

Abstract
TRAPPI is a large complex that mediates the tethering of COPII vesicles to the Golgi (heterotypic tethering) in the yeast Saccharomyces cerevisiae. In mammalian cells, COPII vesicles derived from the transitional endoplasmic reticulum (tER) do not tether directly to the Golgi, instead, they appear to tether to each other (homotypic tethering) to form vesicular tubular clusters (VTCs). We show that mammalian Bet3p (mBet3p), which is the most highly conserved TRAPP subunit, resides on the tER and adjacent VTCs. The inactivation of mBet3p results in the accumulation of cargo in membranes that colocalize with the COPII coat. Furthermore, using an assay that reconstitutes VTC biogenesis in vitro, we demonstrate that mBet3p is required for the tethering and fusion of COPII vesicles to each other. Consistent with the proposal that mBet3p is required for VTC biogenesis, we find that ERGIC-53 (VTC marker) and Golgi architecture are disrupted in siRNA-treated mBet3p-depleted cells. These findings imply that the TRAPPI complex is essential for VTC biogenesis.
Introduction
Vesicle traffic from the ER to the Golgi complex is mediated by the sequential action of the COPII and COPI coat protein complexes (Aridor et al., 1995; Presley et al., 1997; Scales et al., 1997). COPII (coat protein II) vesicles form at the transitional ER (tER), which is a specialized subdomain of the ER that is distinct from the rough ER (Bannykh et al., 1996). These vesicles may tether and fuse to each other (homotypic fusion) to form the vesicular tubular clusters (VTCs), which is a pre-Golgi structure (also called the intermediate compartment) that lies adjacent to the tER (Aridor et al., 1995; Presley et al., 1997; Scales et al., 1997; Stephens et al., 2000; Xu and Hay, 2004). COPI (coat protein I) is then recruited onto VTCs before coated structures move cargo to the Golgi apparatus (Presley et al., 1997; Scales et al., 1997; Ben-Tekaya et al., 2005; Sannerud et al., 2006). Although components of the ER–Golgi trafficking machinery are highly conserved from yeast to man (Ferro-Novick and Jahn, 1994), no equivalent to a pre-Golgi compartment has been identified in the yeast Saccharomyces cerevisiae. COPII vesicle tethering/fusion has been reconstituted in vitro with yeast membrane fractions (Barlowe, 1997; Barrowman et al., 2000). However, in these assays, vesicles released from the ER in vitro are thought to fuse with Golgi membranes (heterotypic fusion).
Tethering factors have been proposed to mediate the initial interaction of a transport vesicle with its acceptor compartment and are implicated in maintaining the identity of organelles (Whyte and Munro, 2002). Tethers are either large oligomeric complexes or long coiled-coil proteins that activate or act downstream of the GTPases (Rabs) that regulate membrane traffic (Pfeffer, 1999; Whyte and Munro, 2002; Sztul and Lupashin, 2006). Most peripherally associate with membranes and are recruited directly onto the compartment on which they function (Whyte and Munro, 2002). Although many tethers have been identified, the mechanism of vesicle tethering has remained elusive. To address questions concerning the specificity of vesicle traffic and organelle identity, we have chosen to focus our studies on the tether called TRAPP. The 10 subunits of TRAPP were initially identified in the yeast S. cerevisiae (Sacher et al., 1998, 2000). TRAPP subunits are found in two complexes (TRAPPI and TRAPPII) that act at different stages of membrane traffic. TRAPPI acts in ER–Golgi traffic, whereas TRAPPII is required for traffic through the early endosome (Sacher et al., 2001; Cai et al., 2005).
Bet3p, which is the most highly conserved TRAPP component, is 54% identical to its mammalian counterpart and stably localizes to Golgi membranes (Sacher et al., 1998; Barrowman et al., 2000). In yeast, the temperature-sensitive bet3-1 mutant blocks ER–Golgi traffic at 37°C (Rossi et al., 1995). Although mammalian Bet3p (mBet3p) has been shown to act in ER–Golgi traffic after COPII vesicle budding and before Rab1, it is unknown if it participates in VTC biogenesis (Loh et al., 2005). We show that mBet3p is enriched at the tER and adjacent VTCs. Microinjected α-mBet3p results in the accumulation of cargo in structures that colocalize with the COPII coat. Furthermore, the inactivation of mBet3p blocks homotypic COPII vesicle tethering in vitro. These findings imply that mBet3p mediates the biogenesis of VTCs by linking COPII vesicles to each other.
Results
The localization of mBet3p is resistant to brefeldin A (BFA)
Previous studies identified a cytosolic pool of mBet3p (Sacher et al., 2000; Loh et al., 2005). However, the enrichment of this protein to a particular intracellular structure was not demonstrated in these studies. To begin to address the role of mBet3p in VTC biogenesis, we prepared polyclonal antibodies to this TRAPP subunit to determine the intracellular compartment where mBet3p resides.
Immunofluorescence microscopy revealed that mBet3p localizes to punctate structures in the perinuclear region of COS-7 cells (Fig. 1 A). This localization was not observed when antibody was pretreated to a nitrocellulose strip containing immobilized recombinant mBet3p (Fig. 1 A). In HeLa cells, the localization of mBet3p was predominantly cytosolic. However, when the cytosolic pool of mBet3p was removed by digitonin before fixation, its perinuclear localization was revealed (unpublished data). The perinuclear localization of mBet3p was also observed in several other mammalian cell lines, including BSC-1 (Fig. 1 B) and NRK cells (Fig. 2). mBet3p was largely found adjacent to ERGIC-53, which is a marker for the pre-Golgi compartment (Schweizer et al., 1988), and COPI (Fig. 1 B). COPI is a heptameric coat complex that is recruited to pre-Golgi and early Golgi structures (Oprins et al., 1993). Interestingly, by immunofluorescence, mBet3p has a punctate appearance rather than a continuous ribbonlike pattern, which is typical of Golgi proteins.
Figure 1.

mBet3p partially colocalizes with markers of the pre-Golgi compartment. (A) Anti-mBet3p antibody specifically recognizes mBet3p. Affinity-purified α-mBet3p was mock treated, or pretreated with His6-mBet3p (mBet3p depleted). The antibody was then used to label endogenous mBet3p in COS-7 cells by indirect immunofluorescence. Nuclei were identified by DAPI staining. (B) Colocalization of endogenous mBet3p (red images) with ERGIC-53 (top, green) and COPI (bottom, green) to pre-Golgi and Golgi membranes in BSC-1 cells. See merged image on right. The insets are magnified in the next row. Bars, 10 μm.
Figure 2.

The localization of mBet3p is largely resistant to BFA. NRK cells were treated with 5 μg/ml BFA for 1 h, and the localization of mBet3p (left), COPI (top right), and Man II (bottom right) were determined by indirect immunofluorescence. Bars, 10 μm.
Although the Golgi disassembles in the presence of the drug BFA, we found that, surprisingly, the localization of mBet3p was largely resistant to BFA (Fig. 2, top and bottom). The observation that the localization of a component of a COPII tethering complex is resistant to BFA prompted us to examine if mBet3p is associated with the tER. The tER is a BFA-resistant subdomain of the ER that specializes in the formation of COPII vesicles (Saraste and Svensson, 1991; Bannykh et al., 1996; Ward et al., 2001). As a marker for the tER we used Sec31p, which is a subunit of the COPII coat (Barlowe et al., 1994 ). Sec31p-labeled structures were most abundant in the vicinity of the Golgi apparatus and largely colocalized with mBet3p (Fig. 3 A and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1).
Figure 3.

The localization of mBet3p is disrupted by reagents that are known to disrupt the tER. (A) The localization of mBet3p (left, red) largely overlaps (Merge) with Sec31p (middle, green) in BSC-1 cells. The insets are magnified on the bottom right. (B) BSC-1 cells, treated for 1 h with 10 μg/ml nocodazole, were fixed and stained with antibodies directed against mBet3p (red) and Sec31p (top, green), and mBet3p (red) and GM130 (bottom, green). The insets are magnified on the right. (C) The localization of mBet3p clusters in response to microinjected Sar1p H79G, which is a GTP-locked mutant. NRK cells were microinjected with 1.5 mg/ml Sar1p H79G. mBet3p (top), Man II (middle), and GM130 (bottom) were visualized by indirect immunofluorescence. All injected cells are marked with an asterisk. Bars, 10 μm.
mBet3p appears to be recruited to the tER region
To address the relationship of mBet3p to the tER, we asked if the localization of mBet3p was perturbed by conditions that are known to disrupt or alter tER sites. When cells are treated with the microtubule-disrupting agent nocodazole, the tER disperses and tER sites cluster near Golgi fragments (Hammond and Glick, 2000). Although the cytosolic pool of mBet3p appeared to increase in the presence of nocodazole, mBet3p largely remained associated with the tER (Fig. 3 B, top) and clustered near the Golgi marker GM130 (Fig. 3 B, bottom). The colocalization of mBet3p with the tER marker Sec31p is illustrated in Video 1 (available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1), which is a 3D reconstruction of confocal images obtained from serial sections. To determine if the localization of mBet3p is changed by other conditions that are known to alter the localization of the tER, we microinjected Sar1p H79G into cells. Sar1p H79G is the constitutively active GTP-bound form of Sar1p that blocks export from the ER (Kuge et al., 1994). In cells expressing Sar1p H79G, ER exit sites have been shown to cluster (Ward et al., 2001). Golgi glycosylation enzymes like mannosidase II (Man II) are recycled back to the ER (Seemann et al., 2000a), whereas the localization of the golgin GM130 remains unchanged (Fig. 3 C; Miles et al., 2001; Ward et al., 2001). Unlike Man II and GM130, mBet3p always clustered with the ER exit sites (Fig. 3 C).
Together, these findings imply that mBet3p is recruited to membranes at or near the tER. Like Sec31p, its localization is resistant to BFA and sensitive to reagents that block export from the ER or disrupt tER sites. To more precisely determine the localization of mBet3p, immuno-EM was performed. For this analysis, the tER was defined as a cluster of Sec31p-labeled (large gold particles) membranes. Immuno-EM demonstrated that COPII vesicles and COPII budding profiles (Fig. 4 A, arrow) contained mBet3p (small gold particles). The observed localization of mBet3p to the tER was significant, as quantitation revealed that the labeling density of mBet3p at the tER was 7.6–22 times higher than the labeling density of mBet3p over the ER and other organelles (Fig. 4 B). Although no significant labeling was observed on Golgi cisternae (Fig. 4 B), mBet3p was found on tubules with a dense lumen in proximity to multivesicular bodies (MVBs). Some of these tubules were connected to MVBs (Fig. 4 B and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1). This observation is consistent with the finding that there is more than one pool of mBet3p (Sacher et al., 2001; Loh et al., 2005). It is also consistent with our recent finding that the yeast TRAPPII complex, which also contains Bet3p, localizes to the early endosome (Cai et al., 2005).
Figure 4.

mBet3p is enriched at the tER and adjacent pre-Golgi membranes. (A) Immuno-EM in NRK cells confirmed that mBet3p (5-nm gold particles) resides on the tER region labeled with Sec31p (10-nm gold particles). The arrow points to a COPII vesicle containing mBet3p. Bar, 100 nm. (B) mBet3p resides on the tER, and not ER, membranes (see Materials and methods for a description of the quantitation). NE, nuclear envelope; PM, plasma membrane; mito, mitochondrial outer membrane; Golgi, Golgi cisternae; endo, endosomal structures.
mBet3p is required for COPII vesicle coisolation in vitro
In vitro transport studies have shown that mBet3p acts after COPII vesicle budding, but before the GTPase Rab1 (Loh et al., 2005). However, the role of mBet3p in VTC biogenesis was not addressed in these earlier studies. Homotypic COPII vesicle tethering and fusion have recently been reconstituted in vitro (Xu and Hay, 2004). Using this in vitro assay, we directly addressed whether mBet3p is required for the interaction of COPII vesicles with each other. For this experiment, COPII vesicle populations that were marked in two different ways were generated from permeabilized cells. One population of vesicles contained Myc-tagged temperature-sensitive vesicular stomatitis virus glycoprotein (ts045 VSV-Myc), and a second population of vesicles contained untagged radiolabeled VSV-G* (Fig. 5 A). After the vesicles were formed in a first-stage incubation, the permeabilized donor cells were removed and the differently marked vesicle populations were combined and incubated together in a second-stage incubation in the presence of cytosol and an ATP-regenerating system. VSV-G*–containing radiolabeled vesicles that interact with VSV-G–Myc vesicles were coisolated from the supernatant with anti-Myc antibody and detected by autoradiography. When the vesicles fused, heterotrimeric VSV-G that contained both VSV-G–Myc and VSV-G* was formed and detected by immunoprecipitation in the presence of detergent (Xu and Hay, 2004). Interestingly, α-mBet3p, but not control IgG, inhibited the coisolation of vesicles and their fusion when added to the second-stage incubation (Fig. 5 B). Furthermore, this inhibition was completely reversed when the antibody was added in the presence of excess recombinant His6-mBet3p (Fig. S2 B). In contrast, antibody directed against the ER–Golgi SNARE syntaxin-5 inhibited the fusion of COPII vesicles without dramatically affecting their coisolation (Fig. 5 B). In support of the proposal that mBet3p mediates homotypic COPII vesicle tethering, Western blot analysis revealed the presence of mBet3p on the reaction product (Fig. 5 C, bottom), nascent VTCs that also contained VSV-G– Myc (Fig. 5 C, top). The localization of mBet3p to nascent VTCs was specific, as mBet3p was not present on the untagged control or when vesicle budding was blocked with the GDP-locked form of Sar1 (Fig. 5 C, bottom).
Figure 5.

mBet3p links COPII vesicles to each other. (A) COPII vesicle coisolation in vitro. Vesicles marked in two different ways were released from permeabilized NRK cells during the stage I incubation. During the stage II incubation, vesicles tethered to each other and subsequently fused. (B) Anti-Bet3p antibody blocks the coisolation of COPII vesicles. Two COPII vesicle populations were formed in vitro in a first-stage incubation, as previously described (Xu and Hay, 2004). The two vesicle populations were then combined and preincubated together for 20 min on ice in the absence or presence of antibody. After the incubation, one sample was kept on ice, while the other samples were incubated at 32°C for 60 min in a second-stage incubation. 1, one population of vesicles was untagged instead of Myc-tagged; 2, incubated on ice for 60 min; 3, complete reaction incubated without antibody; 4, contained α-mBet3p (168 nM; ∼5μg per reaction); 5, contained α-syntaxin-5 (168 nM); 6, contained rabbit IgG (168 nM). (C) Bet3p is present on nascent VTCs formed in vitro. Stage I reactions were performed in the presence or absence of GDP-locked Sar1 T39N. Lane 1, complete reaction; lane 2, complete reaction plus GDP-locked Sar1 T39N (800 nM); lane 3, vesicles were formed from NRK cells expressing untagged VSV-G. Vesicles released during stage I were then incubated in stage II (60 min at 32°C) and immunoisolated with anti-Myc antibody. The final immunoisolates were subjected to Western blot analysis to detect mBet3p (bottom) and VSV-Myc (top). (D) Anti-Bet3p antibody stimulates the release of COPII vesicles from permeabilized cells when added during the stage I incubation. COPII vesicle populations were formed in vitro using permeabilized NRK cells as in B, only the incubation was performed for 90 min (as described in Fig. 1 A of Xu and Hay, 2004). The permeabilized cells were removed by centrifugation, and the released vesicles were immunoisolated with an antibody to the integral membrane protein p24. The VSV-G* in the precipitate was quantified by autoradiography and reported as the percentage of the total VSV-G* in the starting cells. 1, complete reaction without antibody; 2, Sar1 T39N was included in the reaction at a concentration of 1 μM; 3, complete reaction plus 110 nM anti-mBet3 antibody. Error bars are the SEM.
A single-stage transport assay was also performed in the presence of α-mBet3p and vesicle release was monitored from the permeabilized cells. In this experiment, α-mBet3p was added to the cells in vitro before vesicles were formed. Studies in both mammalian cells and yeast have previously demonstrated that blocking the consumption of COPII vesicles increases their release from permeabilized cells in vitro (Barlowe, 1997; Xu and Hay, 2004). Consistent with the finding that COPII vesicles fail to fuse when mBet3p is inactivated, we observed that α-mBet3p stimulated the release of vesicles from the cells (Fig. 5 D). This finding also demonstrates that mBet3p is not required for vesicle budding and is consistent with a previous study that showed α-mBet3p acts after COPII vesicles bud from the ER in vitro (Sacher et al., 1998; Loh et al., 2005).
Accumulated cargo colocalizes with Sec31p when mBet3p is inactivated in vivo
In vitro, α-mBet3p blocks the conversion of VSV-G from an endo H–sensitive ER form to an endo H–resistant Golgi form (Loh et al., 2005). However, the role of mBet3p in vivo has not been addressed. To determine where cargo accumulates when mBet3p is inactivated in vivo, we monitored the trafficking of two different marker proteins, tsO45 VSV-G and the plasma membrane marker CD8. One marker was used to quantitate the trafficking defect when α-mBet3p was injected into cells, and the other marker was used to address where cargo accumulates. Quantitation of tsO45 VSV-G transport in HeLa cells revealed that the inactivation of mBet3p resulted in a nearly 70% decrease in the trafficking of YFP-tagged tsO45 VSV-G to the cell surface (Fig. 6 A).
Figure 6.

Cargo accumulates in structures that colocalize with COPII when mBet3p is inactivated in vivo. (A) The trafficking of YFP-tagged tsO45 VSV-G to the cell surface was quantitated in HeLa cells that were microinjected with α-mBet3p or control IgG, as described in the Materials and methods. Error bars are the SD. (B) Intracellular trafficking was monitored in BSC-1 cells that were injected with cDNA encoding CD8. After CD8 mRNA accumulated, the cells were injected with α-mBet3p or IgG. Subsequent to a 1-h incubation at 37°C, the cells were washed and the synthesis and transport of CD8 was continued for 3 h. Finally, CD8 was chased for 45 min in the presence of cycloheximide. The cells were stained for CD8, Sec31p, or Golgin-84. To visualize Sec31p and Golgin-84, antibodies were directly conjugated to Alexa Fluor 594 and viewed in the red channel. To visualize CD8, the secondary antibody was conjugated to Alexa Fluor 488 and viewed in the green channel. The inset shows the merge of CD8, Sec31p, and Golgin-84 in the α-mBet3p–injected cell. The merged image is on the right. Bar, 20 μm.
To address where cargo accumulates when mBet3p is inactivated, cDNA encoding CD8 was microinjected into the nuclei of BSC-1 cells. The CD8 mRNA was then allowed to accumulate in the presence of cycloheximide before α-mBet3p, or control IgG, was microinjected (Fig. 6 B). After 1 h, the cycloheximide was removed and CD8 was synthesized during a second incubation. Newly synthesized CD8 was then chased out of the ER during a final incubation in the presence of cycloheximide (Pelletier et al., 2000 ). In control cells, CD8 was found at the cell surface and the perinuclear region of the cell (Fig. 6 B; IgG control). Perinuclear CD8 colocalized with Golgin-84 in IgG injected cells (unpublished data). In the presence of α-mBet3p, Golgin-84 localization was fragmented. This finding is consistent with the observation that α-mBet3p disrupts the Golgi (see the next section). When mBet3p was inactivated, CD8 accumulated in spots that surround the nucleus. These spots largely colocalized with Sec31p, but were adjacent to Golgin-84–containing membranes (Fig. 6 B), implying that cargo accumulates in membranes that contain the COPII coat. These results are also consistent with previous studies showing that α-mBet3p blocks the trafficking of VSV-G before it is converted to an endo H–resistant form in the Golgi (Loh et al., 2005). Thus, both in vitro and in vivo trafficking studies support the hypothesis that mBet3p is required for the interaction of COPII vesicles with each other.
tER sites disperse and Golgi architecture is disrupted by perturbation of mBet3p
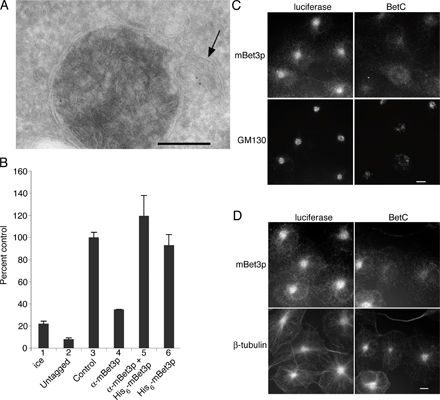
We also determined the consequences of depleting mammalian cells of mBet3p by specifically reducing its expression with siRNA. Two different siRNA duplexes were used for these studies, BetC (Fig. 7 A) and BetA (not depicted). Similar results were obtained with both siRNA duplexes. BetC, but not the control duplex (luciferase), reduced the expression of mBet3p (Fig. 7 A). The loss of mBet3p did not lead to a reduction of Sec31p or the ER protein ER60 (Fig. 7 A). However, the localization of Sec31p was dispersed, and its intense perinuclear localization, which marks the tER, was lost (Fig. 7 B). In addition, pre-Golgi and Golgi compartments were disrupted (Fig. 7 C and Fig. S2 C). The localization of β-tubulin, however, remained unaltered (Fig. S2 D). Similar effects on the Golgi were obtained when cells were micro-injected with α-mBet3p (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1).
Figure 7.

tER sites disperse and Golgi architecture is disrupted by perturbation of mBet3p. Western blot analysis revealed that siRNA targeted to mBet3p (BetC), but not luciferase, resulted in a decrease of mBet3p in COS-7 cells. (A) A >90% decrease of mBet3p, but not Sec31p or the ER protein ER60, was observed. Lane 1, mock-treated cells; lane 2, cells transfected with siRNA targeted to luciferase; lane 3, cells transfected with siRNA targeted to mBet3p. Similar results were obtained with BetA (not depicted) or BetC oligonucleotides. (B) The intense perinuclear localization of Sec31p is lost in cells depleted of mBet3p by RNAi. siRNA targeted to mBet3p (bottom) or luciferase as a control (top) were transfected into COS-7 cells. siRNA-transfected cells were stained with antibodies directed against mBet3p (left) and Sec31p (middle). The inset is magnified on the right. (C) The pre-Golgi compartment is disrupted in COS-7 cells transfected with mBet3-specific siRNA. siRNA targeted to luciferase (left) and mBet3 (right) were transfected into COS-7 cells and stained with α-mBet3p (top) and ERGIC-53 (bottom). (D) Golgi structure was disrupted in siRNA-treated HeLa cells that are depleted of mBet3p. EM analysis was performed on HeLa cells transfected with siRNA targeted to luciferase as a control (top) or mBet3p (BetC, bottom). The empty arrow points to a Golgi stack (top). Vesicles are marked with an arrow (bottom). The arrowhead marks a tubule (bottom). Mini Golgi stacks are labeled G (bottom). Bars: (B and C) 10 μm; (D) 200 nm.
EM analysis confirmed that the depletion of mBet3p disrupts Golgi architecture (Fig. 7 D, compare the Golgi [G] in the top and bottom images), and leads to a dramatic accumulation of vesicles (Fig. 7 D, arrows). Although tER was difficult to identify by EM in the siRNA-treated mBet3p-depleted cells, highly dilated rough ER was observed (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1). Quantification revealed a threefold decrease in the amount of membrane that represents Golgi cisternae. These findings provide in vivo evidence that mBet3p links COPII vesicles to each other at or near the tER, an event that appears to be required for the maintenance of Golgi structure.
Discussion
Although the late stages of ER–Golgi traffic have been visualized by live cell imaging (Presley et al., 1997; Scales et al., 1997), little is known about the homotypic fusion of COPII vesicles and how the pre-Golgi structure is formed from these vesicles. We provide evidence that mBet3p, the most highly conserved component of the TRAPP complex, plays a pivotal role in these events.
Although it was previously shown that the SNARE syntaxin-5 is required for homotypic vesicle fusion (Xu and Hay, 2004), the findings we report here are the first to link the tether TRAPP to VTC biogenesis. Our results imply that mBet3p is recruited to the tER region to mediate homotypic COPII vesicle tethering. Several lines of evidence support this proposal. First, we show that the localization of mBet3p is perturbed by conditions that are known to alter or disrupt the tER, suggesting that mBet3p may be recruited to membranes by one or more components of the tER. Second, mBet3p is required for homotypic COPII vesicle tethering in vitro and the inactivation of mBet3p in vivo leads to the accumulation of cargo in membranes that colocalize with the COPII subunit Sec31p. Finally, a role for mBet3p in VTC biogenesis is supported by data that shows the loss of mBet3p disrupts Golgi structure and the VTC marker ERGIC-53.
Immuno-EM revealed that mBet3p is enriched on more than one membrane, the tER region, and endosomal structures. Surprisingly, although yeast Bet3p was reported to reside on the Golgi (Sacher et al., 1998; Barrowman et al., 2000), we did not observe significant labeling of mBet3p on Golgi cisternae. In the yeast S. cerevisiae, tER, Golgi, and endosomal marker proteins are all found on punctate structures (Rossanese et al., 1999). Yeast Bet3p was originally reported to reside on the Golgi because a significant fraction of the Bet3p containing puncta enlarged in a sec7 mutant (Sacher et al., 1998), a putative Golgi proliferating mutant (Novick et al., 1980). However, our more recent studies suggest that Sec7p and yeast TRAPPII (which contains Bet3p) may reside on endosomal membranes (Cai et al., 2005). The localization of mBet3p to both the tER region and endosomal structures is consistent with studies in yeast showing that the two TRAPP complexes (TRAPPI and TRAPPII) act at different stages of membrane traffic.
Yeast TRAPPI, which contains Bet3p and several other subunits, is required for COPII vesicle tethering (Sacher et al., 1998; Barrowman et al., 2000). Because it is difficult to morphologically distinguish membranes in the ER–Golgi branch of the yeast secretory pathway, no equivalent to a pre-Golgi compartment has been identified, and COPII vesicles are thought to tether directly to the Golgi. In mammalian cells, COPII vesicles do not fuse directly with the Golgi, instead it is thought they fuse with each other to form VTCs (Xu and Hay, 2004). The recent finding that the ER–Golgi intermediate compartment is stable (Ben-Takaya et al., 2005; Sannerud et al., 2006), however, has raised the possibility that homotypic fusion is not exclusive and COPII vesicles may also heterotypically fuse with stable pre-Golgi membranes. Nonetheless, our in vitro and in vivo findings clearly show that mBet3p is required for the biogenesis of VTCs and Golgi membranes. The high degree of homology between yeast and mammalian Bet3p also raises the possibility that yeast COPII vesicles may undergo homotypic tethering and fusion before they fuse with the Golgi. Elucidation of the mechanism of COPII vesicle tethering in yeast and mammalian cells will be needed to resolve this issue. In the future it will also be important to address how mammalian TRAPP mediates subsequent steps in membrane traffic. Experiments are currently in progress to address these questions.
Materials and methods
Antibodies
Monoclonal antibody directed against GM130 was purchased from BD Biosciences. CD8 antibody, monoclonal antibodies directed against the COPI coat (CM1A10), the lumenal domain of VSV-G protein, rodent Man II (53FC3), and Golgin-84 were obtained from G. Warren (Yale University, New Haven, CT). Monoclonal antibody directed against ERGIC-53 was obtained from H.-P. Hauri (University of Basel, Basel, Switzerland). Anti–β-tubulin antibody (sc-5274) was purchased from Santa Cruz Biotechnology, Inc. Anti-ER60 antibody was obtained from P. Kim (University of Cincinnati, Cincinnati, OH). Monoclonal antibody directed against GalT (GTL-2) was obtained from D. Shima (Eyetech Research Center, Woburn, MA). Anti-Sec31p antibody (Shugrue et al., 1999) was obtained from F. Gorelick (Yale University, New Haven, CT), and polyclonal anti–Man II antibody was obtained from M. Farquhar (University of California, San Diego, San Diego, CA). Polyclonal rabbit anti-mBet3p antibody was previously described (Sacher et al., 2000). Rabbit IgG specific to mBet3p was purified on an Affigel column (Bio-Rad Laboratories) preloaded with His6-tagged Bet3p. Bound IgG was eluted with 0.1 M glycine, pH 2.5. For immunofluorescence, fluorescent-labeled secondary antibodies were purchased from Invitrogen.
Fluorescence microscopy
Cells grown on 10-mm coverslips were fixed in cold methanol for 5 min (for GTL-2 and CM1A10 antibodies only), or fixed in PBS with 3.7% formaldehyde for 15 min (for all other antibodies). Samples were quenched with serum-free DME, rinsed with buffer A (PBS with 5 mM EDTA, 0.25% BSA, and 0.05% NP-40) and permeabilized with buffer B (PBS with 5 mM EDTA, 0.25% BSA, 0.05% NP-40, and 0.1% Triton X-100) for 5 min. The samples were then treated with buffer C (PBS with 0.5% BSA) for 15 min. Antibodies were applied, and the samples were incubated at room temperature for 1 h. After four washes in buffer C, the samples were incubated with secondary antibody for 30 min. Four additional washes were performed in buffer C, and the samples were mounted on glass slides in Fluoromount G (Southern Biotech). Images were taken with an Axiophot fluorescence microscope using a 63× oil-immersion objective and a LSM 510 confocal microscope (both from Carl Zeiss MicroImaging, Inc.). Images were captured with a digital camera Orca ER; Hamamatsu Photonics). The images were processed with the OpenLab 3.08 imaging software (Improvision) and Photoshop 7.0 (Adobe).
To deplete α-mBet3p of determinants directed against mBet3p, affinity-purified antibody (1 μg/ml in buffer C) was incubated (with agitation) for 2 h with a nitrocellulose strip containing 60 μg of purified His6-tagged protein. Antibody was mock treated with a blank nitrocellulose strip as a control.
mBet3p was visualized with goat anti–rabbit secondary antibody conjugated to Cy3. GM130, COPI, β-tubulin, and ERGIC-53 were visualized with donkey anti–mouse secondary antibody conjugated to FITC. For double-labeling experiments with mBet3p and Sec31p, the Zenon labeling kit was used (Invitrogen). Samples were first stained with α-mBet3p and then stained for Sec31p, using this kit according to the manufacturer's instructions.
Microinjection experiments
Affinity-purified α-mBet3p, control IgG, or Sar1pDN was injected into COS-7 or NRK cells as previously described (Seemann et al., 2000a). The antibody- injected samples were fixed and stained after 2 h, whereas the Sar1pDN-injected samples were fixed and stained after 6 h.
Homotypic COPII vesicle tethering/fusion assay
Experiments were performed as previously described (Xu and Hay, 2004). The VSV-G–Myc–expressing cells and the pulse-radiolabeled VSV-G*–containing cells were permeabilized by scraping the cells. For the first-stage incubation, each reaction was 1,595 μl and contained 352 μl of water, 55 μl of 0.1 M magnesium acetate, 110 μl of an ATP-regenerating system, 33 μl of 1 M Hepes, pH 7.2, 220 μl of 20/18/50 buffer (20 mM Hepes, pH 7.2, 18 mM CaCl2, and 50 mM EGTA), 550 μl of rat liver cytosol dialyzed into 25/125 buffer (25 mM Hepes, pH 7.2, and 125 mM potassium acetate), and 275 μl of either semi-intact NRK cell population. Reactions were incubated at 32°C for 30 min (stage I incubation), centrifuged at 4,000 g for 1 min, and then centrifuged again at 15,000 g for 1 min. The supernatant, which contained the vesicles, was saved. In the stage II incubation, each reaction was 200 μl and contained 72.5 μl of VSV-G–Myc vesicles, 72.5 μl of VSV-G* vesicles, and 55 μl of 25/125 buffer or purified proteins (antibodies, GST, or GST-Sec23p) dissolved in 25/125 buffer. After a 20-min preincubation on ice, the reactions were incubated at 32°C for 60 min (stage II incubation). For coisolation assays, the final intact vesicle suspensions were processed for immunoisolation using biotinylated anti-Myc antibodies and streptavidin–Sepharose. Bound vesicles were analyzed by SDS-PAGE (8%) and autoradiography, and the coisolated VSV-G* was quantified. For heterotrimer assays, the final vesicle suspensions were incubated with 2% Triton X-100 for 20 min and then centrifuged at 100,000 g for 30 min. The 100,000 g supernatants containing solubilized VSV-G trimers were then processed for immunoprecipitation using biotinylated anti-Myc antibody and streptavidin–Sepharose, and the coprecipitating VSV-G* was quantified.
Trafficking assays
VSV-G transport was assayed as previously described (Diao et al., 2003) with several minor exceptions. HeLa cells were first injected with 1.5 mg/ml α-mBet3p or 1.5 mg/ml control IgG; 2 h later, plasmid encoding 0.2 mg/ml YFP-tagged tsO45 VSV-G was injected. Protein transported to the cell surface was detected with a monoclonal antibody to the lumenal domain of VSV-G that was conjugated to Alexa Fluor 647. Total VSV-G was detected by YFP. The extent of VSV-G transport to the cell surface was determined by the ratio of surface to total measured fluorescence, as previously described (Seemann et al., 2000b). A total of nine cells injected with either α-mBet3p antibody or IgG were quantitated. The trafficking of the plasma membrane marker CD8 was monitored, as previously described (Pelletier et al., 2000). Cells were injected with 1.5 mg/ml α-mBet3p or 1.5 mg/ml IgG after the CD8 plasmid was injected in the presence of 100 μg/ml cycloheximide. The incubation was then continued for 1 h at 37°C.
RNAi
siRNA oligonucleotides specific for human Bet3p were designed according to the method developed by Dharmacon Research, Inc. Two oligonucleotides, BetA (5′-UCACUCCAAGCAUUACUAAUU-3′) and BetC (5′-GGAGACGGUGUGACAGAAAUU-3′) were chosen because they are specific and target to regions that are conserved between human and monkey Bet3 sequences. Duplex RNA was introduced into COS-7 or HeLa cells, using OligofectAMINE (Invitrogen) according to the manufacturer's instructions. 3 d after transfection, the cells were analyzed. In three independent experiments, the reduction of mBet3p (determined by Western blot analysis) ranged from 74 to 99% using BetA, and from 87 to 99% for BetC. Samples were quantitated using Image 1.61/68 K software (National Institutes of Health, Bethesda, MD).
EM analysis
Immuno-EM was performed in NRK cells as previously described (Folsch et al., 2003). Estimation of labeling densities was determined on digital images captured using a charge-coupled device camera (Morada; Soft Imaging System). At a magnification of 43,000×, sections were scanned and 118 fields were captured at random. Transitional elements were defined as tubular vesicular clusters found in the proximity of ER membranes labeled for Sec31p (at least two gold particles). The labeling density for mBet3p was calculated by counting the number of 5-nm gold particles falling on or within 15 nm of membranes, and dividing these numbers by the length of the membranes expressed in micrometers. Length was estimated by counting the number of intersections of membranes with the vertical and horizontal lines of a grid at an estimated distance of 110 nm between lines (Weibel and Bolender, 1979).
HeLa cells were processed for EM analysis as previously described (Lopez et al., 2002), with the following two exceptions: cells were scraped after fixation, and the section thickness was 60 nm. Quantitation of Golgi membrane was done directly at the EM. Sections were scanned horizontally in a random fashion at a magnification of 9,900×, and the number of intersections of membrane with a fixed point were counted. For each sample, >900 intersections with the plasma membrane were counted. The number of intersections with stacked Golgi cisternae was divided by the number of intersections with the plasma membrane (or nuclear envelope). A threefold decrease in the ratio of Golgi to plasma membrane (or Golgi to nuclear envelope) was observed in the mBet3p-depleted sample. HeLa cells were used for this analysis because it was easier to obtain better cryosections with these cells than with COS-7 cells.
Online supplemental material
Figure S1 shows a still from Video 1. Fig. S2 shows that the loss of mBet3p blocks COPII vesicle tethering and disrupts Golgi architecture. Fig. S3 shows that microinjected α-mBet3p, but not control IgG, disrupts the pre-Golgi compartment marked by ERGIC-53 in COS-7 cells. Fig. S4 shows the rough ER is highly dilated in siRNA-treated mBet3p-depleted HeLa cells. Video 1 shows the colocalization of mBet3p and Sec31p in nocodazole-treated cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200603044/DC1.
Supplementary Material
Acknowledgments
We thank Lee Walker for help in the preparation of the manuscript and Yueyi Zhang for technical assistance. We also thank Graham Warren, Fred Gorelick, Marilyn Farquhar, David Shima, Hans-Peter Hauri, Paul Kim, Jaakko Saraste, Derek Toomre, David Stephens, and Jennifer Lippincott-Schwartz for antibodies and plasmids. We also thank Hong Chen for advice on the use of siRNA and Laurence Pelletier for his primary observations, as well as Pete Takizawa, Pietro DeCamilli, Ira Mellman, and Graham Warren, for comments during the preparation of the manuscript. Special thanks to Graham Warren and Pete Takizawa for their advice and stimulating discussions.
This work was supported by the Howard Hughes Medical Institute. Salary support for S. Ferro-Novack and S. Yu were provided by the Howard Hughes Medical Institute. A. Satoh was supported by an American Heart Association scientist development grant. Work at the University of Montana was supported by National Institutes of Health (NIH) grant GM59378 to J.C. Hay and the NIH–Centers of Biomedical Research Excellence Center for Structural and Functional Neuroscience.
Abbreviations used in this paper: MVB, multivesicular body; tER, transitional ER; VTC, vesicular tubular cluster.
References
- Aridor, M., S.I. Bannykh, T. Rowe, and W.E. Balch. 1995. Sequential coupling between COPII and COPI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 131:875–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannykh, S.I., T. Rowe, and W.E. Balch. 1996. The organization of endoplasmic reticulum export complexes. J. Cell Biol. 135:19–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe, C., L. Orci, T Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- Barlowe, C. 1997. Coupled ER to Golgi transport reconstituted with purified cytosolic proteins. J. Cell Biol. 139:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman, J., M. Sacher, and S. Ferro-Novick. 2000. TRAPP stably associates with the Golgi and is required for vesicle docking. EMBO J. 19:862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tekaya, H., K. Miura, R. Pepperkok, and H.P. Hauri. 2005. Live imaging of bidirectional traffic from the ERGIC. J. Cell Sci. 118:357–367. [DOI] [PubMed] [Google Scholar]
- Cai, H., Y. Zhang, M. Pypaert, L. Walker, and S. Ferro-Novick. 2005. Mutants in trs120 disrupt traffic from the early endosome to the late Golgi. J. Cell Biol. 171:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao, A., D. Rahman, D.J. Pappin, J. Lucocq, and M. Lowe. 2003. The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J. Cell Biol. 160:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro-Novick, S., and R. Jahn. 1994. Vesicle fusion from yeast to man. Nature. 370:191–193. [DOI] [PubMed] [Google Scholar]
- Folsch, H., M. Pypaert, S. Maday, L. Pelletier, and I. Mellman. 2003. The AP-1A and AP-1B clathrin adaptor complexes define biochemically and functionally distinct membrane domains. J. Cell Biol. 163:351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, A.T., and B.S. Glick. 2000. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol. Biol. Cell. 11:3013–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuge, O., C. Dascher, L. Orci, T. Rowe, M. Amherdt, H. Plutner, M. Ravazzola, G. Tanigawa, J.E. Rothman, and W.E. Balch. 1994. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J. Cell Biol. 125:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh, E., F. Peter, N.V. Subramaniam, and W. Hong. 2005. Mammalian Bet3 functions as a cytosolic factor participating in transport from the ER to the Golgi apparatus. J. Cell Sci. 118:1209–1222. [DOI] [PubMed] [Google Scholar]
- Lopez, M., C. Huynh, L.O. Andrade, M. Pypaert, and N.W. Andrews. 2002. Role for sialic acid in the formation of tight lysosome-derived vacuoles during Trypanosoma cruzi invasion. Mol. Biochem. Parasitol. 119:141–145. [DOI] [PubMed] [Google Scholar]
- Miles, S., H. McManus, K.E. Forsten, and B. Storrie. 2001. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J. Cell Biol. 155:543–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick, P., C. Field, and R. Schekman. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 21:205–215. [DOI] [PubMed] [Google Scholar]
- Oprins, A., R. Duden, T.E. Kreis, H.J. Geuze, and J.W. Slot. 1993. β-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J. Cell Biol. 121:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, L., E. Jokitalo, and G. Warren. 2000. The effect of Golgi depletion on exocytic transport. Nat. Cell Biol. 2:840–846. [DOI] [PubMed] [Google Scholar]
- Pfeffer, S.R. 1999. Transport-vesicle targeting: tethers before SNAREs. Nat. Cell Biol. 1:E17–E22. [DOI] [PubMed] [Google Scholar]
- Presley, J.F., N.B. Cole, T.A. Schroer, K. Hirschberg, K.J. Zaal, and J. Lippincott-Schwartz. 1997. ER-to-Golgi transport visualized in living cells. Nature. 389:81–85. [DOI] [PubMed] [Google Scholar]
- Rossanese, O.W., J. Soderholm, B.J. Bevis, I.B. Sears, J. O'Connor, E.K. Williamson, and B.S. Glick. 1999. Golgi structure correlates with transitional endoplasmic reticulum organization in Pichia pastoris and Saccharomyces cerevisiae. J. Cell Biol. 145:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, G., K. Kolstad, S. Stone, F. Palluault, and S. Ferro-Novick. 1995. BET3 encodes a novel hydrophilic protein that acts in conjunction with yeast SNAREs. Mol. Biol. Cell. 6:1769–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher, M., Y. Jiang, J. Barrowman, A. Scarpa, J. Burston, L. Zhang, D. Schieltz, J.R. Yates III, H. Abeliovich, and S. Ferro-Novick. 1998. TRAPP, a highly conserved novel complex on the cis-Golgi that mediates vesicle docking and fusion. EMBO J. 17:2494–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher, M., J. Barrowman, D. Schieltz, J.R. Yates III, and S. Ferro-Novick. 2000. Identification and characterization of five new subunits of TRAPP. Eur. J. Cell Biol. 79:71–80. [DOI] [PubMed] [Google Scholar]
- Sacher, M., J. Barrowman, W. Wang, J. Horecka, Y. Zhang, M. Pypaert, and S. Ferro-Novick. 2001. TRAPP I implicated in the specificity of tethering in ER-to-Golgi transport. Mol. Cell. 7:433–442. [DOI] [PubMed] [Google Scholar]
- Sannerud, R., M. Michaël, N. Clément, H.A. Dale, K. Pernet-Gallay, F. Perez, B. Goud, and J. Saraste. 2006. Rab1 defines a novel pathway connecting the pre-Golgi intermediate compartments with the cell periphery. Mol. Biol. Cell. 17:1514–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste, J., and K. Svensson. 1991. Distribution of the intermediate elements operating in ER to Golgi transport. J. Cell Sci. 100:415–430. [DOI] [PubMed] [Google Scholar]
- Scales, S.J., R. Pepperkok, and T.E. Kreis. 1997. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 90:1137–1148. [DOI] [PubMed] [Google Scholar]
- Schweizer, A., J.A. Fransen, T. Bachi, L. Ginsel, and H.P. Hauri. 1988. Identification, by a monoclonal antibody, of a 53-kD protein associated with a tubulo-vesicular compartment at the cis-side of the Golgi apparatus. J. Cell Biol. 107:1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, J., E. Jokitalo, M. Pypaert, and G. Warren. 2000. a. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature. 407:1022–1026. [DOI] [PubMed] [Google Scholar]
- Seemann, J., E.J. Jokitalo, and G. Warren. 2000. b. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol. Biol. Cell. 11:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugrue, C.A., E.R. Kolen, H. Peters, A. Czernik, C. Kaiser, L. Matovcik, A.L. Hubbard, and F. Gorelick. 1999. Identification of the putative mammalian orthologue of Sec31p, a component of the COPII coat. J. Cell Sci. 112:4547–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, D.J., N. Lin-Marq, A. Pagano, R. Pepperkok, and J.P. Paccaud. 2000. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 113:2177–2185. [DOI] [PubMed] [Google Scholar]
- Sztul, E., and V. Lupashin. 2006. Role of tethering factors in secretory membrane traffic. Am. J. Physiol. Cell Physiol. 290:C11–C26. [DOI] [PubMed] [Google Scholar]
- Ward, T.H., R.S. Polishchuk, S. Caplan, K. Hirschberg, and J. Lippincott-Schwartz. 2001. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 155:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel, E.R., and R.P. Bolender. 1979. Stereological Methods. Academic Press, London; New York. 415 pp.
- Whyte, J.R., and S. Munro. 2002. Vesicle tethering complexes in membrane traffic. J. Cell Sci. 115:2627–2637. [DOI] [PubMed] [Google Scholar]
- Xu, D., and J.C. Hay. 2004. Reconstitution of COPII vesicle fusion to generate a pre-Golgi intermediate compartment. J. Cell Biol. 167:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}