2 mediates EMT in response to TGF-β
Mock NMuMG clones treated with an inducer displayed characteristic polarized epithelial morphology (Fig. 3 A). TGF-β caused EMT, as mock cells acquired elongated, fibroblast-like morphology. HMGA2 clones were constitutively elongated and lost cell–cell contacts, suggesting induction of EMT, which was enhanced further by TGF-β (Fig. 3 A). EMT in HMGA2 clones was confirmed by visualizing actin cytoskeleton rearrangements and the loss of ZO-1 and E-cadherin from cell junctions (Fig. 3 B) and by measuring the loss of expression of E-cadherin and Mucin-1 mRNA (Fig. 3 C). Moreover, mRNAs of the mesenchymal markers PAI-1, Timp-3, and Fibronectin-1 were constitutively expressed, and Vimentin was increased to a lesser extent. Immunoblot analysis confirmed E-cadherin protein down-regulation and enhanced expression of mesenchymal N-cadherin in HMGA2 clones (Fig. 3 D). These experiments demonstrate that ectopic HMGA2 causes EMT.
Figure 3.

HMGA2 induces EMT. (A) Phase-contrast microscopy of mock and HMGA2 clones. HMGA2 induction with 10 μM CdCl2 was followed by vehicle (control) or 5 ng/ml TGF-β stimulation for 36 h. (B) Visualization of actin cytoskeleton and the epithelial markers ZO-1 and E-cadherin by immunostaining of mock and HMGA2 clones treated with vehicle (control) or 5 ng/ml TGF-β for 36 h. Bars, 10 μm. (C) RT-PCR analysis of EMT markers. HMGA2 clones were treated as in A. Parental NMuMG cells were treated with 5 ng/ml TGF-β for 36 h. (D) Immunoblot analysis of E- and N-cadherin in cells treated as in A. Molecular mass markers are in bp (C) and kD (D).
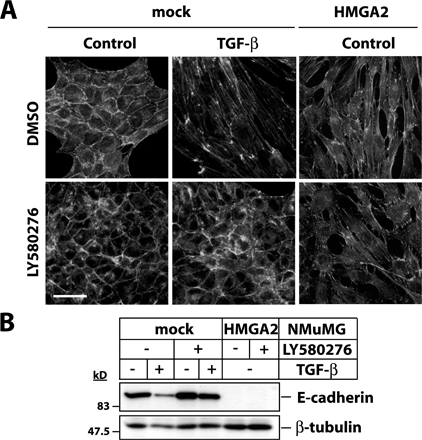
The fact that ectopic HMGA2 mimicked the TGF-β response (Fig. 3, C and D) raises the question of whether HMGA2 activates autocrine TGF-β, leading to EMT. In mock NMuMG cells, the LY580276 inhibitor blocked TGF-β–mediated EMT, as cells kept a cortical actin distribution and did not down-regulate E-cadherin (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200512110/DC1). However, in HMGA2 clones, LY580276 had no effect on the elongated morphology, actin stress fiber network, or lack of E-cadherin. Similar results were obtained with a TGF-β–neutralizing antibody added to the medium of HMGA2 clones for several days (unpublished data). These experiments demonstrate that the profound effects HMGA2 shows on EMT cannot be accounted for by the induction of autocrine TGF-β that signals in a constitutive manner.
Transfection of NMuMG cells with Hmga2 siRNA resulted in an ∼70% decrease in basal and TGF-β–induced Hmga2 mRNA expression (Fig. 4 A). An even stronger reduction was seen of the endogenous HMGA2 protein level (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200512110/DC1). Weak nuclear HMGA2 was seen in control cells; TGF-β dramatically increased the nuclear HMGA2 levels, as the cells became elongated and fibroblast like (Fig. S3). Upon HMGA2 depletion, its nuclear staining was barely detectable. Immunoblot analysis of endogenous HMGA2 protein upon chromatin extraction (Fig. 1 B) was not efficient enough in these transfected cells to quantitatively monitor the degree of protein depletion upon RNAi (unpublished data). We reproducibly observed that the mammary epithelial cells enlarged their diameter by roughly 1.8–2.2-fold when HMGA2 was depleted (Fig. S3). This effect was specific for the Hmga2 siRNA, as we did not observe size changes by control (siLuc) or a panel of siRNAs that target unrelated genes in this cell line (Fig. S3 and unpublished data).
Figure 4.

HMGA2 mediates EMT by TGF-β. (A) Quantitative RT-PCR analysis of Hmga2 expression in NMuMG cells transfected with control (siLuc) or specific siRNA against Hmga2 (siHmga2) and treated with vehicle (white bars) or 5 ng/ml TGF-β (gray bars) for 12 h. (B) Phase-contrast images. (C) Indirect immunofluorescence of ZO-1 and E-cadherin. Bars, 10 μm. (D) Immunoblot analysis of N-cadherin in NMuMG cells transfected as in A and treated with vehicle (control) or 5 ng/ml TGF-β for 36 h. β-Tubulin is loading control. Molecular mass markers are in kD. (E) Quantitative RT-PCR analysis of Fibronectin-1 (FN1) expression in cells treated as in A. Asterisks indicate statistically significant gene expression differences compared with the ground condition (P < 0.05).
TGF-β induced EMT in NMuMG cells transfected with control siRNA. In contrast, cells transfected with Hmga2 siRNA do not undergo EMT. Indeed, these cells maintained polarized morphology, ZO-1, and E-cadherin at their junctions and decreased the TGF-β–inducible levels of N-cadherin and Fibronectin-1 (Fig. 4, B–E; and Fig. S3). Although Hmga2 knockdown restored to a large extent epithelial tight and adherence junctions, we observed only a weak block of total ZO-1 and E-cadherin protein down-regulation by TGF-β (unpublished data). We conclude that endogenous HMGA2 is required for TGF-β–induced EMT.
HMGA2 regulates expression of key regulators of EMT
The zinc-finger transcription factors Snail, Slug, δEF-1/ZEB-1, and SIP-1/ZEB-2 or the basic helix-loop-helix factor Twist repress E-cadherin expression during embryonic EMT (Peinado et al., 2004) and promote tumor cell metastasis or cancer recurrence after therapy (Yang et al., 2004; Moody et al., 2005). The extreme down-regulation of E-cadherin expression in HMGA2 clones (Fig. 3, C and D) prompted us to analyze some of these transcriptional repressors. In parental NMuMG cells, Snail and Slug and, to a lesser extent, Twist mRNA were induced by TGF-β (Fig. 5, A and C). Repressor expression was dramatically up-regulated in HMGA2 clones even in the absence of TGF-β. Similar to regulation of endogenous Snail and Twist mRNA, TGF-β induced Snail and Twist promoter activity (Fig. 5 B). Notably, cotransfection of HMGA2 enhanced Snail and Twist promoter activity to significantly higher levels than TGF-β stimulation alone. As specificity control, the Smad3/Smad4-dependent promoter reporter CAGA12-Luc was not regulated by HMGA2 (unpublished data). Upon RNAi-mediated knockdown of Hmga2, endogenous Snail mRNA induction by TGF-β was reduced by ∼50% (Fig. 5 D). This explains why total E-cadherin and ZO-1 protein levels were still repressible by TGF-β after Hmga2 knockdown (unpublished data). We conclude that partial depletion of endogenous Hmga2 by RNAi is sufficient to restore epithelial differentiation in NMuMG cells and establishment of cell–cell junctions; however, it is not sufficient to block strongly enough Snail induction by TGF-β.
Figure 5.

HMGA2 regulates expression of key regulators of EMT. (A) Quantitative RT-PCR analysis of Snail and Twist expression in parental or HMGA2 clones stimulated (gray bars) or not (white bars) with 5 ng/ml TGF-β for 36 h. (B) Luciferase reporter assays of Snail and Twist promoter constructs in HepG2 cells transfected with mock (−) or HA-hHMGA2 (+) vector and treated with 5 ng/ml TGF-β for 24 h. (C) Quantitative RT-PCR analysis of Slug expression under conditions as in A. (D) Quantitative RT-PCR analysis of Snail expression in NMuMG cells transfected with control (siLuc) or Hmga2 (siHmga2) siRNA and treated with vehicle (white bars) or 5 ng/ml TGF-β (gray bars) for 12 h. Asterisks indicate statistically significant gene expression or promoter activity differences compared with the ground condition (P < 0.05). Expression pattern of Id2 using RT-PCR (E) or protein (F) analysis in mock and HMGA2 clones induced with 10 μM CdCl2 24 h before stimulation with 5 ng/ml TGF-β for 36 h. Molecular mass markers are in bp (E) and kD (F). (G) Diagram of the role of HMGA2 in TGF-β–induced EMT.
TGF-β down-regulates Id2 to induce EMT (Kondo et al., 2004; Kowanetz et al., 2004). Id2 mRNA and protein expression were down-regulated in HMGA2 clones compared with mock cells, and TGF-β further repressed Id2 levels (Fig. 5, E and F). The results demonstrate that HMGA2 regulates Snail, Slug, Twist, and Id2, all key players of EMT. TGF-β induces Snail expression either via Smad3 or via MAPK signaling (Peinado et al., 2003; Saika et al., 2004). Our results add HMGA2 as a novel regulator of Snail expression downstream of Smads. Whether HMGA2 cooperates with Smad3 or MAPK signals to induce Snail is being explored. Twist is another gene target of HMGA2 that is weakly induced by TGF-β. We conclude that TGF-β, via HMGA2, primarily affects the Snail pathway and, to a lesser extent, the Twist pathway.
Concluding remarks
We describe a new target of TGF-β signaling, the nuclear factor HMGA2, and a new transcriptional circuitry that mediates EMT by TGF-β (Fig. 5 G). HMGA2 links TGF-β signaling to major factors of tumor invasiveness and metastasis. This work suggests that HMGA2 acts not only as an architectural chromatin factor as previously thought but also as a gene-specific regulator that responds to signals from extracellular factors.
HMGA2 is overexpressed in a variety of tumors primarily of mesenchymal origin (for review see Sgarra et al., 2004). However, the mechanism of HMGA2 action is not yet known. This study demonstrates for the first time that HMGA2 is involved in EMT. Our results are consistent with the specific presence of HMGA2 at the invasive front of squamous carcinomas (Miyazawa et al., 2004), a place where EMT occurs during cancer progression. On the other hand, overexpression in MCF7 mammary carcinoma cells of HMGA1b, another member of the HMGA family, but not that of the related HMGA1a, led to invasive tumor growth in nude mice (Reeves et al., 2001). Histochemical and transcriptomic analysis of tumor samples from such mice indicated that HMGA1b induced expression of genes with links to EMT.
Our study demonstrates that HMGA2 is necessary and sufficient for TGF-β–induced EMT. Considering the variety of tumors where HMGA2 expression has been detected, it will be interesting to determine in how many of them TGF-β is the upstream inducer of tumor HMGA2 expression and to what extent the signaling network outlined here explains the tumor-promoting properties of TGF-β.
Materials and methods
Cells, adenoviruses, and reagents
Mouse mammary epithelial NMuMG, human hepatocarcinoma HepG2, human MDA-MB-231-Eco cells, and stable NMuMG clones expressing Smad2 SA (mutant with alanines in place of two C-terminal serines that become phosphorylated by receptors) have been described (Kowanetz et al., 2004; Valcourt et al., 2005; Deckers et al., 2006). Adenoviruses expressing control GFP (Ad-GFP) were a gift from B. Vogelstein (Johns Hopkins Medical Institutions, Baltimore, MD); adenoviruses expressing C-terminally HA-tagged constitutively active TGF-β type I receptor (activin receptor-like kinase 5) ALK-5(TD) and HA-tagged kinase-inactive ALK5(KR) receptor were a gift from K. Miyazono (University of Tokyo, Tokyo, Japan) and have been described (Valcourt et al., 2005).
The mouse Hmga2 siRNA (available from GenBank/EMBL/DDBJ under accession no. NM_010441), which was a single RNA oligonucleotide (D-043585-03), and control siRNA against the luciferase reporter vector pGL2 (accession no. X65324) were obtained from Dharmacon Research, Inc. Human HMGA2 cDNA, cloned by PCR from total normal human mRNA in pcDNA3-HA C-terminally of the HA tag using EcoRI–XhoI as restriction sites, resulted in pcDNA3-HA-hHMGA2. The N-terminally HA-tagged hHMGA2 HindIII–XhoI fragment was subcloned in the inducible vector pMEP4 to produce pMEP4-HA-hHMGA2.
The LY580276 inhibitor for the TGF-β type I receptor kinase was a gift from J.M. Yingling (Eli Lilly and Company, Indianapolis, IN; Peng et al., 2005). Recombinant mature TGF-β1 was obtained from PeproTech and TGF-β3 from K.K. Iwata (OSI Pharmaceuticals, Farmingdale, NY). TGF-β1 was used in most experiments and TGF-β3 in experiments with stable NMuMG and MDA-MB-231 clones for Smad shRNAs (Fig. 1 and Fig. S1). TGF-β1 and -β3 have indistinguishable effects on EMT or Hmga2 gene regulation.
Cell counting
Cell monolayers were washed with PBS, trypsinized, and resuspended in PBS, and cell numbers were calculated using a Z1 cell counter (Beckman Coulter). Numbers are plotted as means from triplicate determinations with standard errors.
Cell transfections
pMEP4 and pMEP4-HA-hHMGA2 were transfected into NMuMG using Lipofectamine 2000 (Invitrogen). 2 d after transfection, cells were cultured in 400 μg/ml hygromycin-B (Calbiochem), and individual antibiotic-resistant clones were derived. For HMGA2 induction, cells growing in hygromycin-B were treated with 10 μM CdCl2 (Sigma-Aldrich) for 24 h, and cells were incubated with vehicle or TGF-β for another 24–48 h.
NMuMG cells were transiently transfected with Hmga2 siRNAs using Dharmafect 4 (Dharmacon Research, Inc.). After 2 h of stimulation with TGF-β, cells were retransfected with siRNAs to a final concentration of 50 nM for 10 h (RNA assay) or 36 h (protein/immunofluorescence assay).
NMuMG and MDA-MB-231-Eco clones with Smad knockdown were established after infection with retroviral supernatants derived from cells transfected with pRetroSuper-expressing shRNA against Smad2, -3, or -4 and were provided by M. van Dinther and P. ten Dijke (Leiden University Medical Center, Leiden, Netherlands; Deckers et al., 2006). The shRNA oligonucleotide sequences were 5′-GGAGTGCGCTTGTATTACA-3′ (mouse/ human Smad2), 5′-CGTCAACACCAAGTGCATC-3′ (mouse/human Smad3), and 5′- CCAGCTACTTACCATCATA-3′ (mouse/human Smad4).
RT-PCR
Total DNA-free cellular RNA was extracted with the RNeasy kit (QIAGEN). RT-PCRs were performed as described previously (Kowanetz et al., 2004) and analyzed using specific primers (Tables I and II). Primers for mouse glyceraldehyde-3′-phosphate dehydrogenase (Gapdh) were used for reference. Lack of DNA contamination was verified by omitting reverse transcriptase (−RT). Quantitative real-time PCR reactions were done as described previously (Valcourt et al., 2005). Gene expression levels were determined with the comparative Ct method using Gapdh as reference. The ground condition was set to 1, and expression data are presented as bar graphs of mean values plus standard deviations.
Table I.
Oligonucleotide primers used for semiquantitative RT-PCR analysis of mouse and human genes
| Gene | Primer sequence | Strand | Product size | Temperature | PCR cycle | Reference |
|---|---|---|---|---|---|---|
| bp | °C | |||||
| Id2 | 5′-GCATCCCCCAGAACAAGAAGGT-3′ | + | 451 | 57 | 28 | Kowanetz et al., 2004 |
| 5′-CCAGGCCGGAGAACAAGACAC-3′ | − | |||||
| Timp3 | 5′-CTCCTGTTGCCCAGGATAGC-3′ | + | 350 | 60 | 28 | Valcourt et al., 2005 |
| 5′-CTCTCTCTCCTGCCTTCTCC-3′ | − | |||||
| PAI-1 | 5′-GGGAAAAGGGGCTGTGTGAC-3′ | + | 406 | 57 | 27 | M33960 |
| 5′-GTACACGGTGTGTGGCTGTC-3′ | − | |||||
| Mucin1 | 5′-GATGCAGTTCCCTCCCTCTG-3′ | + | 450 | 60 | 25 | Valcourt et al., 2005 |
| 5′-CCTACCAGAAGTCCTGGCTC-3′ | − | |||||
| Hmga2 | 5′-AACCTTACTGGGTCGGCATC-3′ | + | 419 | 58 | 25 | Valcourt et al., 2005 |
| 5′-GGTGAGGTTTGAGCTCCTTC-3′ | − | |||||
| HMGA2 | 5′-GGGCCAGGAGGTAGTTTCTC-3′ | + | 321 | 60 | 24 | NM_003483 |
| 5′-CCTCGGTGCACCATGTTTGGC-3′ | − | |||||
| Hmga1(a/b) | 5′-CCCAGTGAAGTGCCAACTCCG-3′ | + | 171 | 60 | 24 | NM_016660 NM_001025427 |
| 5′-CCTCAGAGGACTCCTGGGAGA-3′ | − | |||||
| E-cadherin | 5′-GTCAGATCTCCCTGAGTTCG-3′ | + | 391 | 56 | 27 | NM_009864 |
| 5′-GCACCCACACACATACACTC-3′ | − | |||||
| Vimentin | 5′-GAAGGAAGAGATGGCTCGTC-3′ | + | 315 | 57 | 25 | NM_011701 |
| 5′-CTGCACTGTTGCACCAAGTG-3′ | − | |||||
| Fibronectin-1 | 5′-CCCAGACTTATGGTGGCAATTC-3′ | + | 200 | 60 | 26 | NM_010233 |
| 5′-AATTTCCGCCTCGAGTCTGA-3′ | − | |||||
| Gapdh/GAPDH | 5′-ATCACTGCCACCCAGAAGAC-3′ | + | 443 | 57 | 30 | Valcourt et al., 2005 |
| 5′-ATGAGGTCCACCACCCTGTT-3′ | − |
Mouse genes begin with lowercase letters and human genes with capital letters.
Table II.
Oligonucleotide primers used for quantitative RT-PCR and ChIP
| Gene | Primer sequence | Strand | Product size | Reference |
|---|---|---|---|---|
| bp | ||||
| Oligonucleotide primers used for quantitative real-time RT-PCR analysis of mouse genes |
||||
| PAI-1 | 5′-GGGAAAAGGGGCTGTGTGAC-3′ | + | 406 | M33960 |
| 5′-GTACACGGTGTGTGGCTGTC-3′ | − | |||
| Snail | 5′-ACCCCCGCCGGAAGCCCAACT-3′ | + | 127 | NM_011427 |
| 5′-AGCGGCGGGGTTGAGGACCTC-3′ | − | |||
| Slug | 5′-CTCACCTCGGGAGCATACAGC-3′ | + | 146 | NM_011415 |
| 5′-TGAAGTGTCAGAGGAAGGCGGG-3′ | − | |||
| Twist | 5′-CGGGTCATGGCTAACGTG-3′ | + | 196 | NM_011658 |
| 5′-CAGCTTGCCATCTTGGAGTC-3′ | − | |||
| Hmga2 | 5′-AGCAAAAACAAGAGCCCCTCTA-3′ | + | 100 | NM_010441 |
| 5′-ACGACTTGTTGTGGCCATTTC-3′ | − | |||
| Fibronectin-1 | 5′-CCCAGACTTATGGTGGCAATTC-3′ | + | 200 | NM_010233 |
| 5′-AATTTCCGCCTCGAGTCTGA-3′ | − | |||
| Gapdh | 5′-TGTGTCCGTCGTGGATCTGA-3′ | + | 76 | NM_001001303 |
| 5′-CCTGCTTCACCACCTTCTTGA-3′ | − | |||
| Oligonucleotide primers used for quantitative real-time RT-PCR in human cells |
||||
| HMGA2 | 5′-CCCAAAGGCAGCAAAAACAA-3′ | + | 81 | NM_003483 |
| 5′-GCCTCTTGGCCGTTTTTCTC-3′ | − | |||
| GAPDH | 5′-GGAGTCAACGGATTTGGTCGTA-3′ | + | 78 | BC023632 |
| 5′-GGCAACAATATCCACTTTACCAGAGT-3′ | − | |||
| Oligonucleotide primers used to amplify different regions of the Hmga2 promoter |
||||
| Hmga2 promoter −3420/−3320 | 5′-TAATGCGCTTGCCTGAGCTA-3′ | + | 100 | AC153362 |
| 5′-GCTGTCAAATCGGGCATCA-3′ | − | |||
| Hmga2 promoter −495/−245 | 5′-TCCTGGCAGAAACTTCCACTCT-3′ | + | 250 | NM_010441 |
| 5′-TGGAGTGAATTGTGTCCCTTGA-3′ | − | |||
| Hmga2 promoter −194/+4 | 5′-GAGCCTTTGCGGAGAGAGCAA-3′ | + | 200 | NM_010441 |
| 5′-CATCAACACCGGACGTCCA-3′ | − |
Perchlorate extraction of chromatin
NMuMG cells were lysed in 140 mM NaCl, 1.5 mM MgCl2, 10 mM Tris- HCl, pH 8.6, and 0.5% NP-40, supplemented with protease inhibitors. After centrifugation (3 min, 6,000 g, 4°C), nuclear pellets were resuspended, vortexed for 30 s, and rotated at room temperature for 1 h in 5% perchloric acid. Perchloric acid supernatants (5 min, 6,000 g, 25°C) were precipitated by 8 vol cold ethanol and centrifuged (15 min, 10,000 g, 4°C), and pellets were resuspended in lysis buffer and analyzed by immunoblotting.
Immunoblotting and immunofluorescence microscopy
Total protein extracts subjected to SDS-PAGE were analyzed by immunoblotting as described previously (Kowanetz et al., 2004). Mouse monoclonal anti–β-tubulin (T8535) and anti–β-actin (AC-15) were obtained from Sigma-Aldrich; mouse monoclonal anti-HA (12CA5) was obtained from Roche Applied Science; mouse monoclonal anti–E-cadherin (C20820) was obtained from BD Biosciences; rat monoclonal anti–ZO-1 (MAB1520) was obtained from Chemicon; and mouse monoclonal anti–histone H1 (AE-4), anti-Smad2/Smad3 (H-2), and anti-Smad4 (B-8) were obtained from Santa Cruz Biotechnology, Inc. Rabbit polyclonal anti-Hmga2 was raised against a synthetic peptide (CSPSKAAQKKAETIGE, where S maps at residue 59 of mouse Hmga2) and recognizes mouse but not human HMGA2. Secondary anti–mouse IgG and anti–rabbit IgG coupled to horseradish peroxidase were purchased from GE Healthcare. The enhanced chemiluminescence detection system was purchased from Santa Cruz Biotechnology, Inc.
For immunofluorescence, cells were treated as indicated in the figure legends, fixed, and stained with tetramethyl-rhodamine isothiocyanate (TRITC)–labeled phalloidin (Sigma-Aldrich) or with rat anti–ZO-1, mouse anti–E-cadherin, and rabbit anti-HMGA2 antibodies as primary antibodies and TRITC-conjugated goat anti–rat IgG and FITC-conjugated anti–mouse or anti–rabbit IgG antibodies as secondary antibodies (Jackson ImmunoResearch Laboratories) as described previously (Kowanetz et al., 2004). Photomicrographs were obtained by a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.) with a digital camera (C4742-95; Hamamatsu), using Plan-neofluar 40×/0.75 objective lens (Carl Zeiss MicroImaging, Inc.) and photographing at ambient temperature in the absence of oil immersion. Primary images were acquired with the camera's QED software. Image memory content was reduced, and brightness contrast was adjusted using Photoshop 6.0 (Adobe).
Promoter-reporter assays
HepG2 cells were transiently transfected with calcium phosphate and a panel of mouse Hmga2 promoter luciferase constructs as described previously (Rustighi et al., 1999). The Snail and Twist promoter luciferase constructs were provided by A. Cano (Universidad Autónoma de Madrid, Madrid, Spain) and L.R. Howe (Cornell University, New York, NY), respectively. All promoter constructs were cotransfected with the normalization reporter plasmid pCMV–β-Gal and the expression vector pcDNA3 (mock vector) or pcDNA3-HA-hHMGA2 for Snail and Twist promoter analysis or pcDNA3-HA-Smad2 SA for Hmga2 promoter analysis. The enhanced luciferase assay kit from BD Biosciences was used. Normalized promoter activity data are plotted in bar graphs representing mean values from triplicate determinations with standard deviations. Each independent experiment was repeated at least twice.
Chromatin immunoprecipitation (ChIP) assays
The equivalent of 107 cells was used per ChIP reaction. Cross-linking was performed using 1% formaldehyde followed by neutralization with 0.125 M glycine. Cells were lysed in 1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1, and protease inhibitors on ice. DNA was sheared by sonication to 200–1,000 bp in length. Sonicated cell pellets were diluted 10 times in a buffer containing 0.01% SDS and were precleared with protein A–Sepharose in the presence of BSA and salmon sperm DNA before incubation with 5 μg rabbit anti-Smad4 antibody (H-552; Santa Cruz Biotechnology, Inc.) or preimmune rabbit antiserum as a negative control. Protein–DNA complexes were precipitated with protein A–Sepharose in the presence of BSA and salmon sperm DNA. Immunoprecipitated complexes were washed once with 150 mM NaCl, 0.2% SDS, 1% Triton X-100, 2 mM EDTA, and 20 mM Tris-HCl, pH 8.1; once with an identical buffer containing 500 mM NaCl; once with 0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl, pH 8.1; and twice with 10 mM Tris-EDTA. Immunoprecipitated complexes were eluted with 1% SDS and 0.1 M NaHCO3. After reversal of cross-links by heating in 0.2 M NaCl, proteinase K treatment, purification by classical phenol-chloroform extraction, and ethanol precipitation, DNA pellets were resuspended in 20 μl of water. Input material before immunoprecipitation corresponds to one third of the immunoprecipitated material. Input DNA pellets were resuspended in 50 μl of water. PCR was performed using 2 μl of immunoprecipitated or 0.5 μl of input material using Taq DNA polymerase (Invitrogen). Amplification was performed for 30 cycles for immunoprecipitated DNA and 25 cycles for input DNA. Primer sets used to amplify different regions of the Hmga2 promoter are described in Table II.
Statistical analysis
Statistical analysis of real-time PCR quantification and promoter assays was performed by two-tailed paired t test. Significance was considered at P ≤ 0.05.
Online supplemental material
Fig. S1 demonstrates that Hmga2 is a direct gene target of TGF-β signaling and analyzes the effects of siRNA-mediated knockdown of Smads on EMT and Hmga2 expression. Fig. S2 demonstrates that ectopic HMGA2 does not induce autocrine TGF-β signaling. Fig. S3 analyzes the effects of siRNA-mediated knockdown of Hmga2 on its endogenous protein level and localization. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200512110/DC1.
Supplementary Material
Acknowledgments
We thank A. Cano, L.R. Howe, K. Miyazono, P. ten Dijke, M. van Dinther, B. Vogelstein, and J.M. Yingling for valuable reagents and R. Bergström, E. Ihse, M. Kowanetz, and K. Pardali and members of our research group for help and suggestions during the course of this work.
This work was supported by the Ludwig Institute for Cancer Research, the Swedish Cancer Society (project number 4855-B03-01XAC), and the Natural Sciences Foundation of Sweden (project number K2004-32XD-14936-01A). S. Thuault and M. Petersen were supported by the Marie Curie Research Training Network “EpiPlastCarcinoma” under the European Union FP6 program. U. Valcourt was supported by postdoctoral fellowships from the French Association pour la Recherche sur le Cancer and the Swedish Cancer Society (project number 4812-B03-01VAA).
Abbreviations used in this paper: ChIP, chromatin immunoprecipitation; EMT, epithelial–mesenchymal transition; Gapdh, glyceraldehyde-3′-phosphate dehydrogenase; HMG, high mobility group; Id, inhibitor of differentiation; ZO-1, zonula occludens 1.
References
- Berlingieri, M.T., G. Manfioletti, M. Santoro, A. Bandiera, R. Visconti, V. Giancotti, and A. Fusco. 1995. Inhibition of HMGI-C protein synthesis suppresses retrovirally induced neoplastic transformation of rat thyroid cells. Mol. Cell. Biol. 15:1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bhowmick, N.A., and H.L. Moses. 2005. Tumor-stroma interactions. Curr. Opin. Genet. Dev. 15:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckers, M., M. van Dinther, J. Buijs, I. Que, C. Lowik, G. van der Pluijm, and P. ten Dijke. 2006. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 66:2202–2209. [DOI] [PubMed] [Google Scholar]
- Feng, X.-H., and R. Derynck. 2005. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 21:659–693. [DOI] [PubMed] [Google Scholar]
- Hay, E.D. 2005. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 233:706–720. [DOI] [PubMed] [Google Scholar]
- Huber, M.A., N. Kraut, and H. Beug. 2005. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 17:548–558. [DOI] [PubMed] [Google Scholar]
- Kondo, M., E. Cubillo, K. Tobiume, T. Shirakihara, N. Fukuda, H. Suzuki, K. Shimizu, K. Takehara, A. Cano, M. Saitoh, and K. Miyazono. 2004. A role for Id in the regulation of TGF-β-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 11:1092–1101. [DOI] [PubMed] [Google Scholar]
- Kowanetz, M., U. Valcourt, R. Bergström, C.-H. Heldin, and A. Moustakas. 2004. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol. Cell. Biol. 24:4241–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M.O., Y.Y. Wan, S. Sanjabi, A.K. Robertson, and R.A. Flavell. 2006. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24:99–146. [DOI] [PubMed] [Google Scholar]
- Merika, M., and D. Thanos. 2001. Enhanceosomes. Curr. Opin. Genet. Dev. 11:205–208. [DOI] [PubMed] [Google Scholar]
- Miyazawa, J., A. Mitoro, S. Kawashiri, K.K. Chada, and K. Imai. 2004. Expression of mesenchyme-specific gene HMGA2 in squamous cell carcinomas of the oral cavity. Cancer Res. 64:2024–2029. [DOI] [PubMed] [Google Scholar]
- Moody, S.E., D. Perez, T.C. Pan, C.J. Sarkisian, C.P. Portocarrero, C.J. Sterner, K.L. Notorfrancesco, R.D. Cardiff, and L.A. Chodosh. 2005. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 8:197–209. [DOI] [PubMed] [Google Scholar]
- Moustakas, A., and C.-H. Heldin. 2005. Non-Smad TGF-β signals. J. Cell Sci. 118:3573–3584. [DOI] [PubMed] [Google Scholar]
- Peinado, H., M. Quintanilla, and A. Cano. 2003. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 278:21113–21123. [DOI] [PubMed] [Google Scholar]
- Peinado, H., F. Portillo, and A. Cano. 2004. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 48:365–375. [DOI] [PubMed] [Google Scholar]
- Peng, S.B., L. Yan, X. Xia, S.A. Watkins, H.B. Brooks, D. Beight, D.K. Herron, M.L. Jones, J.W. Lampe, W.T. McMillen, et al. 2005. Kinetic characterization of novel pyrazole TGF-β receptor I kinase inhibitors and their blockade of the epithelial-mesenchymal transition. Biochemistry. 44:2293–2304. [DOI] [PubMed] [Google Scholar]
- Reeves, R. 2001. Molecular biology of HMGA proteins: hubs of nuclear function. Gene. 277:63–81. [DOI] [PubMed] [Google Scholar]
- Reeves, R., D.D. Edberg, and Y. Li. 2001. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol. Cell. Biol. 21:575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, A.B., and L.M. Wakefield. 2003. The two faces of transforming growth factor β in carcinogenesis. Proc. Natl. Acad. Sci. USA. 100:8621–8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustighi, A., F. Mantovani, A. Fusco, V. Giancotti, and G. Manfioletti. 1999. Sp1 and CTF/NF-1 transcription factors are involved in the basal expression of the Hmgi-c proximal promoter. Biochem. Biophys. Res. Commun. 265:439–447. [DOI] [PubMed] [Google Scholar]
- Saika, S., S. Kono-Saika, Y. Ohnishi, M. Sato, Y. Muragaki, A. Ooshima, K.C. Flanders, J. Yoo, M. Anzano, C.Y. Liu, et al. 2004. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am. J. Pathol. 164:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarra, R., A. Rustighi, M.A. Tessari, J. Di Bernardo, S. Altamura, A. Fusco, G. Manfioletti, and V. Giancotti. 2004. Nuclear phosphoproteins HMGA and their relationship with chromatin structure and cancer. FEBS Lett. 574:1–8. [DOI] [PubMed] [Google Scholar]
- Valcourt, U., M. Kowanetz, H. Niimi, C.-H. Heldin, and A. Moustakas. 2005. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell. 16:1987–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., S.A. Mani, J.L. Donaher, S. Ramaswamy, R.A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson, and R.A. Weinberg. 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 117:927–939. [DOI] [PubMed] [Google Scholar]
- Zavadil, J., L. Cermak, N. Soto-Nieves, and E.P. Böttinger. 2004. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23:1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}