

Abstract
Clusters of CD59, a glycosylphosphatidylinositol-anchored receptor (GPI-AR), with physiological sizes of approximately six CD59 molecules, recruit Gαi2 and Lyn via protein–protein and raft interactions. Lyn is activated probably by the Gαi2 binding in the same CD59 cluster, inducing the CD59 cluster's binding to F-actin, resulting in its immobilization, termed stimulation-induced temporary arrest of lateral diffusion (STALL; with a 0.57-s lifetime, occurring approximately every 2 s). Simultaneous single-molecule tracking of GFP-PLCγ2 and CD59 clusters revealed that PLCγ2 molecules are transiently (median = 0.25 s) recruited from the cytoplasm exclusively at the CD59 clusters undergoing STALL, producing the IP3–Ca2+ signal. Therefore, we propose that the CD59 cluster in STALL may be a key, albeit transient, platform for transducing the extracellular GPI-AR signal to the intracellular IP3–Ca2+ signal, via PLCγ2 recruitment. The prolonged, analogue, bulk IP3–Ca2+ signal, which lasts for more than several minutes, is likely generated by the sum of the short-lived, digital-like IP3 bursts, each created by the transient recruitment of PLCγ2 molecules to STALLed CD59.
Introduction
In the companion paper (see Suzuki et al. on p. 717 of this issue), we report that single individual Gαi2 and Lyn molecules are dynamically and frequently recruited to CD59 clusters (consisting of three to nine molecules) formed beneath a colloidal gold particle 40 nm in diameter, coated with whole IgG antibody (IgG-gold), as determined by single-molecule tracking. These results are consistent with previous reports showing that clustered glycosylphosphatidylinositol-anchored receptors (GPI-ARs) recruit and activate Gαi and Lyn (and other Src-family kinases [SFKs]; Stefanova et al., 1991; Solomon et al., 1996; Harder et al., 1998). Furthermore, we found that right after the recruitment of Gαi2, the CD59 cluster temporarily stops diffusion, which is an SFK (e.g., Lyn) activity-dependent process termed stimulation-induced temporary arrest of lateral diffusion (STALL). Therefore, we proposed that, when a single Gαi2 molecule is recruited at the CD59 cluster, the recruited Gαi2 molecule would bind to and activate Lyn that was also recruited temporarily to the same CD59 cluster, based on the previous observations in which Gαi2 binds to SFKs and activates them without the need for dephosphorylating the tyrosine residue near the C terminus (Ma et al., 2000; Minshall et al., 2000; Miotti et al., 2000). We also proposed that Gαi2-activated Lyn induces STALL of the CD59 cluster, probably by phosphorylating an as-yet-unknown protein.
In the present paper, we concentrate on the physiological functions of the STALL, rather than the mechanism for inducing the STALL of CD59 clusters. The involvement of raft domains in recruiting signaling molecules (Pierini et al., 2003; del Pozo et al., 2004; Young et al., 2005; Hancock, 2006) is collectively discussed toward the end of the Results in this paper. In another line of earlier studies of GPI-AR signal transduction, the cross-linking of GPI-ARs, e.g., decay accelerating factor (DAF, or CD55) and CD59, was found to trigger the activation of the intracellular inositol-(1,4,5) triphosphate (IP3)–Ca2+ pathway. This is a nonlethal signaling event found in both immune and nonimmune cells (Peiffer et al., 1998; for review see Kimberley et al., 2007), and it involves the hydrolytic generation of IP3 from phosphatidylinositol-bis(4,5)phosphate (PIP2) by PLCγ (Shibuya et al., 1992; Maschek et al., 1993; Peiffer et al., 1998), leading to the release of Ca2+ from the stock in the ER through the IP3 receptor (IP3-dependent calcium channel; Morgan et al., 1993; Stulnig et al., 1997; Pizzo et al., 2002; Omidvar et al., 2006).
Therefore, the next interesting point may be the relationship between the Gαi2–Lyn and IP3–Ca2+ signaling pathways. Previously, Morgan et al. (1993) showed that the inhibition of SFKs blocked the GPI-AR clustering–induced Ca2+ mobilization. Carpenter and Ji (1999) reported that SFK-induced IP3–Ca2+ signaling may be mediated by PLCγ (but not by PLCβ). These results suggest that IgG-gold–induced Lyn activation might occur upstream of IP3 production from PIP2, by PLCγ in the signaling cascade.
Meanwhile, we showed in Suzuki et al. (2007) that Gαi recruitment (and thus Lyn activation) quickly induces STALL. This led us to form the following working hypothesis. Namely, IP3 production from PIP2 may take place exclusively at the CD59 cluster undergoing STALL by recruiting cytoplasmic PLCγ there; therefore, the CD59 cluster undergoing STALL may be a key, albeit temporary (0.57-s lifetime), site for linking the Gαi2-induced Lyn activation to the PLCγ–IP3–Ca2+ signaling pathway. We performed the present research based on this working hypothesis. We examined it by carrying out simultaneous observations of single molecules of GFP-conjugated PLCγ2 (GFP-PLCγ2) and single CD59 clusters. We indeed found that single PLCγ2 molecules are recruited to CD59 clusters, almost exclusively during the STALL periods. Furthermore, the occurrence of STALL is a prerequisite to the bulk increases of the cytoplasmic IP3 and Ca2+ concentrations (Table I).
Table I.
STALL of CD59 clusters and the IP3–Ca2+ responses induced under various conditions
| Probes and pretreatment | Temporal fraction in STALL |
[Ca2+]i increase? | Rise of [IP3]i
(increase in ICyt/IPM) |
Activation of Lyn? |
|---|---|---|---|---|
| % | % | |||
| CD59 monomers (Fab-gold) | 4.5 | No | 3 | No |
| CD59 dimers (IgG-Cy3 probe) | 6.7 | No | ND | ND |
| Addition of C8 (Fab-gold) | 17 | Yes | 33 | Yes |
| Cross-linked by IgG-gold (610 ± 54 IgG-gold particles bound/cell) |
36 | Yes | 34 | Yes |
| Cross-linked by IgG-gold | ||||
| + Cholesterol depletion (4 mM MβCD, 30 min) |
7.6 | No | 1 | No |
| + Cholesterol repletion (10 mM MβCD–cholesterol complex, 30 min) |
28 | Yes | 26 | Yes |
| + Actin depolymerization (50 nM Lat. B, 10 min) |
4.2 | No | −1 | Yes |
| + Inhibition of SFK (10 μM PP2, 5 min) |
8.2 | No | 0 | No |
| + Inhibition of Gαi2 (1.7 nM PTX, 22 h) |
12 | No | 3 | No |
| + Inhibition of PLCγ (1.0 μM U73122, 15 min) |
37 | No | 4 | Yes |
| + Control for U73122 (1.0 μM U73433, 15 min) |
33 | No | 36 | Yes |
| Cross-linked mycCD59TM (560 ± 51 IgG-gold particles bound/cell) |
4.9 | No | 4 | No |
| Cross-linked fluorescein-DOPEa
(670 ± 48 IgG-gold particles bound/cell) |
4.0 | No | ND | No |
All the drug pretreatments were carried out at 37°C. [Ca2+]i, intracellular Ca2+ concentration; [IP3]i, intracellular IP3 concentration.
The DOPE concentrations (0.3–30 μg/ml) and the number of IgG molecules on a gold particle were optimized for highest STALL fraction (see Table I in Suzuki et al., 2007).
Because GPI-ARs have a strong tendency to partition into the low buoyant density fractions on the sucrose density gradient ultracentrifugation, after extraction with Triton X-100 at low temperatures (called detergent-resistant membrane fractions), the so-called raft microdomains may be involved in the signal transduction of GPI-ARs (Brown and Rose, 1992; Munro, 2003; Brugger et al., 2004; Fullekrug and Simons, 2004; Paladino et al., 2004; Simons and Vaz, 2004; Wang et al., 2005). Furthermore, the partitioning of PLCγ into raft microdomains has been suggested (Veri et al., 2001; Rodriguez et al., 2003). We now raise the possibility that the recruitment of PLCγ at CD59 clusters is facilitated by the affinities of both molecules to the so-called raft microdomains (Kenworthy et al., 2004; Mukherjee and Maxfield, 2004; White and Anderson, 2005). The involvement of cholesterol-based special membrane domains or lipid–lipid interactions in the signal transduction of GPI-ARs is collectively discussed at the end of the Results, together with the data presented in Suzuki et al. (2007).
Other critical issues addressed in this paper are the dynamic nature of the recruitment and the very short residency times of the intracellular signaling molecules (PLCγ2, Gαi2, and Lyn) at the CD59 cluster, which are much shorter than the bulk activation durations of these molecules (Mashanov et al., 2004). As described in Suzuki et al. (2007), the residency times of Gαi2 and Lyn (as well as PLCγ2, as shown here) are only 0.1–0.3 s, which was at first surprising. A quantitative understanding of the signaling mechanisms would become possible once the kinetics and the equilibrium of the molecular interactions are known; thus, this issue is fundamentally important. Nevertheless, the lifetimes of the molecular complexes occurring in any signaling pathway are rarely known (Kusumi and Suzuki, 2005).
The single fluorescent molecule tracking used here is particularly suited to tackle this problem. By investigating the recruitment of cytoplasmic PLCγ2 molecules to GPI-AR clusters at the level of single molecules, we were able to track the complicated single-molecule dynamics. In fact, without using single-molecule tracking with sufficient spatiotemporal resolution, transient binding in living cells could not have been found. The lifetimes of the molecular complexes, and the residency time of PLCγ2 at the CD59 clusters, were determined.
Results
CD59 cluster–induced cytoplasmic IP3 and Ca2+ signals mediated by PLCγ, observed by fluorescence imaging of live cells
CD59 is widely expressed in nonimmune cells, as well as in immune cells, and is responsible for triggering nonlethal signaling in both cell types, in addition to controlling complement-induced cell death (for review see Kimberley et al., 2007). The human epithelial T24 cell line, which was extensively used here, expresses CD59, Gαi2, Lyn, and PLCγ2 and was expected to undergo nonlethal signaling responses upon the binding of CD59's natural ligand, C8, which leads to the formation of CD59 clusters (Suzuki et al., 2007), or upon the artificial clustering of CD59, as found in other cell types (Morgan et al., 1993; Stulnig et al., 1997; Peiffer et al., 1998, Kimberley et al., 2007). Therefore, we first established the occurrence of IP3–Ca2+ signaling responses in T24 cells (Figs. 1–3 ), and particularly their amplitude and time courses under the very special conditions of CD59 engagement, which we also used for observing SFK activation (see Fig. 1 a and Fig. 2 in Suzuki et al., 2007): the binding of ∼600 IgG-gold particles, each cross-linking approximately six CD59 molecules on average and inducing the total engagement of ∼3,600 CD59 molecules (Suzuki et al., 2007). We paid special attention to the signal amplitude and on the time courses of the IP3–Ca2+ signal (see Fig. 8).
Figure 1.

Increase of the cytoplasmic IP3 concentrations induced by CD59 clusters, as observed by the epifluorescence imaging of the PH domain (from PLCδ1)–GFP expressed in T24 cells. Typical results are shown. CD59 clustering was induced by the addition of IgG-gold particles or the natural ligand, C8, whereas Fab-gold is a non–cross-linking control. Pretreatment of cells is summarized in the legend to Fig. 2 and Materials and methods.
Figure 3.

Typical time-dependent changes of the cytoplasmic Ca2+ concentrations after the addition of IgG-gold particles or C8 (via the release of Ca2+ from ER through IP3 receptor), as observed by epifluorescence microscopy of fluo-4 in live T24 cells. (a) Typical epifluorescence images of Fluo-4 in the cytoplasm after the addition of IgG-gold. The box in the first image indicates the region of interest (24 × 24 pixels representing 250 μm2), from which the second trace in panel b was obtained. The numbers in the images correspond to the numbers shown in the second trace. (b) The fluorescence intensity of Fluo-4 measured in the box of 24 × 24 pixels, plotted as a function of time. See the legend to Fig. 2 and the Materials and Methods for the various pretreatments of cells. Rhod-2 was used for DOPE cross-linking experiments, to avoid the signal from fluorescein-DOPE. Calcium signals in intact cells and the cells that experienced cholesterol depletion and the subsequent repletion are different in complex and subtle ways. The calcium signal for the latter cells starts at about the same time after stimulation, but the initial rate of the increase of the intracellular Ca2+ concentration ([Ca2+]i)is low, and thus the calcium oscillation starts at ∼3 min later. As usual with pharmacological experiments, the mechanisms for the details of the cellular reactions are difficult to understand. The fourth trace from the bottom is after the application of IgG beads, indicating that these beads are as signal capable as IgG-gold.
Figure 2.

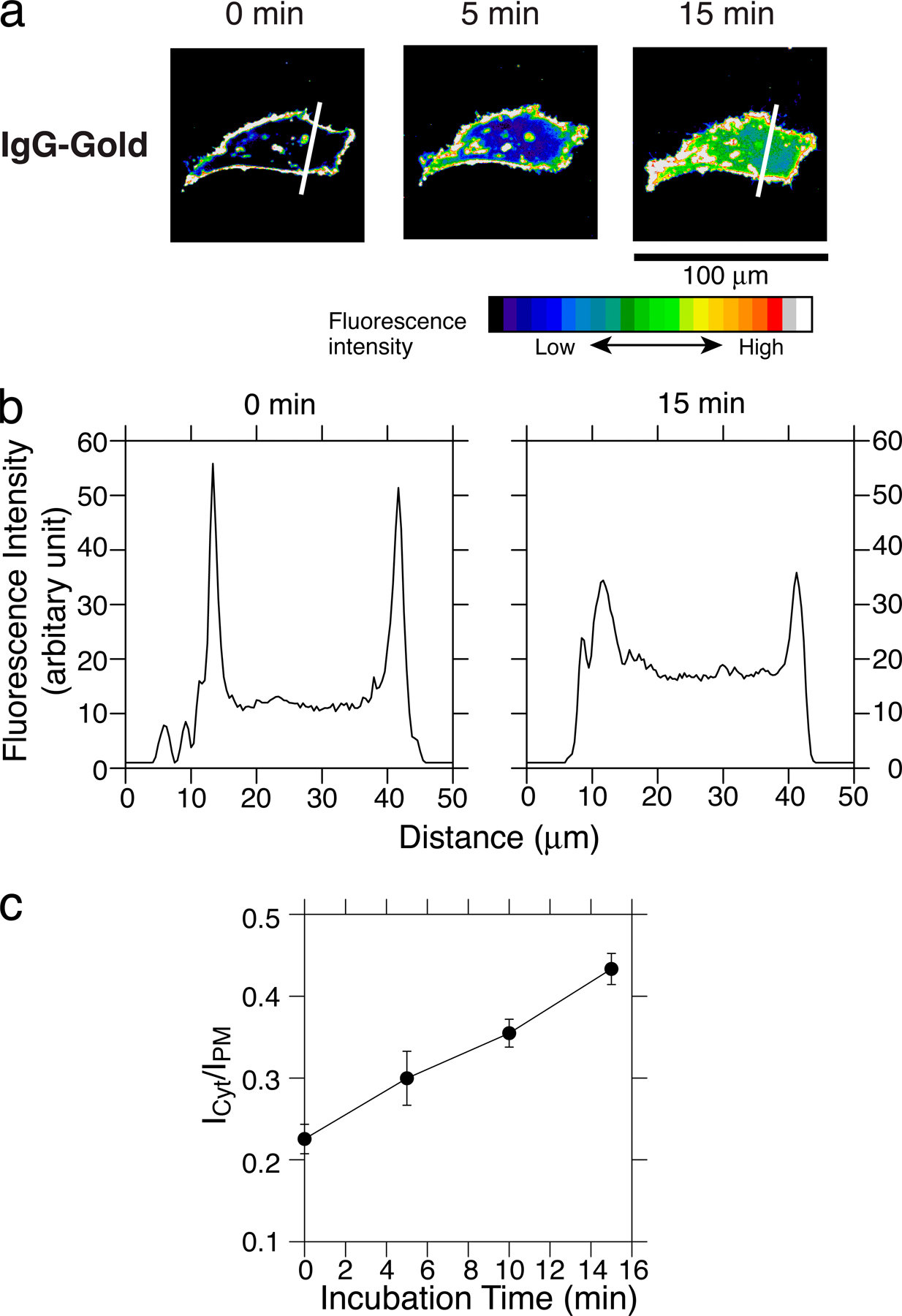
Relative concentration of IP3 in the cytoplasm versus PIP2 on the cytoplasmic surface of the plasma membrane. (a) The fluorescence intensity of the PH domain–GFP, measured along the white lines in Fig. 1 (IgG-gold at 0 [before] and 15 min after IgG-gold addition). The definitions of ICyt and IPM are also shown. (b) The ICyt/IPM ratio provides a convenient measure of the increase of cytoplasmic IP3 versus membrane PIP2. Pretreatment of cells: 1 μM U73122 (PLC blocker), 10 μM PP2, 1.7 nM PTX, 4 mM MβCD, and 50 nM latrunculin B (for 15, 5, 1,320, 30, and 10 min, respectively; 37°C) blocked the increase in the cytoplasmic IP3 concentration, but a 15-min preincubation with 1 μM U73433 (negative control for U73122) did not. The PLC activator, chlorpromazine (CPZ; 30 μM), increased the cytoplasmic IP3 concentration, which was inhibited with U73122, but not with U73343. To avoid overcrowding, the largest error bar (15 min after IgG-gold application; ± SEM) found in the whole set of experiments is shown. Chlorpromazine-related results are only shown up to 5 min because the value is already near the maximum in this parametrization (IPM cannot be defined when IPM is approximately equal to ICyt). The result with IgG beads (50-nm-diameter latex) indicates that these beads used for observing single-molecule recruitment of Lyn, Gαi2, and PLCγ2 are as signal capable as IgG-gold.
Figure 8.

Prolonged bulk signal may be produced by the addition of extremely short, pulse-like recruitment of single molecules. Model A, called the complex integration model, describes the case where each individual signaling event lasts for a length comparable to that for the bulk signaling, which would necessitate a complicated integration of these signals to realize the desired level of the bulk signal. Model B, called the simple summation model, describes the case where each single-molecule event occurs like a digital pulse, as compared with the duration of the bulk signal, which then can simply be added up to generate the desired bulk signal, without the need for complicated integration.
First, we extensively examined GPI-AR cluster–induced IP3 signaling, using live-cell imaging. Although the increase in the cytoplasmic IP3 concentration upon GPI-AR cross-linking has been described in the literature cited above (Shibuya et al., 1992; Maschek et al., 1993; Peiffer et al., 1998), these studies were not conducted in living cells and thus remained somewhat cursory; in addition, we had to know if and how the cross-linking by 600 IgG-gold particles elicited IP3 responses in T24 cells, a cell line that was extensively used here.
The increase in the cytoplasmic IP3 concentration was monitored by observing the partitioning between the cytoplasm (IP3) and the plasma membrane (PIP2) of the pleckstrin homology (PH) domain of PLCδ1 fused to GFP (Hirose et al. 1999; Wang et al., 2004; Fig. 1), as parameterized by the relative cytoplasmic and plasma membrane fluorescence signal intensities (ICyt/IPM) of the PH domain–GFP (Raucher and Sheetz, 2001; Fig. 2; see Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200609175/DC1, for the result using confocal microscopy). Typical time courses of IgG-gold–induced Ca2+ mobilization observed with Ca2+-sensitive fluorescent dyes (see Materials and methods; Minta et al., 1989) are shown in Fig. 3. Both the levels and time courses of the intracellular IP3 and Ca2+ responses to ∼600 bound IgG-gold particles were comparable to those observed after the addition of CD59's natural ligand, C8, at a serum (therefore high) concentration (a cytolytic membrane attack complex unit of 1,000/coverslip; Jasin, 1977), although the Ca2+ signal started somewhat earlier with IgG-gold (Fig. 3).
The involvement of PLCγ was confirmed by the blocking of the IgG-gold–induced IP3 and Ca2+ responses by the pretreatment of cells with the PLCγ inhibitor U73122, but not by the control drug U73343 (Figs. 1–3 ). In addition, the IgG-gold–induced Ca2+ mobilization was inhibited by microinjecting the cells with heparin (Fig. 3), indicating that the rise in the cytoplasmic Ca2+ concentration is due to the release of Ca2+ from ER via the IP3 receptor (Takei et al., 1998). These signaling responses did not occur with the control gold particles (Fab-gold; prepared by conjugating small numbers of Fab fragments of the anti-CD59 antibody to gold particles), which bind to CD59 but do not cross-link it. Our Fab-gold probes were probably bound to single molecules of CD59, as described (see Figs. 1 and 2 and related text in Suzuki et al., 2007). These results, in addition to the CD59-induced SFK activation shown in Suzuki et al. (2007), indicate that CD59 acts as a receptor for nonlethal signals in T24 cells, like that in immune cells.
STALL of CD59 clusters may be caused by their binding to the actin-based membrane skeleton and/or entrapment in a microdomain supported by actin filaments
Individual CD59 clusters induced by IgG-gold particles exhibited alternating periods of apparently simple Brownian diffusion and temporary immobilization, called STALL, as shown in Fig. 4, as well as in Fig. 3 in Suzuki et al. (2007). The lifetimes (the exponential decay times) of each STALL and simple Brownian period are 0.57 and 1.2 s, respectively (see Fig. 3 in Suzuki et al., 2007).
Figure 4.

Clusters of CD59, DAF, and PLAP exhibited STALL in both T24 and NRK cells, within a 48-nm-diameter area. Typical trajectories of IgG-gold–induced clusters, recorded at video rate (33-ms resolution, at which hop movement across the compartment boundaries cannot be resolved), in T24 and NRK cells in culture. Some of the STALL parts of the trajectories are enlarged (bottom). The STALL area sizes are substantially smaller than the median compartment sizes of T24 (110 nm) and NRK (230 nm) cells, shown as square lattices.
Partial actin depolymerization greatly reduced the STALL time fraction of IgG-gold–induced CD59 clusters (Table I). Similar observations were made using cross-linking gold particles for other GPI-ARs, placental alkaline phosphatase (PLAP), and DAF and in other cell types (NRK and PtK2 cells; see Table II in Suzuki et al., 2007). Closer inspection of the trajectories during the STALL periods (Fig. 4) revealed that the CD59 clusters undergo a jittering motion (without macroscopic diffusion), in a limited area of 48 nm in diameter, as estimated by the Gaussian fitting of the determined coordinates during the STALL period (Table II), whereas the median compartment size of the partitioned plasma membrane is 110 nm in the cell type used in this study (T24; Murase et al., 2004; Fig. 4, bottom). Practically the same STALL diameter was found in NRK cells (52 nm; Table II), which have a much greater compartment size (230 nm; Fujiwara et al., 2002; Suzuki et al., 2005; Fig. 4, bottom). This result suggests that STALL is not induced by the corralling effect of the actin-based membrane skeleton that is enhanced by the clustering of CD59 (Iino et al., 2001). STALL may be induced by the binding of CD59 clusters to actin filaments (but indirectly, because they are located in/on the opposite leaflets of the membrane) or by the partitioning of the CD59 clusters into actin-supported microdomains. The jittering motion during STALL may be due to the conformational dynamics of the actin filaments. All of these results are consistent with previous observations suggesting a link between raft domains and actin filaments (Harder and Simons, 1999; Suzuki and Sheetz, 2001; Plowman et al., 2005).
Table II.
The STALL area size determined from the Gaussian fitting of the determined coordinates (2× SD for the diameter) in NRK and T24 cells
| Cells | Median compartment size | Probe | STALL time fraction | STALL diameter (mean ± SEM) | Number of STALLs inspected |
|---|---|---|---|---|---|
| nm | % | nm | |||
| NRK | 230 | IgG-gold | 19 | 52 (±2.1) | 134 |
| T24 | 110 | IgG-gold | 36 | 48 (±2.3) | 139 |
| T24 | 110 | Fab-gold + C8 | 17 | 54 (±2.2) | 81 |
STALL is necessary for inducing IP3–Ca2+ signaling
Here, we summarize six results showing very high correlations between IP3–Ca2+ signaling and STALL of CD59 clusters (Table I). Namely, any treatment that blocked STALL also inhibited IP3 generation and Ca2+ mobilization. We will then show that STALL takes place upstream of IP3 production by PLC, indicating the STALL requirement for IP3–Ca2+ signaling. (1) Treating cells with methyl-β-cyclodextrin (MβCD), which may cause partial depletion of cholesterol, greatly reduced the STALL fraction of CD59 clusters (Table I) and, at the same time, inhibited the IgG-gold–induced production of IP3 (Table I and Figs. 1 and 2) and the intracellular Ca2+ responses (Table I and Fig. 3). (2) Replenishment of cholesterol with cholesterol-loaded MβCD, after partial depletion of cholesterol with MβCD, restored IP3–Ca2+ signaling and reinstated the STALL of CD59 clusters (Figs. 1–3 and Table I). MβCD may have side effects other than cholesterol depletion, as described later, but our point here is the high correlation between IP3–Ca2+ signaling and STALL induction under various conditions. (3) Partial actin depolymerization with latrunculin B, (4) blocking the activities of SFKs with PP2, or (5) blocking Gα's with pertussis toxin (PTX) greatly reduced the STALL of CD59 clusters and, at the same time, blocked the IgG-gold–induced IP3–Ca2+ signaling (Figs. 1–3 and Table I). (6) Fab-gold–CD59, mycCD59TM clusters, and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) clusters induced neither STALL nor IP3–Ca2+ signaling (Table I).
We next examined the effect of inhibiting PLCγ on the STALL of IgG-gold–induced CD59 clusters. The PLCγ blocker U73122, under the conditions where it completely blocked IP3 production (Fig. 2 b, right), did not affect STALL at all (whereas the control reagent U73343 had no effect on either the IP3 concentration or STALL; Table I). This result indicates that STALL occurs upstream of PLCγ-induced IP3 production in the signaling cascade after CD59 engagement and that STALL is required for IP3–Ca2+ signaling.
The STALL site may be the key platform for transducing the CD59 signal to the intracellular IP3 response: single-molecule observations of GFP-PLCγ2 recruitment to CD59 clusters undergoing STALL
Given the STALL requirement for IP3–Ca2+ signaling, we formed the following working hypothesis: the STALL sites may be the places where IP3 is produced from PIP2. Because this reaction is likely catalyzed by the signaling enzyme PLCγ, we investigated whether PLCγ2 is specifically recruited to the STALL sites, i.e., whether the PLCγ2 recruitment to CD59 clusters takes place exclusively when CD59 clusters are undergoing STALL. Thus, we simultaneously observed CD59 clusters and single molecules of GFP-PLCγ2 (Matsuda et al., 2001) in or on the plasma membrane. (PLCγ1 is also expressed in T24 cells used throughout this study, and we expect that PLCγ1 would behave in a way very similar to PLCγ2 because these two molecules behave similarly in a variety of signal transduction pathways [Matsuda et al., 2001].)
For such observations, 50-nm latex particles, rather than 40-nm colloidal gold particles, were used to form CD59 clusters, to avoid the signal from the gold particles (Fig. 5 a). Bright-field microscopy was used to observe the beads, and these two types of particles exhibited practically the same STALL time fractions and durations. In addition, these 50-nm beads were signal capable as much as 40-nm colloidal gold particles, as shown in Fig. 2 b and Fig. 3 b. Fig. 5 b shows a typical trajectory of a CD59 cluster and a GFP-PLCγ2 molecule (also shown in Fig. 5 a), including a period of colocalization (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200609175/DC1). Only during a STALL period of the CD59 cluster was a GFP-PLCγ2 molecule recruited from the cytoplasm for a period of ∼0.3 s (Fig. 5 a; colocalization determined as described by Koyama-Honda et al., 2005). Note that although the GFP-PLCγ2 molecules must be continuously colliding with the cytoplasmic surface of the plasma membrane, the vast majority of them would not stay longer than a 33-ms video frame time, which would make them invisible in video-rate observations. Those recruited at the CD59 clusters undergoing STALL tend to stay for a period of several video frames, thus making themselves visible.
Figure 5.

GFP-PLCγ2 is recruited at CD59 clusters almost exclusively during the STALL period. (a) An image sequence showing superimposed video frames of simultaneous recordings of a CD59 cluster (green spot, which is the bright-field image of a 50-nm bead) and a single molecule of GFP-PLCγ2 (red spot, with Gaussian spatial smoothing). They were colocalized from frame 7 until frame 16 (pink frame numbers), which are within a STALL period. GFP-PLCγ2 suddenly appears from and returns to the cytoplasm. (b) A typical trajectory of a CD59 cluster (black), which includes three STALL periods (three circles). During one of the STALL periods (the blue part of the trajectory), a GFP-PLCγ2 molecule was recruited (magenta trajectory; the CD59 cluster trajectory during the colocalized period is shown in indigo; see Video 1, available at http://www.jcb.org/cgi/conent/full/jcb.200609175/DC1). The colocalized period is completely included within the CD59 cluster's STALL period. (c) Distribution of the time difference between the beginning of GFP-PLCγ2 recruitment and the onset of STALL (time 0 is set at the first frame of STALL). Note that in this graph, each STALL period starts at time 0 but ends at a different time (the pink shading showing STALL is, thus, graded). For the definition of the time difference (lag time), see Fig. 5 a in Suzuki et al. (2007). The yellow bars represent GFP-PLCγ2 molecules with recruitment periods that were completely included within the STALL period, whereas the black bars represent GFP-PLCγ2 molecules whose recruitment periods are not totally included within the STALL period. (d) Distribution of the period of GFP-PLCγ2 colocalization with CD59 clusters (median of 0.25 s), showing transient recruitment of GFP-PLCγ2. As a negative control, single-molecule recruitment of transferrin receptor to the CD59 clusters was observed. No statistically significant recruitment was detected, as described in Suzuki et al. (2007).
Based on many such experiments, we obtained a distribution (histogram) of the time difference (lag time) between the onset of STALL of a CD59 cluster and the recruitment of a single GFP-PLCγ2 molecule to that particular CD59 cluster (Fig. 5 c; see Fig. 5 a in Suzuki et al. [2007] for the definition of the time difference). Time 0 was set at the initiation of the STALL period, and the incidental colocalization was subtracted. This histogram clearly indicates that GFP-PLCγ2 molecules are recruited to CD59 clusters right after the onset of STALL. The yellow bars represent the GFP-PLCγ2 molecules that left the STALLed CD59 clusters before they resumed normal diffusion (i.e., those with recruitment periods completely included within a STALL period). Therefore, this histogram shows that GFP-PLCγ2 recruitment occurs almost exclusively during the STALL periods. Note that, in all of the cases we have observed thus far (see, e.g., Video 1), a CD59 cluster starts showing STALL first, and the recruitment of GFP-PLCγ2 molecules to the CD59 cluster takes place during the STALL period (and not vice versa), consistent with our bulk imaging and biochemical observations indicating that STALL is required for IP3–Ca2+ signaling via PLCγ. These results strongly suggest that the CD59 cluster undergoing STALL is the key, but temporary, platform for the transient recruitment of PLCγ2 molecules and IP3 production and thus for transducing the extracellular CD59 signal to the intracellular signaling network via PLCγ2 recruitment, which eventually leads to the intracellular Ca2+ response.
The duration of GFP-PLCγ2 recruitment was 0.25 s (median, corresponding to 7.5 video frames; Fig. 5 d), which is much shorter than the STALL lifetime (0.57 s; GFP photobleaching/blinking was not responsible for this short duration, as described in Suzuki et al., 2007). As the turnover rates of PLC to produce IP3 from PI(4,5)P2 found in the literature are between 70 and 200 per second (Wahl et al., 1992; Lomasney et al., 1996), assuming a sufficient supply of PIP2 in or around the CD59 cluster undergoing STALL, the recruited PLCγ2 can produce 20–50 molecules of IP3 during the recruitment period of 0.25 s. The biological significance of such transient recruitment is further discussed in the Discussion section (see Fig. 8). How PLCγ recruited to CD59 clusters during the STALL periods is activated remains unknown. Because Lyn is very often (almost constitutively, but dynamically) recruited to the CD59 clusters, Lyn is a good candidate kinase for phosphorylating PLCγ, triggering its activation (Poulin et al., 2005).
Consistency with conventional immunofluorescence observations
The recruitment of PLCγ2 at CD59 clusters was further confirmed by immunofluorescence microscopy of IgG-gold–treated cells (5 min and then fixed; Fig. 6 a). Approximately 20% of the CD59 clusters (IgG-gold particles) were colocalized with PLCγ2, and this association was greatly diminished by cholesterol depletion with MβCD, blocking of SFK activity with PP2, or partial actin depolymerization with latrunculin B (Fig. 6 b), which totally correlated with the STALL occurrence of CD59 clusters (Table I) and IP3–Ca2+ signaling (Figs. 1–3 and Table I). Because the temporal STALL fraction of CD59 clusters (IgG-gold particles) is ∼40%, 20% PLCγ2 immunocolocalization is considered to be consistent with the PLCγ2 recruitment only during the STALL period, considering the binding and visualization efficiency in immunofluorescence staining. Fluorescein-DOPE, a typical nonraft phospholipid preincorporated in the plasma membrane, was not concentrated under the IgG-gold particles (see Fig. 7 a in Suzuki et al., 2007), indicating that membrane concentration and membrane undulations, as reported by Glebov and Nichols (2004), are not involved in the colocalization of the PLCγ2 spots found here.
Figure 6.

Immunofluorescence colocalization of IgG-gold–induced CD59 clusters and PLCγ2 spots. (a) Some of the CD59 clusters formed by IgG-gold particles (green spots; stained with fluorescein-conjugated secondary antibodies to the mouse anti-CD59 antibody attached to the IgG-gold) are colocalized with PLCγ2 spots (red spots). Lookup tables similar to that used for Fig. 1 c in Suzuki et al. (2007) were used. Yellow arrowheads indicate colocalized IgG-gold and anti-PLCγ2 spots. Light green and pink arrowheads indicate IgG-gold and PLCγ2 spots, respectively, which do not show colocalization with each other. (b) The fraction of IgG-gold particles colocalized with the PLCγ2 spots after various pretreatments (mean ± SEM).
Figure 7.

A working model showing how STALL of CD59 clusters may be induced and how it may function as a transient platform to transduce the extracellular CD59 signal to the intracellular IP3–Ca2+ signal (via PLCγ2 recruitment to STALL). The numbers in parentheses correspond to the signaling steps described in the Discussion section.
Discussion
Significance of observing STALL
When larger CD59 clusters are formed by the sequential addition of the primary and secondary antibodies, they are almost always strongly immobilized (see Suzuki et al., 2007) and are capable of activating Lyn and triggering the IP3–Ca2+ pathway. We suggest that the immobilization and signaling mechanisms for large CD59 clusters may be the same as those for IgG-gold–induced CD59 clusters. Because many transient bindings simultaneously occur in large CD59 clusters, the clusters are almost always bound to the actin skeleton by at least one remaining bond, thus keeping the cluster immobilized for long periods. Large CD59 clusters may be strongly signal competent, because they recruit many Gαi2, Lyn, and PLCγ2 molecules at the same time, although each recruitment event lasts for periods as short as those found at IgG-gold–induced clusters. In the present study, the elementary recruitment and signaling steps were dissected by using IgG-gold particles to induce small CD59 clusters and by single-molecule tracking. Furthermore, triggering with IgG-gold resembles the physiological stimulus much more than that with large CD59 clusters: after C8 addition, the Fab-gold particles diffused and underwent STALL, as the IgG-gold particles did (Suzuki et al., 2007). STALL might have further physiological importance in cellular signaling. The external signal may be relayed to the actin membrane skeleton/cytoskeleton at the STALL sites, perhaps inducing local reorganization of the actin filament network (Harder and Simons, 1999; Suzuki and Sheetz, 2001).
Possible involvement of raft microdomains in GPI-AR signaling
In this subsection, we address the possible involvement of raft microdomains in the CD59 signal transduction by combining the results described here with those in Suzuki et al. (2007). For this examination, we used four approaches in this research. First, an artificial transmembrane construct of CD59 (mycCD59TM; CD59 with a myc tag at its N terminus and the transmembrane domain plus the following 12 amino acids of the low-density lipoprotein [LDL] receptor attached at its C terminus) was used to examine the effect of replacing the natural GPI anchor with a transmembrane domain (De Nardo et al., 2002). MycCD59TM clusters, formed by anti–myc-IgG-gold, did not exhibit STALL (Table I) and induced neither Lyn activation (see Table I and Fig. 2 b in Suzuki et al., 2007; Table I) nor IP3–Ca2+ signals (Fig. 2 b, left; Fig. 3; and Table I).
Second, the effect of cross-linking another typical nonraft molecule, the unsaturated phospholipid DOPE, was examined. DOPE clusters induced by IgG-gold did not exhibit STALL (Table I) and induced neither Lyn activation (see Table I and Fig. 2 b in Suzuki et al., 2007) nor Ca2+ mobilization (Fig. 3 and Table I). The Ca2+ mobilization data may appear to be at variance with the results by Wang et al. (2005). Although we cannot resolve this apparent difference, the cross-linked molecules are different, the levels of clustering used here would be much lower than those used by Wang et al. (2005), and they used Jurkat T cells, which may be very sensitive to various external stimulations.
Third, the recruitment frequency of LynN20-GFP (the N-terminal 20-amino-acid sequence of Lyn, containing the binding sites for a palmitoyl and a myristoyl chains, fused at its C terminus to GFP; Pyenta et al., 2001) to CD59 clusters was substantially greater than that for Lyn-GFP to non–cross-linked CD59 or to cross-linked mycCD59TM (negative controls, 3.3-fold). Meanwhile, it was considerably smaller than that of Lyn-GFP to CD59 clusters (positive control, 2.4-fold; see Table IV in Suzuki et al., 2007). These results suggest that both lipid–lipid interactions via Lyn's alkyl chains and protein–protein interactions by way of Lyn's protein part contributed to the recruitment of Lyn to CD59 clusters, consistent with the findings of Gri et al. (2004).
Fourth, the effects of partial cholesterol depletion (with MβCD or saponin) and the subsequent replenishment of cholesterol were examined. Kwik et al. (2003) warned against the use of MβCD because it enhances the PIP2-based F-actin network formation, which could not be recovered by cholesterol replenishment within 12 h. This suggests that if the observed effect of MβCD could be quickly recovered by replenishing cholesterol (generally within 30 min), then the primary effect of MβCD found in that particular experiment probably reflects the direct influence of partial cholesterol depletion.
Cholesterol depletion with MβCD (4 mM for 30 min at 37°C) decreased the temporal STALL fraction of CD59 clusters by a factor of approximately five (Table I) and Lyn activation by a factor of approximately four (see Table I and Fig. 2 c in Suzuki et al., 2007) while virtually blocking the IP3–Ca2+ signaling (Figs. 1–3 and Table I). The subsequent cholesterol replenishment restored STALL and reinstated Lyn phosphorylation in the activation loop (see Table I and Fig. 2 c in Suzuki et al., 2007) as well as IP3–Ca2+ signaling (Figs. 1–3 and Table I; although the initiation of the Ca2+ signal is slower [Fig. 3]). Cholesterol clustering with saponin also reduced STALL (see Table I in Suzuki et al., 2007). Similar observations were made for other GPI-ARs (PLAP and DAF) in other cell types (NRK and PtK2 cells; see Table II in Suzuki et al., 2007). Although the cholesterol replenishment experiments always worked well, drug treatments are always susceptible to side effects and therefore due caution should be paid to the interpretation of the results using MβCD. In addition, single-molecule tracking revealed transient, dynamic recruitment of glycosphingolipid GM1 to CD59 clusters (not depicted, but such transient recruitment is consistent with the lack of colocalization in fixed cells as reported by Fra et al. [1994]), further suggesting the involvement of raft microdomains in the signal transduction of GPI-ARs.
These four lines of results suggest that cholesterol-based raft microdomains or lipid–lipid interactions enhance the recruitment of the cytoplasmic signaling molecules to CD59 clusters. Because the specificity of signaling pathways without any involvement of protein–protein interactions is unlikely (Douglass and Vale, 2005; Holowka et al., 2005; Larson et al., 2005), one of the most critical issues regarding the involvement of raft microdomains in signal transduction in the plasma membrane is whether raft microdomains facilitate or enhance the rate and/or the specificity of molecular recruitment. The aforementioned results, in particular those using LynN20-GFP and mycCD59TM, suggest the occurrence of raft-based facilitation in recruiting Lyn to the CD59 clusters.
A new working hypothesis for the signal transduction of GPI-ARs
Based on the results described in this paper and in Suzuki et al., (2007), we propose the following working model for the signal transduction of GPI-AR, shown in Fig. 7 (also see Video 2, available at http://www.jcb.org/cgi/content/full/jcb.200609175/DC1). We also point out the places in this model where protein interactions may be facilitated by cholesterol-containing (raft) microdomains. There are many unknowns in this working model, but we believe that the present work clarified some of the important processes in the signal transduction steps of GPI-ARs and made it possible to delineate the next investigation steps, as described in this working model. The signaling steps shown in Fig. 7 are described in the following paragraphs.
When CD59 is liganded, it forms larger clusters that resemble those formed by IgG-gold particles (Suzuki et al., 2007; Fig. 7, step 1). Because the subsequent Lyn activation and IP3 production did not occur after cross-linking mycCD59TM or DOPE, and because they are sensitive to cholesterol depletion as well as the subsequent cholesterol replenishment, we raise the possibility that the CD59 cluster may also be a cholesterol-enriched nanodomain (Nicolau et al., 2006).
In the second step, these CD59 clusters undergo slow, apparently simple Brownian diffusion (Fig. 7, step 2a) while frequently and transiently recruiting Lyn, for a median of ∼0.20 s (Fig. 7, step 2b; see Fig. 5 b in Suzuki et al., 2007).
The mechanism for recruiting Lyn is unknown (Fig. 7, step 3). Lipid-based interactions between Lyn and the CD59 cluster raft nanodomains across the bilayer might be involved. Another possibility would be the involvement of an as-yet- unknown transmembrane protein, X, that has certain levels of affinity to the protein moiety of Lyn (and perhaps to Gαi2) and to that of CD59 (avidity to CD59 clusters), as well as an affinity for the raft nanodomains (or to the lipid and lipid- anchoring sequences of Lyn, Gαi2, and CD59; see Table IV in Suzuki et al., 2007). The cholesterol requirements for IP3–Ca2+ signaling reported previously (Veri et al., 2001; Rodriguez et al., 2003) might occur at this stage.
We also found that Gαi2 molecules are transiently recruited to CD59 clusters for ∼0.13 s (median) and that right after the recruitment, the CD59 cluster is engaged in STALL (Fig. 7, step 4; see Fig. 5, d and e, in Suzuki et al., 2007). These results suggest that when the Gαi2 molecule meets Lyn molecules that have also been recruited at the same CD59 cluster, they form a complex at the CD59 cluster, leading to Lyn activation (Ma et al., 2000) and inducing further Lyn activation by phosphorylating its activation loop, perhaps by autophosphorylation (Kmiecik and Shalloway, 1987). As the present work showed that the CD59 cluster acts as a platform for recruiting Gαi2 and Lyn, the next important step is to show their interactions at a single CD59 cluster at the single-molecule level and, at the same time, to detect the activation of Lyn at the level of each individual single molecule right before the onset of STALL. Both Gαi2 and Lyn soon leave the CD59 cluster, and what these activated proteins do after they leave the CD59 cluster is unknown and would be of interest. This is similar to the activation process of H-Ras, reported previously (Prior et al., 2001; for review see Hancock and Parton, 2005). H-Ras may be activated in raft microdomains, but the activated H-Ras leaves them to become incorporated in the domain associated with galectin 1.
In step 5, Lyn activation concurrently induced both STALL of the CD59 cluster and transient PLCγ2 recruitment at the CD59 cluster (median of ∼0.25 s; Fig. 7). Understanding the mechanisms of these processes would be an important next step.
Because PLCγ2 will produce IP3 from PIP2 only at the CD59 cluster that is undergoing STALL (∼0.57 s), the CD59 cluster in STALL is likely to be the key, but transient, platform for relaying the extracellular CD59 signal to the intracellular IP3–Ca2+ signals (Fig. 7, step 6). The residency time of PLCγ2 at the STALLed CD59 cluster is short (∼0.25 s; Fig. 5 d), but it may be sufficient to produce 20–50 IP3 molecules, a calculation based on the turnover rate of the enzyme (Wahl et al., 1992; Lomasney et al., 1996). The direct observation of IP3 production right at the single CD59 cluster undergoing STALL should be performed in future studies.
In step 7, the increase in the IP3 concentration will lead to the release of Ca2+ from the intracellular pool (Fig. 3).
When the present research was undertaken, cell biological and biochemical data accumulated in the literature suggested that (a) the engagement of GPI-ARs may trigger the association of Gαi2 (Solomon et al., 1996), SFKs (Stefanova et al., 1991; Harder et al., 1998), and PLCγ2 (Veri et al., 2001; Rodriguez et al., 2003) with the GPI-AR cluster; (b) Gαi2 may bind to Lyn to activate it (Ma et al., 2000); and (c) Lyn might induce the IP3–Ca2+ pathway via PLCγ recruitment (Morgan et al., 1993). However, the majority of these proposals were based on pharmacological experiments and pull-down assays in the presence of detergents (which jeopardize the legitimacy of the detection of molecular complexes in these assays). Furthermore, these results did not address how these events may occur in a spatiotemporally organized way in living cells (Simons and Vaz, 2004). The single-molecule tracking results obtained in the present study contributed greatly to clarifying this process, by providing a dynamic view of these events as itemized above, although there are still many unknown processes and mechanisms.
Individual, short recruitment of PLCγ2 may produce a unit signal for bulk IP3 signaling
The cytoplasmic IP3 signal, as observed by conventional fluorescence imaging, lasts for at least 15 min, or on the order of 1,000 s (Figs. 1 and 2). Such a bulk signal must be generated by the superposition of the individual events of PLCγ2 recruitment to the plasma membrane, which occurs at the CD59 clusters undergoing STALL, producing 20–50 IP3 molecules for each PLCγ2 recruitment period. The cytoplasmic IP3 signal amplitude (concentration) at a given time may largely be determined by the collective recruitment/activity of thousands of PLCγ2 copies (consumption and long-term accumulation of IP3 might occur). Therefore, the short recruitment period of ∼0.25 s for each single PLCγ2 molecule at the CD59 cluster in STALL, which is at least 4,000-fold shorter than the overall duration of the bulk IP3 signal, might initially seem surprising.
However, such pulse-like occurrences of individual signaling events might address a long-standing problem in the field of cellular signaling, as raised from the viewpoint of communication system engineering (Oliver et al., 1948) or systems biology (Shimizu and Bray, 2001). Here is the enigma. The recruitment/activation of each individual molecule has been believed to last for a duration comparable to (or perhaps 10 times less than that of) the bulk duration. For the sake of simplicity, assume the overall activation duration to be 1,000 s and the durations for recruitment/activation of each individual molecule to be on the order of 100–1,000 s. Generally, as stated at the beginning of this section, the activation level of the bulk signal is determined by the superposition (integration) of the recruitment/activation of thousands of copies of the signaling molecule. However, this integration presents a difficult problem for the cell, which must regulate and maintain the proper, stable level of the overall activation of the signaling event (see Model A [complex integration model] in Fig. 8). This is because the active durations of individual molecules are assumed to be long, on the order of 100–1,000 s for the overall activation period of 1,000 s: to reach the correct level of bulk activation, the activation of the next copy of the molecule must be done on the basis of the correct prediction regarding when the previously activated copies of the molecule are turned off. How the cell can achieve such a complicated integration over many thousands of molecules has remained enigmatic.
In the case of IP3 signaling examined here, each individual elementary process for IP3 production takes place like a quantized burst (∼0.25 s), which is shorter by a factor of at least 4,000 than the bulk activation period. If this were the case, for realizing the proper, stable level of the bulk IP3 concentration, complicated integration would be unnecessary: the simple addition of the IP3 pulses would make the bulk signal, as shown in Model B (simple summation model) in Fig. 8 (for brevity, the signal amplitude by each individual event is assumed to be the same here; Oliver et al., 1948). Therefore, the short lifetime of each recruitment event of PLCγ2 at a CD59 cluster, and thus the briefness of each burst of IP3 production, may be critical for the regulation of the enhanced level of IP3 upon the engagement of CD59.
The analogue amplitude of the bulk IP3 signal is basically proportional to the number of PLCγ2 recruitment events during the unit time (and thus the number of IP3 bursts per unit time). If the rate of IP3 consumption is slow, the bulk amplitude is proportional to the cumulative number of PLCγ2 recruitment until the specified time; in this sense, to obtain the bulk amplitude, the loss of IP3 has to be evaluated. Nevertheless, the digital, pulse-like signal produced by each IP3 production event greatly simplifies the signaling system.
Finally, we would emphasize that the observations of STALL of CD59, the individual recruitment events of single molecules of Gαi2, Lyn, and PLCγ2 to a single CD59 cluster, and their short residency times all became possible for the first time by using single-molecule tracking techniques. They provide unique, valuable information for studies of signal transduction mechanisms.
Materials and methods
Cell culture, drug treatments, and cDNA transfection
T24 (human) and PtK2 (rat kangaroo) epithelial cells and NRK (rat) fibroblastic cells were cultured in HAM's F12 medium (Invitrogen) supplemented with 10% FBS. Transfection was performed using Lipofectamine Plus (Life Technologies) according to the manufacturer's recommendations. The NRK cells were transfected with the cDNA for human CD59 (a gift from M. Tone, Oxford University, Oxford, England). NRK cells express CD59 of their own species, but to carry out experiments under similar clustering conditions, they were transfected with the human cDNA for CD59.
Partial depletion of cholesterol in the plasma membrane was performed by incubating the cells in 4 mM MβCD (Sigma-Aldrich) at 37°C for 30 min (Kilsdonk et al., 1995) or in 60 μg/ml saponin (Sigma-Aldrich) on ice for 15 min (Cerneus et al., 1993). These treatments substantially increased the amount of CD59 recovered in the detergent-soluble fractions in the protocol to prepare detergent-resistant membranes. Replenishment of cholesterol was performed by incubating the cholesterol-depleted cells in 10 mM MβCD–cholesterol complex (1:1) for 30 min at 37°C (Shigematsu et al., 2003). The overall amounts of cholesterol per cell after cholesterol depletion with MβCD and after the subsequent repletion were found to be 66 and 118% of the original amount (SD of ±6%), as determined by a cholesterol quantification kit (Wako). Partial actin depolymerization was performed by incubating the cells in the medium containing 50 nM latrunculin B for 10 min (gifts from G. Marriott, University of Wisconsin–Madison, Madison, WI; Spector et al., 1983). SFKs were inhibited by treating the cells with 10 μM PP2 (Calbiochem) for 5 min at 37°C (Hanke et al., 1996). Heterotrimeric G protein was inhibited by incubating the cells in medium containing 1.7 nM PTX (Calbiochem) at 37°C for 22 h (Gomez-Mouton et al., 2004). The involvement of the IP3 receptor in the GPI-AR–induced Ca2+ mobilization was examined by injecting 10 mg/ml heparin (∼100 femtoliter; Sigma-Aldrich) into the cells (Takei et al., 1998), using a micromanipulator/injector (Eppendorf).
As a control for GPI anchoring, a transmembrane chimeric protein of CD59 was used (mycCD59TM; its cDNA was provided by M. Maio, Instituto Nazionale di Ricovero e Cura a Carattere Scientifico, Ancona, Italy): the CD59 ectodomain was fused with an N-terminal myc tag and a C-terminal LDL receptor transmembrane domain, which additionally contains the 12 amino acids from the N terminus of the cytoplasmic domain of the LDL receptor (and thus lacks the sequence required for internalization via coated pits; De Nardo et al., 2002). IgG-gold particles coated with anti-myc antibody (9E10.2) were used for cross-linking mycCD59TM.
Immunofluorescence colocalization experiments for IgG-gold particles and PLCγ2 spots
Immunofluorescence staining of IgG-gold particles, and their colocalization with immunofluorescent spots of PLCγ2, was examined in the following way. Cells were incubated with IgG-gold for 5 min at 37°C, fixed with 4% paraformaldehyde for 90 min at room temperature, and permeabilized with 0.01% Triton X-100 in PBS for 1 min. After blocking with 5% skim milk for 90 min, the cells were immunostained with the rabbit anti-PLCγ2 antibodies (BD Biosciences). Fluorescein-conjugated goat anti–mouse IgG antibodies were used for the staining of the IgG-gold particles.
Preparation of gold and fluorescent probes, single-particle tracking, single fluorescent molecule tracking, and STALL detection
These are described in detail in Suzuki et al. (2007). STALL (transient confinement zone) was detected in gold-probe trajectories recorded at a 33-ms resolution for a period of 10 s, following the method developed by Simson et al. (1995). The only difference is the definition of the size of the area covered by a CD59 cluster during STALL. It was estimated by the 2D Gaussian fitting of the determined coordinates of the CD59 cluster during the STALL period.
Observation of the recruitment of single molecules of GFP-PLCγ2 to the CD59 cluster
To observe the recruitment of PLCγ2 to CD59 clusters in live cells, T24 cells were transiently transfected with the cDNA for PLCγ2 fused with GFP (at the N terminus of PLCγ2; obtained from M. Katan [The Institute of Cancer Research, London, England] and slightly modified; Matsuda et al., 2001). For the simultaneous tracking of single CD59 clusters and single molecules of PLCγ2, CD59 clusters were formed by using 50-nm latex beads coated with anti-CD59 whole IgG (a gift from V. Horejsi, Academy of Sciences of the Czech Republic, Prague, Czech Republic), because the 40-nm gold particles gave signals that could not be separated from the fluorescence signals from GFP, at the level of single molecules and single particles. These two types of particles exhibited practically the same STALL time fractions and durations. Furthermore, as shown in the left box in Fig. 2 b and the fourth trace from the bottom in Fig. 3 b, these 50-nm beads are capable of inducing intracellular signals as effectively as 40-nm IgG-gold particles.
The bright-field images of the 50-nm latex beads (forming CD59 clusters beneath them) were obtained simultaneously with the images of GFP-tagged single signaling molecules, using the same conditions and instrument described in Suzuki et al. (2007). Determination of colocalization is also described in Suzuki et al. (2007).
Epifluorescence imaging of the increase of IP3 in living cells
The increase in the cytoplasmic IP3 concentration after the addition of IgG-gold particles was observed in cultured T24 cells transfected with the cDNA for the PH domain of PLCδ1 fused with GFP at the C terminus (a gift from K. Hirose, Nagoya University Medical School, Nagoya, Japan; Hirose et al., 1999; Wang et al., 2004). This PH domain binds to both IP3, which is located in the cytoplasm, and PIP2, which is located on the inner leaflet of the plasma membrane. Upon the engagement of CD59, PLCγ starts hydrolyzing PIP2 in the membrane to generate IP3, and thus the cytoplasmic IP3 concentration increases (Hirose et al., 1999). This can be detected as the relative increase in the fluorescence signal of PH domain–GFP in the cytoplasm (ICyt) versus that in the plasma membrane (IPM; Hirose et al., 1999). This change in the fluorescence signal, detected by epifluorescence microscopy, was monitored by measuring the ICyt/IPM ratio, as described by Raucher and Sheetz (2001). The increase in the cytoplasmic IP3 concentration was also observed by confocal fluorescence microscopy, using a microscope (TE300; Nikon) equipped with a spinning-disc confocal scanner system (CSU22; Yokogawa) and a cooled charge-coupled device camera (Cascade 650; Roper Scientific).
Epifluorescence imaging of Ca2+ mobilization in living cells
Intracellular Ca2+ mobilization in living cells was monitored by epifluorescence microscopy, using fluo-4 or rhod-2 (Invitrogen) as a probe (Minta et al., 1989). T24 cells were incubated in HBSS containing 5 μM fluo-4 AM (Invitrogen) for 30 min at room temperature, washed once with fresh HBSS, and incubated with the IgG-gold or Fab-gold particles. When the effect of DOPE cross-linking on Ca2+ mobilization was examined, fluorescein-conjugated DOPE was incorporated in the cells and then cross-linked by IgG-gold particles coated with anti-fluorescein antibodies. Because of the spectral overlap, fluo-4 could not be used to observe the effect of cross-linking (fluorescein-conjugated) DOPE. Therefore, rhod-2 AM (Invitrogen) was used (Minta et al., 1989).
Online supplemental material
Fig. S1 shows the increase of the cytoplasmic IP3 concentration induced by CD59 clusters, as observed by the confocal fluorescence microscopy of the PH domain (from PLCδ1)–GFP expressed in T24 cells. Video 1 shows simultaneous observation of a CD59 cluster and the recruitment of single molecules of GFP-PLCγ2, recorded at video rate. Video 2 provides a model for the signal transduction of CD59, leading to the intracellular IP3–Ca2+ signal. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200609175/DC1.
Supplementary Material
Acknowledgments
We thank V. Horejsi, M. Tone, M. Maio, M. Katan, K. Hirose, and G. Marriott for providing the hybridoma for the production of the anti-CD59 monoclonal antibody, as well as the anti-DAF mouse monoclonal antibody, and the cDNAs for human CD59, CD59TM, and PLCγ2, and the PH domain of PLCδ fused to GFP, and latrunculin B, respectively, and J. Kondo for preparing the figures.
This research was supported in part by National Institutes of Health grants DK44375 and AI14584 (to M. Edidin) and grants-in-aid for scientific research and those on priority areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to A. Kusumi).
Abbreviations used in this paper: DAF, decay accelerating factor; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; GPI-AR, glycosylphosphatidylinositol-anchored receptor; IP3, inositol-(1,4,5) triphosphate; LDL, low-density lipoprotein; MβCD, methyl-β-cyclodextrin; PH, pleckstrin homology; PIP2, phosphatidylinositol-bis(4,5)phosphate; PLAP, placental alkaline phosphatase; PTX, pertussis toxin; SFK, Src-family kinase; STALL, stimulation-induced temporary arrest of lateral diffusion.
References
- Brown, D.A., and J.K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- Brugger, B., C. Graham, I. Leibrecht, E. Mombelli, A. Jen, F. Wieland, and R. Morris. 2004. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 279:7530–7536. [DOI] [PubMed] [Google Scholar]
- Carpenter, G., and Q. Ji. 1999. Phospholipase C-γ as a signal-transducing element. Exp. Cell Res. 253:15–24. [DOI] [PubMed] [Google Scholar]
- Cerneus, D.P., E. Ueffing, G. Posthuma, G.J. Strous, and A. van der Ende. 1993. Detergent insolubility of alkaline phosphatase during biosynthetic transport and endocytosis. Role of cholesterol. J. Biol. Chem. 268:3150–3155. [PubMed] [Google Scholar]
- del Pozo, M.A., N.B. Alderson, W.B. Kiosses, H.H. Chiang, R.G. Anderson, and M.A. Schwartz. 2004. Integrins regulate Rac targeting by internalization of membrane domains. Science. 303:839–842. [DOI] [PubMed] [Google Scholar]
- De Nardo, C., E. Fonsatti, L. Sigalotti, L. Calabro, F. Colizzi, E. Cortini, S. Coral, M. Altomonte, and M. Maio. 2002. Recombinant transmembrane CD59 (CD59-TM) confers complement resistance to GPI-anchored protein defective melanoma cells. J. Cell. Physiol. 190:200–206. [DOI] [PubMed] [Google Scholar]
- Douglass, A.D., and R.D. Vale. 2005. Single-molecule microscopy reveals plasma microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 121:937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, T., K. Ritchie, H. Murakoshi, K. Jacobson, and A. Kusumi. 2002. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullekrug, J., and K. Simons. 2004. Lipid rafts and apical membrane traffic. Ann. N. Y. Acad. Sci. 1014:164–169. [DOI] [PubMed] [Google Scholar]
- Fra, A.M., E. Williamson, K. Simons, and R.G. Parton. 1994. Detergent-insoluble glycolipid microdomains in lymphocytes in the absence of caveolae. J. Biol. Chem. 269:30745–30748. [PubMed] [Google Scholar]
- Glebov, O.O., and B.J. Nichols. 2004. Lipid raft proteins have a random distribution during localized activation of the T-cell receptor. Nat. Cell Biol. 6:238–243. [DOI] [PubMed] [Google Scholar]
- Gomez-Mouton, C., R.A. Lacalle, E. Mira, S. Jimenez-Baranda, D.F. Barber, A.C. Carrera, and A.C. Martinez. 2004. Dynamic redistribution of raft domains as an organizing platform for signaling during cell chemotaxis. J. Cell Biol. 164:759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gri, G., B. Molon, S. Manes, T. Pozzan, and A. Viola. 2004. The inner side of T cell lipid rafts. Immunol. Lett. 94:247–252. [DOI] [PubMed] [Google Scholar]
- Hancock, J.F. 2006. Lipid rafts: contentious only from simplistic standpoints. Nat. Rev. Mol. Cell Biol. 7:456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, J.F., and R.G. Parton. 2005. Ras plasma membrane signalling platforms. Biochem. J. 389:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke, J.H., R.L. Dow, P.S. Changelian, W.H. Brissette, E.J. Weringer, B.A. Pollok, and P.A. Connelly. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 271:695–701. [DOI] [PubMed] [Google Scholar]
- Harder, T., and K. Simons. 1999. Clusters of glycolipid and glycosylphosphatidylinositol-anchored proteins in lymphoid cells: accumulation of actin regulated by local tyrosine phosphorylation. Eur. J. Immunol. 29:556–562. [DOI] [PubMed] [Google Scholar]
- Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose, K., S. Kadowaki, M. Tanabe, H. Takashima, and M. Iino. 1999. Spatiotemporal dynamics of inositol 1,4,5-triphosphate that underlies complex Ca2+ mobilization patterns. Science. 284:1527–1530. [DOI] [PubMed] [Google Scholar]
- Holowka, D., J.A. Gosse, A.T. Hammond, X. Han, P. Sengupta, N.L. Smith, A. Wagenknecht-Wiesner, M. Wu, R.M. Young, and B. Baird. 2005. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim. Biophys. Acta. 1746:252–259. [DOI] [PubMed] [Google Scholar]
- Iino, R., I. Koyama, and A. Kusumi. 2001. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 80:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin, H.E. 1977. Absence of the eighth component of complement in association with systemic lupus erythematosus-like disease. J. Clin. Invest. 60:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy, A.K., B.J. Nichols, C.L. Remmert, G.M. Hendrix, M. Kumar, J. Zimmerberg, and J. Lippincott-Schwartz. 2004. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 165:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilsdonk, E.P., P.G. Yancey, G.W. Stoudt, F.W. Bangerter, W.J. Johnson, M.C. Phillips, and G.H. Rothblat. 1995. Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 270:17250–17256. [DOI] [PubMed] [Google Scholar]
- Kimberley, F.C., B. Sivasankar, and B.P. Morgan. 2007. Alternative roles for CD59. Mol. Immunol. 44:73–81. [DOI] [PubMed] [Google Scholar]
- Kmiecik, T.E., and D. Shalloway. 1987. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 49:65–73. [DOI] [PubMed] [Google Scholar]
- Koyama-Honda, I., K. Ritchie, T. Fujiwara, R. Iino, H. Murakoshi, R.S. Kasai, and A. Kusumi. 2005. Fluorescence imaging for monitoring the colocalization of two single molecules in living cells. Biophys. J. 88:2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi, A., and K. Suzuki. 2005. Toward understanding the dynamics of membrane-raft-based molecular interactions. Biochim. Biophys. Acta. 1746:234–251. [DOI] [PubMed] [Google Scholar]
- Kwik, J., S. Boyle, D. Fooksman, L. Margolis, M.P. Sheetz, and M. Edidin. 2003. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. USA. 100:13964–13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson, D.R., J.A. Gasse, D.A. Holowka, B.A. Baird, and W.W. Webb. 2005. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell Biol. 171:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomasney, J.W., H.-F. Cheng, L.P. Wang, Y.S. Kuan, S.M. Liu, S.W. Feslk, and K. King. 1996. Phosphatidylinositol 4,5-bisphosphate binding to the pleckstrin homology domain of phospholipase C-δ1 enhances enzyme activity. J. Biol. Chem. 271:25316–25326. [DOI] [PubMed] [Google Scholar]
- Ma, Y.C., J. Huang, S. Ali, W. Lowry, and X.Y. Huang. 2000. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 102:635–646. [DOI] [PubMed] [Google Scholar]
- Maschek, B.J., W. Zhang, P.M. Rosoff, and H. Reiser. 1993. Modulation of the intracellular Ca2+ and inositol triphosphate concentrations in murine T lymphocytes by the glycosylphosphatidylinositol-anchored protein sgp-60. J. Immunol. 150:3198–3206. [PubMed] [Google Scholar]
- Mashanov, G.I., D. Tacon, M. Peckham, and J.E. Molloy. 2004. The spatial and temporal dynamics of pleckstrin homology domain binding at the plasma membrane measured by imaging single molecule in live mouse myoblasts. J. Biol. Chem. 279:15274–15280. [DOI] [PubMed] [Google Scholar]
- Matsuda, M., H.F. Paterson, R. Rodriguez, A.C. Fensome, M.V. Ellis, K. Swann, and M. Katan. 2001. Real time fluorescence imaging of PLCγ translocation and its interaction with the epidermal growth factor receptor. J. Cell Biol. 153:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall, R.D., C. Tiruppathi, S.M. Vogel, W.D. Niles, A. Gilchrist, H.E. Hamm, and A.B. Malik. 2000. Endothelial cell-surface gp60 activates vesicle formation and trafficking via Gi-coupled Src kinase signaling pathway. J. Cell Biol. 150:1057–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minta, A. J.P. Kao, and R.Y. Tsien. 1989. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 264:8171–8178. [PubMed] [Google Scholar]
- Miotti, S., M. Bagnoli, A. Tomassetti, M.I. Colnaghi, and S. Canevari. 2000. Interaction of folate receptor with signaling molecules lyn and Gαi-3 in detergent-resistant complexes from the ovary carcinoma cell line IGROV1. J. Cell Sci. 113:349–357. [DOI] [PubMed] [Google Scholar]
- Morgan, B.P., C.W. van den Berg, E.V. Davies, M.B. Hallett, and V. Horejsi. 1993. Cross-linking of CD59 and of other glycosyl phosphatidylinositol-anchored molecules on neutrophils triggers cell activation via tyrosine kinase. Eur. J. Immunol. 23:2841–2850. [DOI] [PubMed] [Google Scholar]
- Mukherjee, S., and F.R. Maxfield. 2004. Membrane domains. Annu. Rev. Cell Dev. Biol. 20:839–866. [DOI] [PubMed] [Google Scholar]
- Munro, S. 2003. Lipid rafts: elusive or illusive? Cell. 115:377–388. [DOI] [PubMed] [Google Scholar]
- Murase, K., T. Fujiwara, Y. Umemura, K. Suzuki, R. Iino, H. Yamashita, M. Saito, H. Murakoshi, K. Ritchie, and A. Kusumi. 2004. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys. J. 86:4075–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolau, D.V., Jr., K. Burrage, R.G. Parton, and J.F. Hancock. 2006. Identifying optimal lipid raft characteristics required to promote nanoscale protein-protein interactions on the plasma membrane. Mol. Cell. Biol. 26:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, B.M., J.R. Pierce, and C.E. Shannon. 1948. The philosophy of PCM. Proceedings of the I.R.E. 36:1324–1331. [Google Scholar]
- Omidvar, N., E.C. Wang, P. Brennan, M.P. Longhi, R.A. Smith, and B.P. Morgan. 2006. Expression of glycosylphosphatidylinositol-anchored CD59 on target cells enhances human NK cell-mediated cytotoxicity. J. Immunol. 176:2915–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino, S., D. Sarnataro, R. Pillich, S. Tivodar, L. Nitsch, and C. Zurzolo. 2004. Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. J. Cell Biol. 167:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer, I., A.L. Servin, and M.-F. Bernet-Camard. 1998. Piracy of decay-accelerating factor (CD55) signal transduction by the diffusely adhering strain Escherichia coli C1845 promotes cytoskeletal F-actin rearrangements in cultured human intestinal INT407 cells. Infect. Immun. 66:4036–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini, L.M., R.J. Eddy, M. Fuortes, S. Seveau, C. Casulo, and F.R. Maxfield. 2003. Membrane lipid organization is critical for human neutrophil polarization. J. Biol. Chem. 278:10831–10841. [DOI] [PubMed] [Google Scholar]
- Pizzo, P., E. Giurisato, M. Tassi, A. Benedetti, T. Pozzan, and A. Viola. 2002. Lipid rafts and T cell receptor signaling: a critical re-evaluation. Eur. J. Immunol. 32:3082–3091. [DOI] [PubMed] [Google Scholar]
- Plowman, S., C. Muncke, R.G. Parton, and J.F. Hancock. 2005. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 102:15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin, B., F. Sekiya, and S.G. Rhee. 2005. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc. Natl. Acad. Sci. USA. 102:4276–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior, I., A. Harding, J. Yan, J. Sluimer, R.G. Parton, and J.F. Hancock. 2001. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 3:368–375. [DOI] [PubMed] [Google Scholar]
- Pyenta, P.S., D. Holowka, and B. Baird. 2001. Cross-correlation analysis of inner-leaflet-anchored green fluorescent protein co-redistributed with IgE receptors and outer leaflet lipid raft components. Biophys. J. 80:2120–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucher, D., and M.P. Sheetz. 2001. Phospholipase C activation by anesthetics decreases membrane-cytoskeleton adhesion. J. Cell Sci. 114:3759–3766. [DOI] [PubMed] [Google Scholar]
- Rodriguez, R., M. Matsuda, A. Storey, and M. Katan. 2003. Requirements for distinct steps of phospholipase Cgamma2 regulation, membrane-raft- dependent targeting and subsequent enzyme activation in B-cell signalling. Biochem. J. 374:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya, K., T. Abe, and T. Fujita. 1992. Decay-accelerating factor functions as a signal transducing molecule for human monocytes. J. Immunol. 149:1758–1762. [PubMed] [Google Scholar]
- Shigematsu, S., R.T. Watson, A.H. Khan, and J.E. Pessin. 2003. The adipocyte plasma membrane caveolin function/structural organization is necessary for the efficient endocytosis of GLUT4. J. Biol. Chem. 278:10683–10690. [DOI] [PubMed] [Google Scholar]
- Shimizu, T.S., and D. Bray. 2001. Computational cell biology—The stochastic approach. In Foundations of Systems Biology. H. Kitano, editor. MIT Press, Cambridge, MA. 213–232.
- Simons, K., and W.L. Vaz. 2004. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 33:269–295. [DOI] [PubMed] [Google Scholar]
- Simson, R., E.D. Sheets, and K. Jacobson. 1995. Detection of temporary lateral confinement of membrane proteins using single-particle tracking analysis. Biophys. J. 69:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon, K.R., E.R. Christopher, and R.W. Finberg. 1996. The association between glycosylphosphatidylinositol-anchored proteins and heterotrimeric G protein a subunits in lymphocytes. Proc. Natl. Acad. Sci. USA. 93:6053–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector, I., N.R. Shochet, Y. Kashman, and A. Groweiss. 1983. Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science. 219:493–495. [DOI] [PubMed] [Google Scholar]
- Stefanova, I., V. Horejsi, I.J. Ansotegui, W. Knapp, and H. Stockinger. 1991. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 254:1016–1019. [DOI] [PubMed] [Google Scholar]
- Stulnig, T.M., M. Berger, T. Sigmund, H. Stockinger, V. Horejsi, and W. Waldhausl. 1997. Signal transduction via glycosyl phosphatidylinositol-anchored proteins in T cells is inhibited by lowering cellular cholesterol. J. Biol. Chem. 272:19242–19247. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., and M.P. Sheetz. 2001. Binding of cross-linked glycosylphosphatidylinositol-anchored proteins to discrete actin-associated sites and cholesterol-dependent domains. Biophys. J. 81:2181–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K., K. Ritchie, E. Kajikawa, T. Fujiwara, and A. Kusumi. 2005. Rapid hop diffusion of a G-protein-coupled receptor in the plasma membrane as revealed by single-molecule techniques. Biophys. J. 88:3659–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K.G.N., T.K. Fujiwara, F. Sanematsu, R. Iino, M. Edidin, and A. Kusumi. 2007. GPI-anchored receptor clusters transiently recruit Lyn and Gα for temporary cluster immobilization and Lyn activation: single-molecule tracking study 1. J. Cell Biol. 177:717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei, K., R.M. Shin, T. Inoue, K. Kato, and K. Mikoshiba. 1998. Regulation of nerve growth mediated by inositol 1, 4, 5-triphosphate receptors in growth cones. Science. 282:1705–1708. [DOI] [PubMed] [Google Scholar]
- Veri, M.C., K.E. DeBell, M.C. Seminario, A. DiBaldassarre, I. Reischl, R. Rawat, L. Graham, C. Noviello, B.L. Rellahan, S. Miscia, et al. 2001. Membrane raft-dependent regulation of phospholipase Cgamma-1 activation in T lymphocytes. Mol. Cell. Biol. 21:6939–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, M.I., G.A. Jones, S. Nishibe, S.G. Rhee, and G. Carpenter. 1992. Growth factor stimulation of phospholipase C-gamma 1 activity. J. Biol. Chem. 267:10447–10456. [PubMed] [Google Scholar]
- Wang, T.Y., R. Leventis, and J.R. Silvius. 2005. Artificially lipid-anchored proteins can elicit clustering-induced intracellular signaling events in Jurkat T-lymphocytes independent of lipid raft association. J. Biol. Chem. 280:22839–22846. [DOI] [PubMed] [Google Scholar]
- Wang, Y.J., W.H. Li, J. Wang, K. Xu, P. Dong, X. Luo, and H.L. Yin. 2004. Critical role of PIP5KIγ87 in InsP3-mediated Ca2+ signaling. J. Cell Biol. 167:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, M.A., and R.G. Anderson. 2005. Signaling networks in living cells. Annu. Rev. Pharmacol. Toxicol. 45:587–603. [DOI] [PubMed] [Google Scholar]
- Young, R.M., X. Zheng, D. Holowka, and B. Baird. 2005. Reconstitution of regulated phosphorylation of FcɛRI by a lipid raft-excluded protein-tyrosine phosphatase. J. Biol. Chem. 280:1230–1235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}