

Abstract
Mouse chromocenters are clusters of late-replicating pericentric heterochromatin containing HP1 bound to trimethylated lysine 9 of histone H3 (Me3K9H3). Using a cell-free system to initiate replication within G1-phase nuclei, we demonstrate that chromocenters acquire the property of late replication coincident with their reorganization after mitosis and the establishment of a global replication timing program. HP1 dissociated during mitosis but rebound before the establishment of late replication, and removing HP1 from chromocenters by competition with Me3K9H3 peptides did not result in early replication, demonstrating that this interaction is neither necessary nor sufficient for late replication. However, in cells lacking the Suv39h1,2 methyltransferases responsible for K9H3 trimethylation and HP1 binding at chromocenters, replication of chromocenter DNA was advanced by 10–15% of the length of S phase. Reintroduction of Suv39h1 activity restored the later replication time. We conclude that Suv39 activity is required for the fine-tuning of pericentric heterochromatin replication relative to other late-replicating domains, whereas separate factors establish a global replication timing program during early G1 phase.
Introduction
In mammalian cells, DNA replication takes place in a defined spatio-temporal order. In general, euchromatic domains reside in the interior of the nucleus and replicate in early S phase, whereas heterochromatic domains localize to the nuclear periphery or near nucleoli and replicate late (Schwaiger and Schubeler, 2006; Zink, 2006). Although this spatio-temporal organization has been appreciated for some time, its functional significance is not understood. Because chromatin is assembled at the replication fork, temporal segregation could provide an important regulatory opportunity (McNairn and Gilbert, 2003). Indeed, reporter genes microinjected into mammalian nuclei at different times are assembled into different types of chromatin (Zhang et al., 2002), and the replication timing and subnuclear position of some genes is developmentally regulated (Hiratani et al., 2004; Perry et al., 2004; Williams et al., 2006).
By introducing G1-phase nuclei into a cell-free replication system, we previously demonstrated that the replication timing program is established at a discrete point during early G1 phase termed the timing decision point (TDP; Dimitrova and Gilbert, 1999; Li et al., 2001, 2003). Intriguingly, the subnuclear spatial repositioning of chromosomal domains as well as the clustering of synchronously firing replication origins occurs during this same brief window of time. A similar early G1-phase event may regulate subnuclear position and replication timing in budding yeast (Raghuraman et al., 1997; Heun et al., 2001a). What has not been clear is whether the replication program for constitutive heterochromatin is also reestablished in each cell cycle. In this study, we have examined the establishment of late replication for mouse pericentric heterochromatin.
Chromocenters contain a large central core of pericentric heterochromatin consisting of γ-satellite DNA repeats packaged into chromatin that contains histone H3 trimethylated at lysine 9 (Me3K9H3; Peters et al., 2003; Rice et al., 2003). Trimethylation, which is performed by the Suv39h1,2 histone methyltransferases, creates a high affinity (albeit context dependent) binding site for HP1α and -β, which become concentrated within chromocenters (Bannister et al., 2001; Lachner et al., 2001; Stewart et al., 2005). HP1 localization to chromocenters is a logical candidate for a replication timing determinant given the parallels between HP1 proteins and budding yeast silent chromatin (Sir) proteins (Jones et al., 2000; Wang et al., 2000). Sir proteins concentrate at clusters of telomeres anchored to the nuclear periphery. Telomere clustering creates a sink for Sir proteins, sequestering them at the periphery and preventing them from silencing the active genome (Gartenberg et al., 2004). Telomeres replicate late, and Sir proteins are required for the late replication of some yeast replication origins (Stevenson and Gottschling, 1999; Zappulla et al., 2002).
We find that the replication timing program of chromocenters is reestablished coincident with the reorganization of pericentric heterochromatin into chromocenters. Thus, the TDP affects many types of chromatin simultaneously. However, Suv39h1,2-mediated trimethylation of K9H3 and the interaction of HP1 with chromatin were neither necessary nor sufficient for the establishment of late replication at the TDP. Instead, Suv39 was required for a partial delay of chromocenter replication relative to other late-replicating domains, demonstrating that a global timing program is established independently from additional factors that fine-tune the replication program.
Results
Late replication of chromocenters is established during early G1 phase
Mouse chromocenters are replicated in the second half of S phase (Guenatri et al., 2004; Wu et al., 2005). To determine whether the replication timing of chromocenters is established during early G1 phase, mouse C127 cells were synchronized in mitosis and released into G1 phase for different time periods (G1 1, 2, and 3 h). Intact nuclei were introduced into Xenopus laevis egg extracts, and DNA synthesized in vitro was pulse labeled with biotin-dUTP at various times after the initiation of replication. FISH with a mouse γ-satellite (major satellite) DNA probe was used to visualize pericentric heterochromatin, and the colocalization of FISH signals with replicated DNA, which was identified by staining with labeled avidin, was monitored as an indication of chromocenter replication (Fig. 1). With G1 1- and 2-h nuclei, chromocenters were replicated at the earliest detectable signs of DNA synthesis, indicating a lack of temporal specificity. In contrast, replication of chromocenters within G1 3-h nuclei was significantly delayed, indicating that the late replication program for chromocenters was established between 2 and 3 h after mitosis. Because the overall rate of replication was identical between all three populations of nuclei (Fig. 1 d), the early replication of chromocenters in G1 1- and 2-h nuclei was not simply caused by an increased rate of DNA synthesis.
Figure 1.

The replication timing program of chromocenters is established 2–3 h after mitosis. (a) Nuclei isolated from cells synchronized at different times during G1 phase were introduced into Xenopus egg extract, and DNA synthesized in vitro was pulse labeled with biotin-dUTP at 30, 60, and 120 min thereafter. Labeled nuclei were subjected to FISH with a γ-satellite DNA probe (green in b) and counterstained with Texas red streptavidin (red in b). Chromocenter replication was visualized as the colocalization of γ-satellite DNA with biotin-dUTP. (b) Optical sections through the center of each nucleus were obtained by dual-color confocal laser scanning microscopy. Separate green and red images and their merges are shown on the left (colocalization in yellow). Colocalizing pixels were then imaged in white, and the coefficient of colocalization (percentage of γ-satellite signal that colocalized with biotin signal) was quantified for each nucleus (value shown). (c) Box plot of the coefficient of colocalization for >100 nuclei per time point. Horizontal bars represent the 10th, 25th, 50th (median), 75th, and 90th percentiles, and the 25–75th percentiles are presented as gray boxes. Shown are the results from a single experiment. Similar results were obtained in two additional experiments in which anti-Me3K9H3 antibodies were used to localize chromocenters. (d) Aliquots of nuclei from panel c were introduced into Xenopus egg extract supplemented with α-[32P]dATP, and the percentage of input DNA replicated at the indicated times was determined. The means and SEM (error bars) of all three independent experiments are shown.
This window of time (2–3 h after mitosis) is later than the previously characterized TDP in CHO cells (∼1–2 h after mitosis; Dimitrova and Gilbert, 1999; Li et al., 2001, 2003). To determine whether this difference was the result of cell type–specific differences in a global TDP or to a distinctly later establishment for replication timing of constitutive heterochromatin, we evaluated the global spatio-temporal replication program in C127 (Fig. 2). Asynchronously growing cells were pulse labeled with BrdU and chased for a period of time (4–5 h) that was optimized to obtain the highest percentage of mitotic cells in late S phase during the BrdU pulse (∼50% of mitotic figures display BrdU label, with >95% of the label from late S phase). Nuclei from cells released into G1 phase for 1, 2, or 3 h were introduced into Xenopus egg extract, and the sites of earliest DNA synthesis in vitro (first 30 min) were monitored by biotin-dUTP incorporation (Fig. 2 b). Colocalization of late-replicating BrdU label with biotin-labeled early in vitro DNA synthesis indicates a lack of temporal specificity. In these experiments, the presence or absence of colocalization could be clearly distinguished manually using a dual (red/green) filter (scoring for yellow coloration), obviating the need for the cumbersome colocalization analysis performed in Fig. 1. This difference may be caused by the enhanced preservation of 3D structure in nuclei that have not been denatured for FISH analysis. As shown in Fig. 2 c, most G1 1- and 2-h but very few 3-h nuclei displayed yellow foci, demonstrating that the overall replication timing program in mouse C127 cells is established between 2 and 3 h after mitosis. This later G1-phase timing for the TDP in C127 versus CHO cells may be the result of a 2-h longer G1 phase in these cells relative to CHO cells (unpublished data).
Figure 2.

Global replication timing and subnuclear repositioning. (a) Asynchronous cultures were pulse labeled with BrdU for 30 min, and mitotic cells were harvested 4–5 h later to create a population of cells in which late-replicating sequences were tagged with BrdU. At 1, 2, and 3 h after release into G1 phase, nuclei were introduced into Xenopus egg extract. The earliest DNA to replicate in vitro was pulse labeled with biotin-dUTP, and labeled nuclei were stained with Texas red streptavidin (red in b) and anti-BrdU antibodies (green in b). In parallel, aliquots of the same cells were fixed and stained with anti-BrdU antibody to visualize the subnuclear repositioning of labeled domains making up the easily identifiable late S peripheral spatial replication pattern. (b) Exemplary confocal images from G1 1- and 3-h nuclei displayed as in Fig. 1. (c) The percentage of BrdU-labeled nuclei displaying precocious synthesis of late-replicating sequences in vitro was scored manually as nuclei displaying yellow foci using a dual red/green filter. (d) Percentage of nuclei from the same cells that had repositioned late-replicating BrdU-labeled chromosome domains to the nuclear periphery. Under these conditions, ∼50% of BrdU-labeled cells display the label at the periphery. Data in panels c and d show the mean and SD (when >1%; error bars) of two independent experiments in which >100 nuclei for each time point were scored.
To determine the time at which chromosome domains become repositioned after mitosis, late S BrdU label was tracked in G1 nuclei. Because a large percentage of late S-phase DNA synthesis takes place at the nuclear periphery, we quantified the percentage of nuclei that had repositioned the BrdU label to the nuclear periphery. As show in Fig. 2 d, with G1 1- and 2-h nuclei, <10% of BrdU-positive cells displayed the label in a peripheral pattern, whereas this percentage reached nearly 50% (a plateau level equivalent to that obtained at much later times in the cell cycle) by 3 h after mitosis. We conclude that although different cell types may reestablish replication timing at slightly different times after mitosis, the TDP is nonetheless coincident with the repositioning of chromosome domains and simultaneously affects many different types of chromatin.
The HP1–methyl K9H3 interaction is not sufficient to establish late replication
Although the large blocks of pericentric AT-rich satellite DNA are readily visible by FISH or DAPI staining throughout the cell cycle, we could nonetheless observe their reorganization into more regularly shaped structures during the TDP transition (Fig. 3 a). Because K9H3 trimethylation and binding of HP1 proteins are implicated in the assembly of heterochromatin (Grewal and Rice, 2004), we examined the presence of Me3K9H3 and HP1 proteins within chromocenters as cells pass through the TDP. Me3K9H3 was concentrated within DAPI-dense regions before the TDP (Fig. 3 a), and the total amount of Me3K9H3 in cells was unchanged during this time (Fig. 3 b). Because we and others have shown that Me3K20H4 is also enriched at pericentric heterochromatin (Kourmouli et al., 2004; Schotta et al., 2004) and some studies suggest that this modification may be cell cycle regulated (Fang et al., 2002; Rice et al., 2002), we also examined its abundance during the TDP, but no change was detected. Therefore, these two histone modifications within pericentric heterochromatin are not sufficient to establish late replication.
Figure 3.

Me3K9H3–HP1 association takes place before chromocenter assembly and the TDP. (a) Confocal images of γ-satellite DNA (γ-sat) by FISH and deconvolution images of HP1β or Me3K9H3 for G1 1- (before TDP) and 3-h (after TDP) nuclei. Immunofluorescence images were counterstained with DAPI, and the enrichment of HP1 and Me3K9H3 at chromocenters (DAPI-dense DNA; pseudocolored in red) is revealed in the merged images. (b) Whole cell extracts (WCE) prepared from equal numbers of G1-phase cells were analyzed by immunoblotting with anti-Me3K9H3, Me3K20H4, or anti-HP1β antibodies. The blot used to probe Me3K9H3 was reprobed with anti-histone H3 antibody as a loading control. (c) Protocol for chromatin extraction. (d) Aliquots of cells from panel b in mitosis (M phase), G1 1 h (before TDP), and G1 3 h (after TDP) were extracted as in panel c with the indicated salt concentration, and fractions representing equal cell numbers (half the cell number for M phase) were analyzed by immunoblotting with anti-HP1β antibody. No differences in salt lability were observed for HP1β between pre- and post-TDP cells. Virtually identical results were obtained when parallel immunoblots were probed for HP1α. Similar results were obtained in three independent experiments.
Previous experiments in Drosophila melanogaster (Pak et al., 1997) and mammalian cells (Fischle et al., 2005; Hirota et al., 2005) have demonstrated that the majority of HP1 dissociates from chromatin during mitosis and reassociates thereafter, making HP1 an interesting candidate for a protein involved in resetting replication timing at the TDP. We confirmed by both immunofluorescence and live cell imaging of GFP-tagged HP1 proteins that HP1α and -β were largely dispersed during mitosis (unpublished data). However, all detectable HP1α and -β rebound to chromatin by anaphase (not depicted) and could clearly be seen concentrated within the DAPI-dense regions in G1 1-h nuclei (Fig. 3 a). To determine whether any change in the affinity of HP1 proteins for chromatin coincided with the TDP, we extracted soluble cellular proteins from pre- and post-TDP cells with nonionic detergent at various salt concentrations (Fig. 3, c and d). These results revealed that approximately half of HP1 proteins were soluble or readily dissociated from metaphase chromatin. However, by 1 h after mitosis, all detectable HP1α and -β were very tightly associated with chromatin, with no detectable change in affinity at the TDP (Fig. 3 d). We conclude that the HP1–Me3K9H3 interaction in pericentric heterochromatin takes place before the TDP and, therefore, is not sufficient to establish the late replication timing program of chromocenters.
HP1 is not necessary for late replication of chromocenters
To investigate whether HP1 association is necessary for late replication, we took advantage of the cell-free nature of our system to remove HP1 from post-TDP chromatin before in vitro replication using a peptide mimicking the methylated H3 tail (Bannister et al., 2001). Nuclei from cells synchronized 3 h after mitosis were incubated with a trimethylated peptide consisting of the first 20 amino acids of histone H3 (Fig. 4 a). As controls, aliquots of the same nuclei were incubated with either the unmethylated form of the same peptide or no peptide. Incubation with the trimethylated but not the control peptide resulted in the solubilization of 30–40% of total HP1 protein (Fig. 4 b) and the removal of almost all detectable HP1 at chromocenters (Fig. 4 c). In fact, HP1 remained bound to chromatin surrounding chromocenters but was selectively removed from the DAPI-dense chromocenters themselves. This indicates that Me3K9H3 is a primary binding site for HP1 in chromocenters, whereas HP1 at other sites is bound to other components of chromatin known to tether HP1 (Polioudaki et al., 2001; Singh and Georgatos, 2002). These nuclei were then introduced into a Xenopus egg extract, and the colocalization of the earliest in vitro DNA synthesis with γ-satellite DNA was evaluated. Depletion of HP1 at chromocenters had no significant effect on the timing of these domains or the total rate of in vitro DNA synthesis (Fig. 4, d and e).
Figure 4.

Removal of HP1 proteins from chromatin does not permit early replication of chromocenters. Nuclei prepared from C127 cells synchronized at 3 h after mitosis were incubated for 2 h on ice in the presence of 50 μg/ml of either methylated or unmethylated peptide or buffer alone. (a) Peptide used for HP1 competition. The underlined amino acid is the attachment site for the methyl group. (b) Aliquots of nuclei were extracted, and fractions were analyzed by immunoblotting as in Fig. 3 (0.15 M NaCl). Adding additional methylated peptide or a combination of chromoshadow peptides (Smothers and Henikoff, 2000) and methyl-H3 peptides did not remove additional HP1 proteins (not depicted). WCE, whole cell extract. (c) Aliquots of the same nuclei were stained with anti-HP1 antibody (red) and counterstained with DAPI (pseudocolored green). The insets show higher magnification images of one chromocenter each. HP1 was not detected in the core of the chromocenters after extraction with methylated peptide, although the less DAPI-dense chromatin surrounding the chromocenters retained some HP1. (d) Additional aliquots of the same nuclei were introduced into Xenopus egg extract, and the replication timing of chromocenters was evaluated as in Fig. 1 except that the analysis was restricted to the 30-min time point. Shown is a box plot (displayed as in Fig. 1) for >200 nuclei from three independent experiments (40–80 nuclei/experiment). (e) Aliquots of the Xenopus egg extract reaction were supplemented with α-[32P]dATP, and the percentage of the total input genomic DNA synthesized was evaluated at each of the indicated time points as in Fig. 1. The means and SEM (error bars) for three independent experiments are shown.
Loss of Suv39h1,2 activity advances the time of chromocenter replication
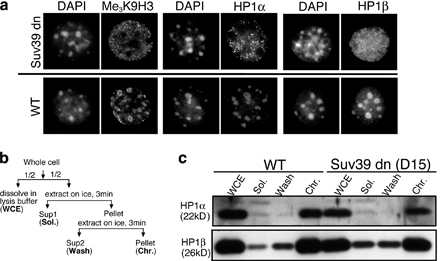
The aforementioned experiments demonstrate that the HP1– Me3K9H3 interaction is not sufficient for establishing the late replication of chromocenters. This was surprising in light of recent links between chromatin structure and replication timing in both mammalian (Li et al., 2004; Takebayashi et al., 2005) and yeast systems (for review see Donaldson, 2005) and the role of HP1 proteins in the formation of pericentric heterochromatin (Maison and Almouzni, 2004). To address whether this interaction has any role in chromocenter replication timing, we examined chromocenter replication in cells lacking Suv39h1 and Suv39h2 (Peters et al., 2001). Mouse embryonic fibroblasts (MEFs) derived from mice lacking both of these enzymes (double null; Suv39dn) have <30% of the total amount of cellular Me3K9H3 and no detectable Me3K9H3 within chromocenters (Peters et al., 2003; Rice et al., 2003). In these cells, HP1 proteins remain tightly bound to chromatin but are depleted from chromocenters (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200601113/DC1). This is consistent with the results in Fig. 4 indicating that HP1 is tethered to chromocenters through Me3K9H3 but binds to other chromatin sites via other mechanisms. Reintroduction of Suv39 activity by stable transfection with a Suv39h1 expression vector (Rice et al., 2003) partially restores Me3K9H3 and HP1 at chromocenters (Fig. S2), providing an “add-back” control to verify that any differences are the result of the loss of Suv39 activity.
To determine the timing of chromocenter replication in these three cell lines, we used a retroactive synchrony method that is commonly used to analyze replication timing of specific gene sequences (Hiratani et al., 2004). This method avoids the need for cumbersome cell line–specific synchronization methods that can perturb the cell cycle. After pulse-labeling nascent DNA with BrdU, cells were retroactively sorted by flow cytometry into populations in different stages of S phase (Fig. 5 a). Genomic DNA was isolated from each fraction, and nascent (BrdU substituted) DNA was immunoprecipitated with anti-BrdU antibodies. Aliquots of these nascent strand preparations were immobilized on nylon filters and hybridized with probes containing either the major or minor satellite DNA repeats (Fig. 5 b) that characterize pericentric and centromeric DNA, respectively (Peters et al., 2003). As controls, we monitored the replication of α- and β-globin genes (not depicted), which are early and late replicating, respectively, and mitochondrial DNA (Fig. 5 b), which replicates throughout the cell cycle and is equally represented in nascent DNA preparations from all cell cycle times (Bogenhagen and Clayton, 1977; James and Bohman, 1981; Magnusson et al., 2003). As shown in Fig. 5 c, minor satellite DNA replicated at a distinctly earlier time during S phase than major satellite, but we could detect no significant difference in the replication program of these DNA sequences in either Suv39dn MEFs or the rescued add-back cell line.
Figure 5.

Late replication of major and minor satellite DNA is independent of Suv39h1,2. (a) Each of the three indicated cell lines (wild type [WT], Suv39dn knockout [D15], and a derivative of D15 in which a Suv39h1 cDNA expression vector was stably integrated into the genome [D15 + Suv39h1; cell line described in Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200601113/DC1]) was pulse labeled with BrdU, stained for DNA content with propidium iodide, and separated by flow cytometry into four S-phase populations with increasing DNA content. (b) DNA was isolated from each fraction of each cell line, BrdU-substituted (nascent) DNA was immunoprecipitated with an anti-BrdU antibody, and aliquots of each of these nascent DNA preparations were immobilized on a nylon filter and hybridized with major and minor satellite DNA probes as well as with mouse mitochondrial DNA that is known to replicate throughout the cell cycle (Bogenhagen and Clayton, 1977; James and Bohman, 1981; Magnusson et al., 2003). (c) For each fraction, the total counts per minute for major and minor hybridizations were first normalized to that of mitochondrial DNA to control for any variation in sample preparation, and the percentage of the total signal from all four fractions of the cell cycle that was present in any given fraction was plotted. The mean values and SEM (error bars) for five (wild type), four (D15), and three (D15 + Suv39h1) independent experiments are shown.
The molecular analyses in Fig. 5 confirm the in vitro studies in Fig. 4 and demonstrate that Suv39h1,2 and the HP1–Me3K9H3 interaction are neither necessary nor sufficient for late replication of pericentric heterochromatin. However, we did notice a slight but not statistically significant advance in the replication time of major satellite DNA in D15 (Fig. 5 c). Small changes in replication timing are better revealed by cell-based assays. For example, the small differences in replication timing of imprinted and immunoglobulin genes can be detected with cell-based assays (Mostoslavsky et al., 2001; Singh et al., 2003) but not molecular analyses (Zhou et al., 2002). Thus, we evaluated the replication time of chromocenters in individual cells using a pulse-chase-pulse method that also does not require cell synchronization (Wu et al., 2005). Cells were pulse labeled with 5′-chloro-2′-deoxyuridine (CldU), chased for different lengths of time, and subsequently pulse labeled with 5′-iodo-2′-deoxyuridine (IdU). Sites of CldU and IdU incorporation were detected by immunofluorescence with CldU- and IdU-specific antibodies (Aten et al., 1992). The change in distribution of these two labels within individual nuclei after different chase times reveals the temporal order of these replication patterns. Exemplary merged CldU and IdU images in Suv39dn MEFs are shown in Fig. 6 a. The overall spatial arrangement and temporal order of the typical six mouse replication patterns (Wu et al., 2005), which are described in detail in Fig. 6 b, were largely unchanged between wild-type and Suv39dn MEFs, demonstrating that Me3K9H3 is not necessary for maintaining the overall spatio-temporal replication program. However, careful inspection of the patterns in Suv39dn MEFs revealed that a true pattern III was not observed (Fig. 6 b). Rather, it appeared as if chromocenter replication (normally specific to pattern IV) was already taking place in cells that were otherwise characteristic of pattern III.
Figure 6.

Suv39h1,2 knockout advances the replication timing of chromocenters. (a) The pulse-chase-pulse method to define the temporal order of spatial replication patterns. MEFs were labeled for 10 min with CldU, chased for various lengths of time, labeled for 10 min with IdU, and stained with fluorescent antibodies specific to CldU (green) and IdU (red). Exemplary confocal images of the dynamic changes in replication patterns observed with increasing chase times (from 0 min through 10 h) in D15, which were similar in all lines, are shown. (b) Displaying only the IdU stain within nuclei that display early CldU patterns reveals the temporal order in which each of the replication patterns take place, which were similar to other mouse cell lines (Wu et al., 2005). In brief, DNA synthesis begins at many small, discrete foci in the internal euchromatic region of the nucleus, excluding the nucleoli (and associated chromocenters) and nuclear periphery (pattern I). In pattern II, replication continues throughout the euchromatic region but is also observed in the perinucleolar and nuclear periphery regions so that a clear demarcation of the nucleoli is no longer apparent. Pattern III is characterized by decreasing euchromatic foci in the interior and increased replication foci at the nuclear (and nucleolar) periphery. Shortly thereafter, most euchromatic (small internal) foci have finished replication, and DNA synthesis begins within the chromocenters (pattern IV). Next, chromocenter replication is completed, whereas some replication at the nuclear periphery continues, coinciding with the replication of a few internal but nonpericentric domains (pattern V). Finally, a few large clusters of foci are observed in both the interior and periphery of the nuclei (pattern VI). All six of these patterns were observed in their proper temporal order in the wild-type MEFs, but in D15 the chromocenter replication pattern IV appeared to merge with pattern III, making it difficult to distinguish between these patterns (i.e., chromocenters were seen to begin replication coincident with the decrease in internal foci labeling). (c) The percentage of cells displaying early CldU-labeled patterns (pattern I) that also showed IdU-labeled chromocenters at each chase time was scored. At least 200 nuclei were scored for each data point. Because the total length of S phase varied (7–10 h) from experiment to experiment, each data point was normalized to the length of S phase, which was measured as the time it takes for 50% of cells to progress from CldU-labeled pattern I into G2 phase (unlabeled IdU), to give a relative chase time as the percentage of S phase (chase time/length of S × 100). After this normalization, the relative chase time for each data point is no longer the same for each experiment, so multiple experiments cannot be averaged. Instead, all data points from all experiments are shown, and a sixth order polynomial curve was fit to each dataset. Experiments were repeated four (wild type), six (D15), and two (D15 + Suv39h1) times. (d) Data on the rising end of each curve in panel c were plotted independently, and a linear curve as well as the slope and y intercept for each dataset are shown. This reveals a very similar slope for all cell lines, with the y intercept advanced by 15% in D15 versus wild-type cells.
To specifically evaluate the time of chromocenter replication relative to the global replication program, we calculated the length of time it took for cells to progress from spatial replication patterns characteristic of early S phase to the time of chromocenter replication (Fig. 6, c and d). This was quantified as the percentage of nuclei that had CldU-labeled early S-phase patterns and IdU-labeled chromocenters at different chase times. These results revealed that chromocenter replication was advanced in the Suv39dn MEFs relative to wild-type MEFs by 10–15% of S phase. Reintroduction of Suv39 activity in the add-back cell line mostly restored this slight delay in chromocenter replication (Fig. 6, c and d).
In the course of the experiments described in Fig. 6, we recognized the presence of a prominent body of chromatin that was intensely labeled with BrdU during pattern III DNA synthesis and replicated synchronously with chromocenters only in Suv39dn mutant MEFs (Fig. 6). This body also stained intensely with an antibody specific to trimethylated lysine 27 of histone H3 (Fig. 7 a), a modification that is highly enriched in the inactivated late-replicating X chromosome (Plath et al., 2003; Silva et al., 2003). Given that these MEFs were derived from a female mouse embryo, it is very likely that this body is the late-replicating inactive X chromosome (Xi), and, for the purposes of discussion, we will refer to it as the Xi. Importantly, in wild-type MEFs (Fig. 7, b and d) and in Suv39dn MEFs rescued by Suv39h1 add-back (Fig. 7 d), chromocenter replication took place distinctly after replication of the Xi. In contrast, Suv39dn MEFs replicated chromocenters simultaneously with replication of the Xi (Fig. 7, c and d) in 75% of Xi-labeled cells. Because the Xi is not enriched for Me3K9H3 (Kohlmaier et al., 2004) and chromocenters can be seen to replicate in an otherwise pattern III along with the last remnants of the small internal euchromatic foci (Fig. 6 b), we interpret these results as an advance of chromocenter replication rather than a delay in Xi replication. These results provide an internally controlled reference and, together with the data in Fig. 6, demonstrate that the Suv39-mediated trimethylation of H3K9 is responsible for a 10–15% delay in replication time.
Figure 7.

Chromocenters replicate after the Xi in wild-type cells but coincident with the Xi in Suv39dn. (a) A large synchronously replicating body (indicated with arrowheads) is highly enriched for Me3K27H3, a defining characteristic of the Xi. The Xi does not colocalize with DAPI-dense bodies that define chromocenters. (b) In wild-type (WT) cells, the Xi replicates during a distinctly different spatial replication pattern (pattern III) than do the chromocenters (pattern IV). In pulse-chase-pulse experiments like those shown in Fig. 6, 2 h of chase time are necessary to traverse from CldU-labeled Xi to IdU-labeled chromocenters. (c) In contrast to wild-type cells, in D15 cells, the Xi (arrowheads) can be seen to replicate synchronously with the replication of chromocenters in a single BrdU pulse (i.e., with no chase). (d) The percentage of cells displaying simultaneous colocalization of the Xi and chromocenters with BrdU label (after a 10-min pulse of BrdU) was scored for wild type, D15, and the D15 derivative rescued by the expression of a Suv39h1 cDNA. More than 100 cells showing Xi replication were scored for each cell line. The means and SD (error bars) for two independent experiments are shown.
Discussion
We demonstrate that the property of late S-phase replication for chromocenters is established during early G1 phase coincident with the reassembly of centromere clusters known as chromocenters. This event was coincident with the reestablishment of a global replication timing program, demonstrating that constitutive heterochromatin is subject to the same dismantling and reassembly of replication timing components during the cell cycle as previously shown for other chromatin domains. Surprisingly, trimethylation of K9H3 and the high affinity binding of HP1 to pericentric heterochromatin were neither necessary nor sufficient for late replication. However, in cells lacking Suv39, replication timing of chromocenters was slightly advanced relative to wild-type and Suv39-rescued cells. These results reveal separate global versus fine-tuning mechanisms that regulate replication timing at pericentric heterochromatin.
Replication timing and the assembly of subnuclear domains
Our results demonstrate that a delay in the replication of pericentric heterochromatin is imposed at a discrete point during early G1 phase, which is coincident with the subnuclear organization of chromocenters and consistent with the existence of a global TDP (Dimitrova and Gilbert, 1999; Li et al., 2001, 2003). This is the first demonstration of a TDP for constitutive heterochromatin and suggests that an overall replication time for all chromatin domains is established simultaneously. The close temporal link between the establishment of late replication and reassembly of subnuclear domains in different cell lines and in budding yeast (Raghuraman et al., 1997; Heun et al., 2001a,b) points to some aspect of nuclear reorganization as the replication timing determinant. In this study, we show that this determinant is resistant to extensive changes in chromatin structure.
HP1 is a major chromatin component of chromocenter chromatin. Because we could not detect any change in the affinity of HP1 for chromatin at this time, the TDP determinant must be independent of the HP1–Me3K9H3 interaction. Moreover, loss of HP1 from chromocenters in Suv39dn mutants did not result in a shift of chromocenter replication to early S-phase replication but caused a partial advance in replication timing. DNA methylation, a prevalent modification of major satellite DNA, is also unlikely to explain early G1-phase establishment of replication timing because this covalent DNA modification is not removed during mitosis, and Suv39dn mutants have significantly reduced DNA methylation at the major satellite DNA (Lehnertz et al., 2003). Finally, Suv39dn mutants did not show any obvious alterations in the number or overall structure of assembled chromocenters (unpublished data). Together, these results suggest that some aspect of the subnuclear domain structure of chromocenters is necessary for their overall late replication and is assembled at the TDP, but this event is independent of DNA methylation, trimethylation of K9H3 and H4K20, and HP1 proteins. Instead, these major chromatin components are responsible for fine-tuning replication timing relative to other domains, revealing at least two levels of control over replication timing.
Replication timing and mouse centromeres
Our results demonstrate that mouse centromeres replicate earlier than flanking heterochromatin. Consistent with these results, earlier replication of centromeric chromatin relative to flanking heterochromatin has been observed in Drosophila cells (Ahmad and Henikoff, 2001; Sullivan and Karpen, 2001), and human centromeres have been shown to replicate asynchronously with flanking heterochromatin (Blower et al., 2002; Sullivan and Karpen, 2004). However, our results contradict a recent study reporting that mouse centromeric chromatin replicates after pericentric chromatin (Guenatri et al., 2004), which was based upon the colocalization of BrdU with major and minor satellite DNA probes. In our hands, it was very difficult to interpret results using this method. Because chromocenters contain more than one centromere and different centromeres replicate at different times over the course of 2 h (Wu et al., 2005), immunofluorescence approaches can reveal cases in which the pericentric heterochromatin of one chromosome replicates before the centromeric chromatin of a different chromosome within the same chromocenter. In fact, a more recent study found that chromatin containing mouse kinetochore proteins replicated earlier and more broadly throughout S phase than pericentric heterochromatin (Weidtkamp-Peters et al., 2006), which is consistent with our molecular analyses (Fig. 5).
Suv39 activity is directly responsible for the trimethylation of K9H3 and HP1 association with chromocenters and is also required for DNA methylation (Lehnertz et al., 2003) and trimethylation of H4K20 (Kourmouli et al., 2004; Schotta et al., 2004) in pericentric heterochromatin. Thus, it is surprising that a mutation with such a profound effect on chromatin structure should have only an incremental effect on replication timing. However, this incremental difference is similar to the timing differences between homologues of imprinted and immunoglobulin genes (Mostoslavsky et al., 2001; Singh et al., 2003), and it is intriguing to speculate that this replication differential may contribute to centromere identity. Centromeres and pericentric heterochromatin have distinctly different protein compositions (Sullivan and Karpen, 2004). Moreover, neither HP1 nor Me3K9H3 are present within centromeric chromatin (Lehnertz et al., 2003; Peters et al., 2003), suggesting that Suv39 does not modify centromeric chromatin. Our results suggest that the differential in replication timing of pericentric relative to centromeric chromatin may be diminished in Suv39h1,2 mutants. Moreover, Suv39dn mice show impaired viability, and those that survive are growth retarded and have an increased tumor risk (Peters et al., 2001). Cells from these mice display severe chromosomal abnormalities indicative of centromere malfunctions (Peters et al., 2001; Guenatri et al., 2004). Thus, our results raise the possibility that the relative replication times of centromere and flanking chromatin could be important for centromere structure and function.
Materials and methods
Cell culture and synchronization
Mouse C127 cells were cultured and synchronized in mitosis as described previously (Gilbert and Cohen, 1987; for review see Wu et al., 1997). MEFs were cultured as described previously (Peters et al., 2001), except with 10% FBS. Under these conditions, the lengths of S phase (measured as shown in Fig. 6) were 7.2–9.4 h for wild type, 8.6–10.2 h for D15, and 9–9.7 h for D15 + Suv39h1. The Suv39h1-rescued D15 cell line was provided by J. Rice (University of Southern California, Los Angeles, CA).
Replication in Xenopus egg extracts
Chromocenter replication timing. Intact nuclei were prepared from G1-phase C127 cells and introduced into Xenopus egg extract as described previously (Wu et al., 1997; Dimitrova and Gilbert, 1998). Where indicated, nuclei were incubated with peptides in the described transport buffer containing 1.5% BSA. Peptides (Cowell et al., 2002) were dissolved in distilled water at 10 mg/ml, and aliquoted stocks were stored at −70°C. At each time point, aliquots of reactions were removed and pulse labeled with 50 μM biotin-16-dUTP (Sigma-Aldrich) for 5–10 min. Reactions were stopped by diluting 1:10 in cold nuclear isolation buffer, and nuclei were adhered to glass slides and fixed as previously described (Li et al., 2001). Pericentric heterochromatin was detected by FISH with a γ-satellite DNA probe, and biotin-dUTP incorporation was detected with Texas red–conjugated streptavidin (GE Healthcare). The γ-satellite plasmid containing eight copies of the 234-bp satellite repeat (Lundgren et al., 2000) was labeled by nick translation with digoxigenin-11–dUTP (Life Technologies), and FISH was performed as previously described (Li et al., 2001).
Global replication timing.
Asynchronously growing cells were pulse labeled with 15 μg/ml BrdU for 30 min and incubated in medium containing 50 ng/ml nocodazole for 4–5 h before mitotic shake-off. At the indicated G1-phase time points, intact nuclei were incubated in Xenopus egg extract. At various time points in vitro, aliquots of nuclei were removed and pulse labeled with biotin-dUTP, and the colocalization of BrdU and biotin was detected as described previously (Dimitrova and Gilbert, 1999).
The percentage of input DNA replicated was determined in Xenopus egg extracts supplemented with α-[32P]dATP by acid precipitation as described previously (for review see Wu et al., 1997). Note that the rate of DNA synthesis in vitro is more rapid than in vivo because of a more synchronous firing of replication origins, resulting in a compressed S phase that nonetheless follows the same temporal order (Dimitrova and Gilbert, 1998; Li et al., 2003).
Dynamic analysis of spatio-temporal patterns using IdU and CldU
The pulse-chase-pulse protocol has been described in detail previously (Dimitrova and Gilbert, 1999; Wu et al., 2005). Note that the 10–15% replication timing advance was not reported in a study of embryonic stem cells that were deficient in Suv39 (Wu et al., 2005), possibly because chromocenter domains are not as prominent in these embryonic stem cells.
Immunolocalization and Western blotting
Immunofluorescence was performed as described previously (Wu et al., 2005). Rat anti-HP1β antibody (Wreggett et al., 1994) was diluted 1:200 in blocking buffer (3% BSA in PBS/0.5% Tween). Rabbit anti-2xMe3K9H3 (Peters et al., 2003) was diluted 1:1,000. AlexaFluor488- or -594–conjugated secondary antibodies (Invitrogen) were diluted ∼1:300 to 1:400. Preparation of whole cell extracts, chromatin isolation, and Western blotting were performed as described previously (Okuno et al., 2001).
Microscopy
Stained specimens were observed with a microscope (Labophot-2; Nikon) equipped with a 100× 1.4 NA planApo oil immersion objective (Nikon), and epifluorescence images were collected with a CCD camera (SPOT RT Slider; Diagnostic Instruments). Deconvolution of stacked images collected at 0.5-μM intervals with QED Image software (Media Cybernetics) was processed with AutoDeblur software (AutoQuant Imaging, Inc.) using the Adaptive Blind setting. Confocal sections were obtained with a confocal microscope (MRC-1024; Bio-Rad Laboratories) mounted on a microscope (Eclipse 600; Nikon). Colocalization analysis was performed with LaserPix software (Bio-Rad Laboratories) as described previously (Wu et al., 2005). Selected images were assembled using Adobe Photoshop.
Molecular analysis of replication timing
BrdU labeling, cell sorting, and immunoprecipitation of BrdU-labeled DNA was performed as described previously (Hiratani et al., 2004). BrdU-labeled DNA from equal numbers of cells was immobilized on nylon membranes and hybridized with major (provided by N. Dillon, Medical Research Council, Clinical Sciences Centre, London, United Kingdom; Lundgren et al., 2000) and minor (pCR4 Min5-1; provided by T. Jenuwein, Research Institute of Molecular Pathology, Vienna, Austria; Lehnertz et al., 2003) satellite DNA probes as well as a mouse mitochondrial probe (p501-1; provided by T. Brown, National Institutes of Health, Bethesda, MD; Brown and Clayton, 2002). Probes were labeled by the random-priming method (Invitrogen). Membranes were hybridized and washed as described previously (for review see Wu et al., 1997), and relative counts per minute were obtained by phosphorimaging analysis (Molecular Dynamics). Values for major and minor satellite DNA were normalized to values for mitochondrial DNA hybridization, and the relative hybridization signal was presented as a percentage of the sum of these normalized values across all cell cycle fractions.
Online supplemental material
Figs. S1 and S2 characterize the solubility of HP1 in Suv39dn MEFs and the restoration of HP1 binding to chromocenters when Suv39h1 activity is reintroduced into the Suv38h1,2-deficient MEFs. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200601113/DC1.
Supplementary Material
Acknowledgments
We dedicate this manuscript to the memory of John W. Newport, who passed away during its preparation. We thank T. Jenuwein for MeH3 antibodies and pCR4, N. Dillon for the major satellite DNA plasmid, T. Brown for p501-1, and J. Rice for the Suv39h1-rescued D15 cell line. We also thank F. Li, A. McNairn, I. Hiratani, T. Yokochi, J. Lu, B. Knox, and P. Kane for helpful discussions and J. Chen, N. Gonchoroff, and National Science Foundation (NSF) REU undergraduates A. Terry, J. Affortunato, J. Tranchell, M. Panning, and B. Niessen for technical support.
This work was supported by NSF grant MCB-0077507 and by the National Institutes of Health grant GM-57233 to D.M. Gilbert.
D.M. Gilbert's present address is Department of Biology, Florida State University, Tallahassee, FL 32306.
Abbreviations used in this paper: CldU, 5′-chloro-2′-deoxyuridine; IdU, 5′-iodo-2′-deoxyuridine; MEF, mouse embryonic fibroblast; TDP, timing decision point.
References
- Ahmad, K., and S. Henikoff. 2001. Centromeres are specialized replication domains in heterochromatin. J. Cell Biol. 153:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten, J.A., P.J.M. Bakker, J. Stap, G.A. Boschman, and C.H.N. Veenhof. 1992. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. J. 24:251–259. [DOI] [PubMed] [Google Scholar]
- Bannister, A.J., P. Zegerman, J.F. Partridge, E.A. Miska, J.O. Thomas, R.C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 410:120–124. [DOI] [PubMed] [Google Scholar]
- Blower, M.D., B.A. Sullivan, and G.H. Karpen. 2002. Conserved organization of centromeric chromatin in flies and humans. Dev. Cell. 2:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen, D., and D.A. Clayton. 1977. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 11:719–727. [DOI] [PubMed] [Google Scholar]
- Brown, T.A., and D.A. Clayton. 2002. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′- dideoxycytidine. Nucleic Acids Res. 30:2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell, I.G., R. Aucott, S.K. Mahadevaiah, P.S. Burgoyne, N. Huskisson, S. Bongiorni, G. Prantera, L. Fanti, S. Pimpinelli, R. Wu, et al. 2002. Heterochromatin, HP1 and methylation at lysine 9 of histone H3 in animals. Chromosoma. 111:22–36. [DOI] [PubMed] [Google Scholar]
- Dimitrova, D., and D. Gilbert. 1998. Regulation of mammalian replication origin usage in Xenopus egg extracts. J. Cell Sci. 111:2989–2998. [DOI] [PubMed] [Google Scholar]
- Dimitrova, D.S., and D.M. Gilbert. 1999. The spatial position and replication timing of chromosomal domains are both established in early G1-phase. Mol. Cell. 4:983–993. [DOI] [PubMed] [Google Scholar]
- Donaldson, A.D. 2005. Shaping time: chromatin structure and the DNA replication programme. Trends Genet. 21:444–449. [DOI] [PubMed] [Google Scholar]
- Fang, J., Q. Feng, C.S. Ketel, H. Wang, R. Cao, L. Xia, H. Erdjument-Bromage, P. Tempst, J.A. Simon, and Y. Zhang. 2002. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 12:1086–1099. [DOI] [PubMed] [Google Scholar]
- Fischle, W., B.S. Tseng, H.L. Dormann, B.M. Ueberheide, B.A. Garcia, J. Shabanowitz, D.F. Hunt, H. Funabiki, and C.D. Allis. 2005. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 438:1116–1122. [DOI] [PubMed] [Google Scholar]
- Gartenberg, M.R., F.R. Neumann, T. Laroche, M. Blaszczyk, and S.M. Gasser. 2004. Sir-mediated repression can occur independently of chromosomal and subnuclear contexts. Cell. 119:955–967. [DOI] [PubMed] [Google Scholar]
- Gilbert, D.M., and S.N. Cohen. 1987. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S-phase of the cell cycle. Cell. 50:59–68. [DOI] [PubMed] [Google Scholar]
- Grewal, S.I., and J.C. Rice. 2004. Regulation of heterochromatin by histone methylation and small RNAs. Curr. Opin. Cell Biol. 16:230–238. [DOI] [PubMed] [Google Scholar]
- Guenatri, M., D. Bailly, C. Maison, and G. Almouzni. 2004. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun, P., T. Laroche, M.K. Raghuraman, and S.M. Gasser. 2001. a. The positioning and dynamics of origins of replication in the budding yeast nucleus. J. Cell Biol. 152:385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun, P., T. Laroche, K. Shimada, P. Furrer, and S.M. Gasser. 2001. b. Chromosome dynamics in the yeast interphase nucleus. Science. 294:2181–2186. [DOI] [PubMed] [Google Scholar]
- Hiratani, I., A. Leskovar, and D.M. Gilbert. 2004. Differentiation-induced replication-timing changes are restricted to AT-rich/long interspersed nuclear element (LINE)-rich isochores. Proc. Natl. Acad. Sci. USA. 101:16861–16866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, T., J.J. Lipp, B.H. Toh, and J.M. Peters. 2005. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438:1176–1180. [DOI] [PubMed] [Google Scholar]
- James, T.W., and R. Bohman. 1981. Proliferation of mitochondria during the cell cycle of the human cell line (HL-60). J. Cell Biol. 89:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.O., I.G. Cowell, and P.B. Singh. 2000. Mammalian chromodomain proteins: their role in genome organisation and expression. Bioessays. 22:124–137. [DOI] [PubMed] [Google Scholar]
- Kohlmaier, A., F. Savarese, M. Lachner, J. Martens, T. Jenuwein, and A. Wutz. 2004. A chromosomal memory triggered by xist regulates histone methylation in x inactivation. PLoS Biol. 10.1371/journal.pbio.0020171. [DOI] [PMC free article] [PubMed]
- Kourmouli, N., P. Jeppesen, S. Mahadevhaiah, P. Burgoyne, R. Wu, D.M. Gilbert, S. Bongiorni, G. Prantera, L. Fanti, S. Pimpinelli, et al. 2004. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J. Cell Sci. 117:2491–2501. [DOI] [PubMed] [Google Scholar]
- Lachner, M., D. O'Carroll, S. Rea, K. Mechtler, and T. Jenuwein. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 410:116–120. [DOI] [PubMed] [Google Scholar]
- Lehnertz, B., Y. Ueda, A.A. Derijck, U. Braunschweig, L. Perez-Burgos, S. Kubicek, T. Chen, E. Li, T. Jenuwein, and A.H. Peters. 2003. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 13:1192–1200. [DOI] [PubMed] [Google Scholar]
- Li, F., J. Chen, M. Izumi, M.C. Butler, S.M. Keezer, and D.M. Gilbert. 2001. The replication timing program of the Chinese hamster beta-globin locus is established coincident with its repositioning near peripheral heterochromatin in early G1 phase. J. Cell Biol. 154:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F., J. Chen, E. Solessio, and D.M. Gilbert. 2003. Spatial distribution and specification of mammalian replication origins during G1 phase. J. Cell Biol. 161:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., R. Santoro, K. Koberna, and I. Grummt. 2004. The chromatin remodeling complex NoRC controls replication timing of rRNA genes. EMBO J. 24:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren, M., C.M. Chow, P. Sabbattini, A. Georgiou, S. Minaee, and N. Dillon. 2000. Transcription factor dosage affects changes in higher order chromatin structure associated with activation of a heterochromatic gene. Cell. 103:733–743. [DOI] [PubMed] [Google Scholar]
- Magnusson, J., M. Orth, P. Lestienne, and J.W. Taanman. 2003. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp. Cell Res. 289:133–142. [DOI] [PubMed] [Google Scholar]
- Maison, C., and G. Almouzni. 2004. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5:296–304. [DOI] [PubMed] [Google Scholar]
- McNairn, A.J., and D.M. Gilbert. 2003. Epigenomic replication: linking epigenetics to DNA replication. Bioessays. 25:647–656. [DOI] [PubMed] [Google Scholar]
- Mostoslavsky, R., N. Singh, T. Tenzen, M. Goldmit, C. Gabay, S. Elizur, P. Qi, B.E. Reubinoff, A. Chess, H. Cedar, and Y. Bergman. 2001. Asynchronous replication and allelic exclusion in the immune system. Nature. 414:221–225. [DOI] [PubMed] [Google Scholar]
- Okuno, Y., A.J. McNairn, N. den Elzen, J. Pines, and D.M. Gilbert. 2001. Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J. 20:4263–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak, D.T., M. Pflumm, I. Chesnokov, D.W. Huang, R. Kellum, J. Marr, P. Romanowski, and M.R. Botchan. 1997. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell. 91:311–323. [DOI] [PubMed] [Google Scholar]
- Perry, P., S. Sauer, N. Billon, W.D. Richardson, M. Spivakov, G. Warnes, F.J. Livesey, M. Merkenschlager, A.G. Fisher, and V. Azuara. 2004. A dynamic switch in the replication timing of key regulator genes in embryonic stem cells upon neural induction. Cell Cycle. 3:1645–1650. [PubMed] [Google Scholar]
- Peters, A.H., D. O'Carroll, H. Scherthan, K. Mechtler, S. Sauer, C. Schofer, K. Weipoltshammer, M. Pagani, M. Lachner, A. Kohlmaier, et al. 2001. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 107:323–337. [DOI] [PubMed] [Google Scholar]
- Peters, A.H., S. Kubicek, K. Mechtler, R.J. O'Sullivan, A.A. Derijck, L. Perez-Burgos, A. Kohlmaier, S. Opravil, M. Tachibana, Y. Shinkai, et al. 2003. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell. 12:1577–1589. [DOI] [PubMed] [Google Scholar]
- Plath, K., J. Fang, S.K. Mlynarczyk-Evans, R. Cao, K.A. Worringer, H. Wang, C.C. de la Cruz, A.P. Otte, B. Panning, and Y. Zhang. 2003. Role of histone H3 lysine 27 methylation in X inactivation. Science. 300:131–135. [DOI] [PubMed] [Google Scholar]
- Polioudaki, H., N. Kourmouli, V. Drosou, A. Bakou, P.A. Theodoropoulos, P.B. Singh, T. Giannakouros, and S.D. Georgatos. 2001. Histones H3/H4 form a tight complex with the inner nuclear membrane protein LBR and heterochromatin protein 1. EMBO Rep. 2:920–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman, M., B. Brewer, and W. Fangman. 1997. Cell cycle-dependent establishment of a late replication program. Science. 276:806–809. [DOI] [PubMed] [Google Scholar]
- Rice, J.C., K. Nishioka, K. Sarma, R. Steward, D. Reinberg, and C.D. Allis. 2002. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 16:2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, J.C., S.D. Briggs, B. Ueberheide, C.M. Barber, J. Shabanowitz, D.F. Hunt, Y. Shinkai, and C.D. Allis. 2003. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell. 12:1591–1598. [DOI] [PubMed] [Google Scholar]
- Schotta, G., M. Lachner, K. Sarma, A. Ebert, R. Sengupta, G. Reuter, D. Reinberg, and T. Jenuwein. 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaiger, M., and D. Schubeler. 2006. A question of timing: emerging links between transcription and replication. Curr. Opin. Genet. Dev. 16:177–183. [DOI] [PubMed] [Google Scholar]
- Silva, J., W. Mak, I. Zvetkova, R. Appanah, T.B. Nesterova, Z. Webster, A.H. Peters, T. Jenuwein, A.P. Otte, and N. Brockdorff. 2003. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell. 4:481–495. [DOI] [PubMed] [Google Scholar]
- Singh, N., F.A. Ebrahimi, A.A. Gimelbrant, A.W. Ensminger, M.R. Tackett, P. Qi, J. Gribnau, and A. Chess. 2003. Coordination of the random asynchronous replication of autosomal loci. Nat. Genet. 33:339–341. [DOI] [PubMed] [Google Scholar]
- Singh, P.B., and S.D. Georgatos. 2002. HP1: facts, open questions, and speculation. J. Struct. Biol. 140:10–16. [DOI] [PubMed] [Google Scholar]
- Smothers, J.F., and S. Henikoff. 2000. The HP1 chromo shadow domain binds a consensus peptide pentAm. Curr. Biol. 10:27–30. [DOI] [PubMed] [Google Scholar]
- Stevenson, J.B., and D.E. Gottschling. 1999. Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev. 13:146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, M.D., J. Li, and J. Wong. 2005. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol. Cell. Biol. 25:2525–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, B., and G. Karpen. 2001. Centromere identity in Drosophila is not determined in vivo by replication timing. J. Cell Biol. 154:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, B.A., and G.H. Karpen. 2004. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat. Struct. Mol. Biol. 11:1076–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi, S., K. Sugimura, T. Saito, C. Sato, Y. Fukushima, H. Taguchi, and K. Okumura. 2005. Regulation of replication at the R/G chromosomal band boundary and pericentromeric heterochromatin of mammalian cells. Exp. Cell Res. 304:162–174. [DOI] [PubMed] [Google Scholar]
- Wang, G., A. Ma, C.M. Chow, D. Horsley, N.R. Brown, I.G. Cowell, and P.B. Singh. 2000. Conservation of heterochromatin protein 1 function. Mol. Cell. Biol. 20:6970–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidtkamp-Peters, S., H.P. Rahn, M.C. Cardoso, and P. Hemmerich. 2006. Replication of centromeric heterochromatin in mouse fibroblasts takes place in early, middle, and late S phase. Histochem. Cell Biol. 125:91–102. [DOI] [PubMed] [Google Scholar]
- Williams, R.R., V. Azuara, P. Perry, S. Sauer, M. Dvorkina, H. Jorgensen, J. Roix, P. McQueen, T. Misteli, M. Merkenschlager, and A.G. Fisher. 2006. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 119:132–140. [DOI] [PubMed] [Google Scholar]
- Wreggett, K.A., F. Hill, P.S. James, A. Hutchings, G.W. Butcher, and P.B. Singh. 1994. A mammalian homologue of Drosophila heterochromatin protein 1 (HP1) is a component of constitutive heterochromatin. Cytogenet. Cell Genet. 66:99–103. [DOI] [PubMed] [Google Scholar]
- Wu, J.R., G. Yu, and D.M. Gilbert. 1997. Origin-specific initiation of mammalian nuclear DNA replication in a Xenopus cell-free system. Methods. 13:313–324. [DOI] [PubMed] [Google Scholar]
- Wu, R., A.V. Terry, P.B. Singh, and D.M. Gilbert. 2005. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol. Biol. Cell. 16:2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulla, D.C., R. Sternglanz, and J. Leatherwood. 2002. Control of replication timing by a transcriptional silencer. Curr. Biol. 12:869–875. [DOI] [PubMed] [Google Scholar]
- Zhang, J., F. Xu, T. Hashimshony, I. Keshet, and H. Cedar. 2002. Establishment of transcriptional competence in early and late S phase. Nature. 420:198–202. [DOI] [PubMed] [Google Scholar]
- Zhou, J., O.V. Ermakova, R. Riblet, B.K. Birshtein, and C.L. Schildkraut. 2002. Replication and subnuclear location dynamics of the immunoglobulin heavy-chain locus in B-lineage cells. Mol. Cell. Biol. 22:4876–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink, D. 2006. The temporal program of DNA replication: new insights into old questions. Chromosoma. 10.1007/s00412-006-0062-8. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}