

Abstract
None of the 28 identified point mutations in tafazzin (Taz1p), which is the mutant gene product associated with Barth syndrome (BTHS), has a biochemical explanation. In this study, endogenous Taz1p was localized to mitochondria in association with both the inner and outer mitochondrial membranes facing the intermembrane space (IMS). Unexpectedly, Taz1p does not contain transmembrane (TM) segments. Instead, Taz1p membrane association involves a segment that integrates into, but not through, the membrane bilayer. Residues 215–232, which were predicted to be a TM domain, were identified as the interfacial membrane anchor by modeling four distinct BTHS mutations that occur at conserved residues within this segment. Each Taz1p mutant exhibits altered membrane association and is nonfunctional. However, the basis for Taz1p dysfunction falls into the following two categories: (1) mistargeting to the mitochondrial matrix or (2) correct localization associated with aberrant complex assembly. Thus, BTHS can be caused by mutations that alter Taz1p sorting and assembly within the mitochondrion, indicating that the lipid target of Taz1p is resident to IMS-facing leaflets.
Introduction
The mitochondrion is central for pathways of ATP production, metabolite synthesis and degradation, lipid metabolism, and iron–sulfur cluster assembly (Koehler, 2004). Mitochondrial dysfunction contributes to a broad range of neural and muscular diseases, including the X-linked disease Barth syndrome (BTHS; Barth et al., 1983, 1999, 2004). BTHS is characterized by cardiac and skeletal myopathies, delayed growth until puberty, and cyclic neutropenia. The disease presents in infants and, if undiagnosed, is frequently fatal because of cardiac failure or sepsis. The human tafazzin (TAZ) gene, located on Xq28 and expressed at high levels in cardiac and skeletal muscle, was recognized as mutated in BTHS patients (Bione et al., 1996). To date, ∼28 different mutations resulting in single amino-acid changes in the Taz protein have been identified in BTHS patients (a comprehensive database of TAZ mutations is available on the Barth Syndrome Foundation website [http://www.barthsyndrome.org]). Aside from mutations resulting in either complete loss of Taz protein expression or expression of a severely truncated Taz, a biochemical explanation for the defect associated with any identified BTHS point mutation has not been provided.
Bioinformatics studies showed that tafazzin was similar to acyltransferases, suggesting that BTHS might be caused by an acyltransferase deficiency (Neuwald, 1997). Analysis of fibroblasts derived from BTHS patients demonstrated decreased steady-state levels of the mitochondrial-specific phospholipid cardiolipin (CL), although the biosynthetic rate of CL was normal (Vreken et al., 2000). CL is hypothesized to obtain its final composition of fatty acyl groups via a remodeling process (Schlame and Rustow, 1990). According to this model, the final step in CL biosynthesis occurs when newly synthesized CL is deacylated to form monolyso-CL (MLCL) and subsequently reacylated with polyunsaturated fatty acyl chains, forming mature CL. Because the CL contained in patient samples was deficient in the mature tetralinoleoyl form of CL, which is the predominant form in normal cardiac muscle (Schlame et al., 2003), the defect associated with BTHS was suggested to occur during the process of CL remodeling.
Yeast contains an orthologue of TAZ1 and has proven effective as a BTHS model (Vaz et al., 2003; Gu et al., 2004; Ma et al., 2004). Yeast lacking taz1 (Δtaz1) arrest growth at 37°C in ethanol media and have decreased CL content. Importantly, as observed in patient samples, the predominant mature CL acyl species of wild-type (wt) cells (C18:1 and C16:1) are replaced with immature, saturated fatty acids. In addition, MLCL, which is the predicted intermediate in the remodeling cycle, accumulates in the Δtaz1 yeast strain. Epitope-tagged Taz1p constructs have been localized to mitochondria (Ma et al., 2004; Testet et al., 2005) and suggested to reside in the outer membrane (OM), facing the IMS (Brandner et al., 2005). However, this localization has not been confirmed for endogenous Taz1p and is difficult to reconcile with CL enrichment in the inner membrane. Interestingly, a recent study in the yeast Saccharomyces cerevisiae suggests that Taz1p might additionally function as a lyso-phosphatidylcholine (PC) acyltransferase (Testet et al., 2005). In fact, analyses of Δtaz1 yeast and samples derived from BTHS patients have shown that PC, in addition to CL, is altered (Schlame et al., 2003; Testet et al., 2005; Xu et al., 2005). Therefore, the true functions of Taz1p, and, thus, the molecular basis for the pathologies observed in BTHS patients, are at present unknown.
In this study, yeast was used to determine if Taz1p does localize to mitochondria; and, if so, in which submitochondrial compartment it resides. The detailed subcellular and submitochondrial localization of Taz1p presented herein shed new insight into the mechanism of Taz1p function. Moreover, a group of authentic BTHS point mutations that occur in an identified membrane anchor of Taz1p are characterized and provide the first molecular explanations for any of the numerous mutations identified to date that are linked to this important human disease. Strikingly, mutations in one membrane anchor result in two distinct biochemical fates for the characterized mutant tafazzins, which are all, nonetheless, nonfunctional. Specifically, for three of the characterized mutants, nonfunctional Taz1p is mislocalized to the matrix side of the inner membrane, indicating that its target lipid localizes to the IMS-sided leaflets of the membrane. Surprisingly, the fourth characterized BTHS Taz1p mutant localized appropriately within the mitochondrion, but, presumably because of an altered association with membranes, assembled into aberrant complexes. Thus, proper Taz1p sorting and assembly is critical for Taz1p activity, and the defect associated with a cluster of BTHS patients is caused by the missorting or misassembly of the mutated Taz1p.
Results
Taz1p is a membrane-associated mitochondrial resident
To gain insight into Taz1p functions, we sought to confirm the mitochondrial localization of endogenous Taz1p and, if endogenous Taz1p is a mitochondrial resident, to determine its submitochondrial localization. To this end, we raised a polyclonal antiserum in rabbits against the recombinant, full-length yeast His6Taz1 protein (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200605043/DC1). The specificity of the resulting antiserum was confirmed by immunoblot analysis of whole-cell extracts derived from a wt S. cerevisiae strain and an isogenic strain in which the endogenous TAZ1 gene was deleted (Δtaz1). The polyclonal antisera detected Taz1p in wt, but not Δtaz1, extracts at the predicted molecular mass of 44 kD (Fig. S1).
To determine the subcellular localization of Taz1p, a series of differential centrifugations were performed on dounced homogenates derived from the wt strain, and the resulting fractions were analyzed by immunoblotting with antisera specific for assorted organellar markers (Fig. 1 A). Taz1p did not cofractionate with the ER marker Sec62p, the ribosomal marker α-nascent polypeptide–associated chain, or the cytosolic marker hexokinase. Based on cofractionation with the ADP/ATP carrier (AAC), Taz1p was demonstrated to be a mitochondrial resident. Additionally, we determined the subcellular localization using immunoelectron microscopy. In representative sections (Fig. 1 B), Taz1p localized to mitochondria, associating with the inner membrane (IM; Fig. 1 B, arrowheads) and, occasionally, with the OM (Fig. 1 B, arrows). Nuclei and cytoplasmic matrix were devoid of significant labeling (Fig. 1 B1). In the Δtaz1 strain, Taz1p was not detected (not depicted).
Figure 1.

Taz1p is a mitochondrial resident. (A) Fractions were prepared from the wt strain through a series of differential centrifugations. 50 μg of each fraction was separated by SDS-PAGE and analyzed by immunoblot using antisera specific for the indicated subcellular organelle. n = 2. (B) Taz1p localizes to the IM and OM in immunogold-labeled ultrathin cryosections of the parental wt strain. n, nucleus; ne, nuclear envelope; pm, plasma membrane; m, mitochondria. Arrows, OM; arrowheads, IM. Bars, 0.1 μm.
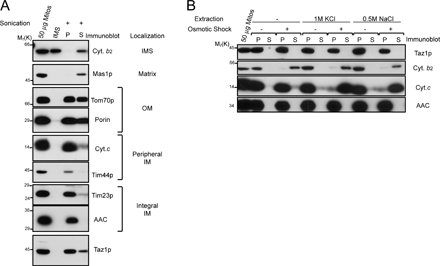
Because Taz1p is hypothesized to function as an acyltransferase, we investigated the nature, if any, of the membrane association of Taz1p with wt mitochondria. Sonication (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200605043/DC1) discriminates between soluble proteins residing within the IMS (cytochrome b 2), the matrix (Mas1p), and proteins that are integrally or peripherally associated with either the mitochondrial OM or IM. That Taz1p remained largely in the pellet fraction after sonication provides evidence that it is membrane associated, but does not reveal the nature of this membrane association. To determine if Taz1p associates with mitochondrial membranes peripherally through electrostatic interactions, mitochondria or mitoplasts, which were generated by placing mitochondria in hypoosmotic conditions resulting in OM rupture (a process termed osmotic shock), were washed with either 1 M KCl or 0.5 M NaCl (Fig. S2). Cytochrome b 2 was released after osmotic shock, independent of the addition of high salt. In contrast, cytochrome c, which is known to be peripherally anchored to the IM through electrostatic interactions with CL and/or phosphatidylglycerol (Tuominen et al., 2002), was only released after a high-salt wash of mitoplasts, but not of intact mitochondria. Taz1p, like the integral membrane protein AAC, remained in the pellet fraction after every tested treatment.
To distinguish between a peripheral and an integral membrane association, mitochondria were incubated with 0.1 M Na2CO3 at increasing pH; integral membrane proteins remain in the pellet after centrifugation, whereas peripheral membrane and soluble proteins release into the supernatant. Taz1p and Tim23p fractionated similarly, suggesting that Taz1p may be an integral membrane protein (Fig. 2 A). Importantly, the observation that the peripherally associated cytochrome c was released at every tested pH further confirms the conclusion that Taz1p is not a peripheral membrane protein.
Figure 2.

Taz1p nonperipherally associates with the IM, OM, and contact sites. (A) Wt mitochondria were analyzed by alkali extraction using 0.1 M carbonate at the indicated pH values. Equal volumes of the pellet (P) and TCA-precipitated supernatant (S) fractions were resolved by SDS-PAGE, transferred to nitrocellulose, and immunoblotted for the indicated mitochondrial markers. n = 3. (B) Sonicated mitochondrial membrane vesicles were prepared from wt mitochondria for fractionation on linear sucrose gradients (0.85–1.8 M). Fractions were collected from heavy (fraction 1; bottom) to light density (fraction 16; top), and 5 μg of each fraction was immunoblotted as indicated. Chemiluminescent images were also collected and two exposures per blot were quantified. For each individual mitochondrial marker, the amount in each fraction is expressed as the percentage of the sum of the signals for that marker in all of the fractions. For both the IM and OM, the mean ± SD of three different markers of each compartment are presented (IM represents AAC, Cox2p, and cytochrome c 1; OM represents OM45p, porin, and Tom70p). The immunoblots and derived data are from a single representative experiment. n = 3.
To gain insight as to which mitochondrial membrane Taz1p associates with, we used a fractionation technique that allows the separation of IM, OM, and so-called contact sites, which are areas where the IM and OM are connected (Pon et al., 1989). Sonicated submitochondrial particles were separated over a linear sucrose gradient, fractions were collected from bottom to top (Fig. 2 B, fraction 1 and 16, respectively), and equal amounts of protein derived from each fraction were analyzed by immunoblot (Fig. 2 B). Three distinct peaks corresponding to IM (heavy density; revealed by the presence of IM proteins, AAC [depicted], Cox2p, and cytochrome c 1), contact sites (intermediate density; contains detectable IM and OM markers), and OM (light density; enriched in OM45 [Fig. 2 B], porin, and Tom70p) were identified after quantitation of the immunoblots. Interestingly, Taz1p was present in each of the three peaks, indicating that it is localized in all three mitochondrial membrane compartments. Therefore, Taz1p is a new member of an emerging class of mitochondrial-resident proteins that have a dual localization to the IM and OM.
Taz1p is an integral interfacial membrane protein lining the IMS
Because Taz1p is a nonperipherally associated membrane protein, we used a variety of TM prediction programs to identify potential membrane-spanning domains (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200605043/DC1). The programs did not predict the same regions, but two stretches, 26–46 and 215–232, were consistently predicted in the 381–amino acid protein. Collectively, these different prediction methods suggest that Taz1p may contain from zero to two membrane-spanning domains.
To decipher which, if any, of these potential TM domains exist, three Taz1p constructs were generated with an epitope tag placed at either termini (MycTaz and TazHA for N- or C-terminal–tagged Taz1p, respectively) or between the two putative TM domains (TazMycTaz; Fig. 3 A). After transformation in the Δtaz1 strain (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200605043/DC1), each construct rescued a growth defect of the Δtaz1 strain on galactose medium at 37°C (Fig. S3), as well as Taz1p function, as demonstrated by the lack of accumulation of MLCL in these strains (Fig. 3, B and C). That each construct localized to mitochondria indicates that the immediate N or C termini are not required for mitochondrial targeting. Lastly, the membrane association of each construct in mitochondria was investigated by alkali extraction (Fig. 3 D). Quantitation of the alkali extraction profiles demonstrated that each construct behaved in a manner indistinguishable from endogenous Taz1p (Fig. 3 E).
Figure 3.

Three epitope-tagged Taz1p constructs are functional. (A) Schematics of the three constructs, with potential TM domains indicated. (B) After steady-state labeling with 32Pi, phospholipids were extracted from the indicated strains, separated by TLC, and revealed by phosphoimaging. The migration of phospholipids is indicated (PI, phosphatidylinositol; PS, phosphatidylserine; PA, phosphatidic acid). (C) The relative abundance of MLCL was determined for each strain. The amount of MLCL in each strain is expressed as a percentage of the total phospholipids in each strain. Mean ± SEM. n = 6. Δtaz1 yeast accumulate significant amounts of MLCL relative to wt yeast (P ≤ 0.001), as determined by t-test. Control Δtaz1 yeast transformants ([Vector Alone]) accumulate significant amounts of MLCL relative to Δtaz1 yeast transformed with wt Taz1p ([WT Taz1p]) or any of the epitope-tagged constructs (P < 0.001) as determined by one way analysis of variance (ANOVA), with Holm–Sidak pairwise comparisons. (D) Purified mitochondria from the indicated yeast strains were analyzed by alkali extraction as before, except that Taz1p was identified by immunoblotting with monoclonal antibodies specific for the appropriate epitope tag. (E) Quantitation was performed as previously described. The percentage of Taz1p present in the derived supernatants after carbonate extraction was determined as follows: S/(S + P) × 100, where S is the volume of Taz1p detected in the supernatant at a given pH and P is the volume associated with the pellet at the same pH. Mean ± SD. n = 3.
Insight into the topology of Taz1p was obtained by ascertaining the proteinase K accessibility of endogenous Taz1p (Fig. 4 A) and the three tagged Taz1p constructs (Fig. 4, C–E) in intact mitochondria (Fig. 4, A–E, lanes 2–5), osmotically shocked mitoplasts (Fig. 4, A–E, lanes 6–9), and 0.1% Triton X-100–solubilized mitochondria (Fig. 4, A–E, lanes 10–13). As expected, Tom70p, which is an OM protein, was readily degraded by proteinase K added to intact mitochondria (Fig. 4, A–E, lanes 2–5); Tim54p, which is an integral IM protein facing the IMS, was accessible to added proteinase K after OM rupture by osmotic shock (Fig. 4, A–E, lanes 6–9); and α-ketoglutarate dehydrogenase (KDH), which is a matrix resident, was only degraded by proteinase K when the mitochondria were solubilized with Triton X-100 (Fig. 4, A–E, lanes 10–13). Interestingly, 2–3 fragments (Fig. 4 A, gray arrows) could be detected in wt-derived samples after each experimental condition in the absence of added proteinase K (Fig. 4 A, lanes 2, 6, and 10). Each of these bands is not detected in samples prepared from Δtaz1 mitochondria (Fig. 4 B, background bands highlighted with asterisks). The same three fragments, migrating slightly slower because of the appended epitope tags, are detected in MycTaz- (Fig. 4 C) and TazMycTaz- (Fig. 4 D), but not in TazHA- (Fig. 4 E), derived samples, implying that these fragments are generated through the removal of increasingly larger portions of the C terminus. Upon further characterization (unpublished data), these fragments are generated during the TCA precipitation step used to completely inactivate proteinase K. Therefore, the presence of these bands reflects the persistence of full-length Taz1p at the end of an indicated incubation; and, conversely, their absence reflects a loss in full-length Taz1p caused by proteinase K–mediated degradation.
Figure 4.

Taz1p is an interfacial membrane protein facing the IMS. Intact mitochondria, mitochondria subjected to osmotic shock (mitoplasts), or mitochondria solubilized with 0.1% Triton X-100 were incubated alone or in the presence of the indicated concentration of proteinase K. Equivalent amounts of each sample (designated by the indicated quantity of starting material) were resolved by SDS-PAGE and immunoblotted as indicated. The source of the mitochondria for each image is wt (A), Δtaz1 (B), MycTaz (C), TazMycTaz (D), and TazHA (E). The three gray arrows highlight Taz1p fragments generated during the TCA precipitation step performed to inactivate proteinase K, which involves the removal of increasing amounts of the C terminus. The white arrow, which designates an ∼27-kD fragment generated upon addition of low amounts of proteinase K to mitoplasts, lacks at least the N-terminal 154 amino acids, and contains the C terminus, which is stabilized in 0.1% Triton X-100. The black arrow reveals an ∼24-kD fragment only generated upon addition of high concentrations of proteinase K to wt mitoplasts. To visualize the Taz1p fragments, overexposed immunoblots are presented, except for D, where the indicated exposure lengths for the anti-Myc immunoblots were spliced together using Photoshop. Asterisks in A and B highlight background bands identified in the Δtaz1 mitochondria immunoblots (B). (F) Drawing summarizing the localization and membrane association of Taz1p. n = 3.
No diminution in the full-length Taz1p signal was observed upon addition of increasing amounts of proteinase K to intact mitochondria (Fig. 4, A and C–E, lanes 3–5). In wt mitoplasts, the addition of low amounts of proteinase K results in a decrease in detectable full-length Taz1p and the appearance of an ∼27-kD fragment (Fig. 4 A, lanes 7 and 8, white arrow). Addition of high concentrations of proteinase K results in the loss of the ∼27-kD Taz1p fragment and the appearance of a novel ∼24-kD protected band (Fig. 4 A, lanes 8–9, black arrow). It is worth noting that the full-length Taz1p detected after addition of proteinase K to mitoplasts reflects the proportion of mitochondria remaining intact after the osmotic shock reaction. To detect the Taz1p fragments, overexposed images of the Taz1p immunoblots are presented (for lighter exposure see Fig. 6 E). Importantly, neither of these bands is detected in samples prepared from Δtaz1 (Fig. 4 B), MycTaz (Fig. 4 C), or TazMycTaz (Fig. 4 D) mitoplasts (lanes 6–9), demonstrating that these fragments are generated by the removal of at least the N-terminal 155 amino acids of Taz1p (the first amino acid downstream of the integrated tag in TazMycTaz). In TazHA mitoplasts (Fig. 4 E), the addition of increasing amounts of proteinase K results in the sequential appearance and disappearance of an ∼29-kD fragment. Thus, the final protected ∼24-kD fragment observed in wt mitoplasts (Fig. 4 A, black arrow) results from the proteinase K–mediated removal of the C-terminal ∼27 amino acids of Taz1p.
Figure 6.

Altered membrane association of the BTHS Taz1p mutants results in two fates: matrix mistargeting or aberrant complex assembly. Mitochondria isolated from the indicated strains were analyzed by sonication (A) or alkali extraction (B), as previously described. (C) Quantitation and data analyses were performed as described in Fig. 3 E. The asterisks indicate a statistically significant increase in the release into the supernatant of each of the BTHS Taz1p mutants, relative to endogenous Taz1p (P < 0.001) as determined by one-way ANOVA, with Holm–Sidak pairwise comparisons. Mean ± SD. n = 3. (D) 100 μg mitochondria from the indicated strain was solubilized in 1.5% (wt/vol) digitonin and subjected to blue native–PAGE (6–16% acrylamide), and then Taz1p was detected by immunoblotting. The black, green, and blue arrows highlight 160-, 220-, and 280-kD Taz1p-containing complexes identified in wt and [WT Taz1p] mitochondrial extracts. The red arrow highlights a 460-kD complex distinctly observed in mitochondrial extracts derived from the G230R Taz1p mutant. n = 4. (E) Mitochondria derived from the indicated strains were treated exactly as described in Fig. 4. n = 3. For simplicity, only one set of control immunoblots is presented in B and E. The controls for every source of mitochondria are provided in Fig. S4. Fig. S4 is available at http://www.jcb.org/cgi/content/full/jcb.200605043/DC1.
Interestingly, 0.1% Triton X-100 stabilizes a core structure of Taz1p, which is ∼27 kD and resists degradation, even at 100 μg/ml proteinase K (Fig. 4 A, lane 13, white arrow). This band is not detected in samples prepared from Δtaz1- (Fig. 4 B), MycTaz- (Fig. 4 C), or TazMycTaz-solubilized (Fig. 4 D) mitochondria (lanes 11–13). In TazHA-solubilized mitochondria, a ∼29-kD band is readily detected at 10 μg/ml proteinase K. However, upon addition of 10× more proteinase K, this fragment is much fainter, suggesting that the appended C-terminal HA tag is not included in the final 0.1% Triton X-100–stabilized Taz1p core structure. Collectively, both termini of Taz1p are exposed to the IMS. Moreover, given that the banding profile for the MycTaz and TazMycTaz constructs are identical and that Taz1p is not a peripheral membrane protein, we conclude that Taz1p is a so-called integral interfacial protein, associating with mitochondrial membranes by protruding into, but not through, the lipid bilayer (Fig. 4 F).
A cluster of BTHS mutations alter the normal integral interfacial membrane association of Taz1p
To date, 28 distinct mutations resulting in single amino acid changes in Taz1p have been identified in BTHS patients, 21 of which occur at residues that are either conserved or identical in the yeast orthologue. Interestingly, a cluster of four such conserved BTHS mutations reside within the second predicted TM domain, amino acid residues 215–232 of Taz1p (Fig. 5 A). This predicted TM domain is located within the Triton X-100–stabilized, tightly folded Taz1p structural element and is an attractive candidate for mediating the integral interfacial association of Taz1p with membranes. To test the hypothesis that yeast Taz1p residues 215–232 are an integral interfacial membrane anchor and that the BTHS mutants occurring within the orthologous region of human Taz1p inactivate this function, each of the four mutations were modeled in yeast Taz1p and individually expressed in the Δtaz1 yeast strain. Compared with Δtaz1 yeast transformed with wt Taz1p (WT Taz1p), the expression of the BTHS mutations was either drastically (V223D, V224R, and I226P) or slightly (G230R) reduced (Fig. 5 B). Moreover, none of these BTHS mutants rescued the growth defect of the Δtaz1 yeast strain (not depicted) or prevented the accumulation of MLCL, which is a hallmark of loss of Taz1p activity (Fig. 5, D and E). As three of the four Barth syndrome mutations occur at conserved, but not identical, residues in the yeast orthologue, a “humanized” yeast Taz1p was generated, containing the human residues at all three positions. Importantly, the humanized yeast Taz1p was expressed at levels similar to wt Taz1p expressed in Δtaz1 yeast (Fig. 5 C), rescued the growth defect of the Δtaz1 strain (not depicted), and prevented the accumulation of MLCL (Fig. 5, D and E). Thus, all four BTHS mutations, when modeled in yeast Taz1p, are nonfunctional.
Figure 5.

Yeast Taz1p harboring authentic BTHS mutations that occur in the putative interfacial membrane anchor of Taz1p localize to mitochondria, but are nonfunctional. (A) ClustalW alignment of the putative interfacial membrane anchor (boxed) of Taz1p from human, mouse, and S. cerevisiae. The BTHS mutations are indicated at the top, with mutations occurring at conserved and identical residues provided in green and red, respectively. (B) The relative expression of the four different BTHS mutants (three clones/mutant) was determined from whole-cell extracts by immunoblotting for Taz1p (bottom) with KDH serving as a loading control (top). (C) The same as B, except that three clones derived from a humanized yeast Taz1p were analyzed next to the three Taz1p mutants harboring single BTHS mutations occurring at conserved residues. (D and E) Steady-state 32P labeling and analyses were performed as described in Fig. 3 (B and C). Δtaz1 yeast accumulate significant amounts of MLCL relative to wt yeast (P ≤ 0.001), as determined by t-test. All of the BTHS mutants, with the notable exception of the humanized Taz1p, demonstrate a statistically significant accumulation of MLCL relative to Δtaz1 transformed with wt Taz1p ([WT Taz1p]; P < 0.001) as determined by one-way ANOVA, with Holm–Sidak pairwise comparisons. Mean ± SEM. n = 4, except for humanized Taz1p, where n = 3. (F) Fractions were prepared from the indicated yeast strains through a series of differential centrifugations. 50 μg of each fraction was separated by SDS-PAGE and analyzed by immunoblot using antisera specific for the indicated subcellular organelle. n = 2.
One possibility for the inability of each of these mutations to rescue Taz1p function is that they fail to localize properly to mitochondria. However, all four mutations were exclusively localized to mitochondria (Fig. 5 F), demonstrating that this cluster of BTHS mutations does not result in the inactivation of a mitochondrial targeting signal in Taz1p.
The membrane association of each of the mutants was investigated. Surprisingly, all four BTHS mutants retained the ability to associate with mitochondrial membranes based on their continued presence in the pellet fraction after sonication (Fig. 6 A). Identical to wt Taz1p, high-salt washing of intact mitochondria or osmotically swollen mitoplasts failed to strip any of the four mutants off of the mitochondrial membranes (unpublished data). However, when the membrane association of each of the mutants was assessed by alkali extraction, all four BTHS mutants were significantly more extractable by 0.1 M Na2CO3, pH 10.9 and 11, than wt Taz1p (Fig. 6, B and C, red arrows). Therefore, whereas each of the BTHS mutants retains some capacity to associate with mitochondrial membranes, the nature of that membrane association is altered. This is consistent with the hypothesis that residues 215–232 of yeast Taz1p represent a membrane anchor.
To determine if the altered membrane association of each of the BTHS mutants resulted in a different submitochondrial localization, the compartment in which each mutant resides was assessed using a proteinase K protection assay (Fig. 6 E). Intriguingly, the three BTHS mutants occurring in the middle of the postulated membrane anchor are mislocalized to the mitochondrial matrix because they are not susceptible to protease digestion during osmotic shock (Fig. 6 E, red arrows); rather, the mutants are only digested by protease when Triton X-100 is added to disrupt the mitoplasts. Thus, for these three BTHS mutants, the failure to rescue Taz1p function in the Δtaz1 yeast strain is explained by a mislocalization within the mitochondrion. In contrast, the BTHS mutation occurring more toward the edge of the predicted membrane anchor, G230R, is resident to IMS-facing mitochondrial membranes, similar to wt Taz1p.
Given that the G230R mutant displayed an altered membrane association and was unable to rescue Taz1p function in the Δtaz1 yeast strain, the assembly of the G230R Taz1p mutant into macromolecular complexes was assessed by blue native–PAGE after solubilization of mitochondria with 1.5% (wt/vol) digitonin. Importantly, wt Taz1p overexpressed in the Δtaz1 yeast strain provided an identical profile of Taz1p complexes as endogenous Taz1p, with the expected increase in intensity of each detected complex; thus, overexpression of Taz1p, per se, does not alter its complex assembly. Specifically, wt Taz1p migrated on blue native gels as a broad smear ranging from ∼45–140 kD, with three larger 160-, 220-, and 280-kD complexes evident (Fig. 6 D, black, green, and blue arrows, respectively); critically, all of these Taz1p complexes were not detected in mitochondrial extracts derived from either Δtaz1 yeast or Δtaz1 yeast transformed with empty vector (Fig. 6 D, Vector Alone). In stark contrast to the three mislocalized mutants, the G230R Taz1p mutant migrated as a broad and intense smear from ∼45 to 400 kD; however, a discrete and unique 460-kD complex was also observed (Fig. 6 D, red arrow). Thus, a single point mutation that alters the membrane association of Taz1p, but not its submitochondrial localization, results in the inappropriate assembly of G230R Taz1p into aberrant protein complexes or, instead, freezes G230R Taz1p into protein complexes that are normally dynamic and transient in nature. In conclusion, these data demonstrate that Taz1p residues 215–232 are, in fact, an integral interfacial membrane anchor and provide the first mechanistic explanations for a series of BTHS mutations.
Discussion
More than a CL acyltransferase?
In this study, using a new anti-Taz1p antiserum, we have demonstrated that endogenous Taz1p is a normal resident of mitochondria, consistent with the hypothesis that it functions as a CL acyltransferase. Moreover, we show that Taz1p associates with all mitochondrial membranes facing the IMS. The conclusion that Taz1p associates with the inner leaflet of the OM and the outer leaflet of the IM and, thus, effectively lines the IMS is based on three separate observations. First, Taz1p was only susceptible to added proteinase K upon osmotic shock of the OM, demonstrating that it resides within the IMS. Second, Taz1p was associated with both the IM and OM, as assessed by immunoelectron microscopy (Fig. 1 B). Third, Taz1p was localized to the IM, OM, and contact sites after separation of these compartments using a linear sucrose gradient (Fig. 2 B). Our conclusion that Taz1p is localized to the IM, as well as to the OM, contrasts with recent results in which it was concluded that an epitope-tagged Taz1p was found exclusively in association with the OM (Brandner et al., 2005), which is a localization that is difficult to reconcile with the vast enrichment of CL in the IM. However, this conclusion was drawn although their IM and OM resolved in immediately adjacent fractions. In addition, a recent paper describing the proteome of purified OM vesicles failed to identify Taz1p (Schmitt et al., 2005). That Taz1p was identified in another proteomic study using whole mitochondria (Sickmann et al., 2003) indicates that Taz1p can be identified in a proteomics-based approach and that the failure to identify it in the OM may reflect its relatively low abundance in this compartment.
Is it surprising that Taz1p is localized on both the IM and OM of mitochondria? As only two other mitochondrial proteins, Mgm1p and Fzo1p, have been demonstrated to have this dual-membrane topology (Fritz et al., 2001; Sesaki et al., 2003), the simple answer is yes. CL is highly enriched in the IM, and the expectation was that Taz1p would, as a CL acyltransferase, reside in the IM. However, CL has been additionally detected in both contact sites and the OM (Ardail et al., 1990; Simbeni et al., 1991), although the proportion of CL associated with these two membrane compartments relative to the IM is, at present, controversial. Quite possibly, our determination that Taz1p resides on the IM and OM of wt mitochondria reflects the relative distribution of its putative target, CL.
However, recent work has suggested that, in yeast, Taz1p may additionally function as a lyso-PC acyltransferase (Testet et al., 2005). That this might not be a yeast-specific phenomenon is suggested by the observation that the molecular composition of PC and phosphatidylethanolamine (PE) is altered in specimens from BTHS patients (Schlame et al., 2003; Xu et al., 2005). Moreover, a pathway of CL remodeling identified in rat liver and human lymphoblast mitochondria was recently described, in which PC and PE acted as the acyl donor for either CL or MLCL (Xu et al., 2003). Importantly, this transacylation pathway was decreased in lymphoblasts derived from BTHS patients. PC constitutes approximately half of all mitochondrial phospholipids and is slightly enriched in the OM, relative to the IM (Voelker, 2004). Additionally, the OM has long been known to be enriched in a lyso-PC acyltransferase activity (Sarzala et al., 1970; Waite et al., 1970). Therefore, if Taz1p were to act as an acyltransferase for both CL and PC, then the observed dual localization might reflect the relative distribution of PC between the IM and OM.
Two fates associated with mutations in one membrane anchor
As Taz1p was not removed by high salt and exhibited an alkali extraction profile distinct from the peripherally associated cytochrome c and similar to the integrally associated Tim23p, it was expected that Taz1p is an integral membrane protein. Instead, a series of functional epitope-tagged constructs allowing the unambiguous determination of the membrane topology of Taz1p revealed that Taz1p does not contain any TM domains (up to two TM domains were predicted by the different programs). Our conclusion is supported by the fact that the regions of Taz1p on either side of the first potential TM domain both face the IMS, as does the extreme C terminus of Taz1p. Thus, we concluded that Taz1p is a monotopic integral interfacial membrane protein, which is an emerging class of membrane proteins that includes the alternative oxidase of plants and prostaglandin H2 synthase-1 (Andersson and Nordlund, 1999; Nina et al., 2000). Members of the monotopic integral interfacial membrane protein class are proposed to associate with membranes by protruding into, but not completely through, a lipid bilayer (Fig. 4 F). Worth briefly considering is that although alkali extraction, in combination with sonication and high-salt washes, can clearly identify peripheral membrane proteins, it cannot distinguish between classical integral membrane proteins, such as Tim23 with four TM domains, and nonclassical membrane proteins, such as Taz1p, which associate with membranes presumably through TM-like loops into the lipid bilayer.
An attractive candidate for mediating the integral interfacial association of Taz1p with the membrane was the second region (residues 215–232) predicted to be a TM domain. The importance of this domain in Taz1p function was first suggested by the observation that a cluster of four BTHS mutations occur within the orthologous region of human Taz1p. Modeling each of these mutations in yeast Taz1p results in a loss of Taz1p function. Consistent with the assignment of residues 215–232 as an interfacial membrane anchor, each of these BTHS mutants exhibited an altered association with mitochondrial membranes (Fig. 6 C). Perhaps the most interesting aspect of this cluster of mutations was that there were two different consequences of mutations within this defined region. Three of the BTHS mutants were mislocalized to the mitochondrial matrix. As each of these mutations occurs in the middle of the predicted membrane anchor, it is tempting to speculate that they result in the inactivation of a stop–transfer signal that normally prevents the transport of Taz1p across the IM. Implicit in this observation is that the import of Taz1p into mitochondria normally involves an interaction with one of the translocases of the IM. This lends further weight to our conclusion that Taz1p includes the IM as one of its resident compartments. Thus, BTHS can be caused by Taz1p missorting within the mitochondrion (Fig. 7 B).
Figure 7.

Model of Taz1p function and the defect associated with the two distinct classes of BTHS Taz1p variants. (A) Taz1p (blue) associates with IMS-facing membranes via a TM-like loop. Here, it transfers fatty acyl groups (light green) to MLCL (gray) and/or lyso-PC (light blue), forming CL and PC, respectively. (B) The V223D, V224R, and I226P Taz1p mutations inactivate a putative stop–transfer signal (red squiggle) resulting in Taz1p mistargeting to the mitochondrial matrix. In this compartment, Taz1p is unable to function (red X), potentially caused by the absence of its physiological lipid target within the matrix. (C) The stop–transfer activity is preserved in the G230R BTHS mutant; however, possibly because of charge interactions between the positively charged Arg and negatively charged phospholipid headgroups, the mutant Taz1p exhibits an altered association with mitochondrial membranes. This results in aberrant complex assembly (unidentified components depicted in yellow, purple, and dark green) and loss of function (red X).
The fourth BTHS mutant, G230R, localized appropriately to membranes lining the IMS, but assembled into abnormal complexes or abnormally stable complexes. Because this mutation occurs near the edge of the membrane anchor and involves the acquisition of a positive charge, we suggest that the membrane anchor is pulled partially out of the membrane by interactions between the positively charged Arg and negatively charged phospholipid headgroups (Fig. 7 C). Therefore, although the stop–transfer activity of this region is intact, the association of G230R with mitochondrial membranes facing the IMS is altered, leading to aberrant complex formation and loss of Taz1p function.
Finally, the conclusion that Taz1p is a monotopic, integral, interfacial membrane protein that lines the IMS indicates that the acyltransferase activity of Taz1p is mechanistically performed in the context of only those membrane leaflets facing the IMS (Fig. 7 A). Thus, the final distribution of remodeled CL and/or PC within mitochondrial membranes would require trafficking between leaflets of a bilayer subsequent to Taz1p-mediated acylation. Future detailed investigations such as these will provide important insights into the mitochondrial dysfunction associated with BTHS and potential targets for treating this disease.
Materials and methods
Cloning and recombinant protein expression
Tafazzin was cloned into pBSK after PCR, using genomic DNA isolated from the wt GA74-6A strain as a template and 5′ and 3′ primers hybridizing ∼300 bp upstream of the predicted start translation and 430 bp downstream of the stop translation codon of TAZ1. This construct, termed pBSK.Taz, acted as the template in all subsequent cloning procedures involving tafazzin. To generate Taz1p containing an N-terminal His6 tag, the entire open reading frame was cloned into the pET28a vector (Novagen) in frame and downstream of the His6 tag and thrombin cleavage site provided by the vector. His6Taz was induced in BL21-CodonPlus(DE3)-RIL Escherichia coli (Stratagene) and purified under native conditions using Ni2+-agarose (QIAGEN) as per the manufacturer's instructions. Tafazzin with an in-frame C-terminal HA tag was generated using a 5′ primer that hybridized ∼300 bp upstream of the start translation codon and a 3′ primer containing a stop translation codon, the sequence for the HA tag, and the 19 bps immediately upstream of the endogenous stop codon. To generate tafazzin with an in-frame N-terminal Myc tag, overlap extension (Ho et al., 1989) was performed using a primer set that placed a Kozak and Myc tag sequence in frame with the first predicted amino acid of tafazzin, still downstream of the same ∼300 upstream of the endogenous start site used in the previous construct. To insert a Myc tag between the two potential TM domains, overlap extension was performed using a primer set that inserted the first seven amino acids of the myc tag sequence between amino acids 154 and 155 of tafazzin, with the final two amino acids (Asp and Leu) of the Myc tag provided by amino acids 155 and 156 of tafazzin. The series of BTHS point mutations were also generated by overlap extension. Each construct was cloned into pRS425. The sequence of every construct was verified by sequencing. The sequences of all primers are available upon request.
Yeast strains
The wt parental S. cerevisiae yeast strains used were as follows: GA74-6A (MAT α, his3-11,15, leu2, ura3, trp1, ade8, rho + , mit +) and GA74-1A (MAT a, his3 -11,15, leu2, ura3, trp1, ade8, rho + , mit +). The Δtaz1His1.5 (MAT a, leu2, ura3, trp1, ade8, Δtaz1∷HISMX6) strain was constructed by replacing the entire open reading frame of TAZ1 with the His3MX6 marker using a PCR-mediated one-step gene replacement strategy (Wach et al., 1994). The Δtaz1His1.5 strain was transformed and selected as previously described (Gietz et al., 1992) with the empty vector, pRS425, WT Taz1p, or the aforementioned epitope-tagged Taz1p or BTHS Taz1p mutant constructs, all cloned into pRS425.
Antibodies
Antibodies were raised in rabbits using the yeast His6Taz. Yeast whole-cell extracts were prepared as previously described (Yaffe and Schatz, 1984). Most of the antibodies used in this work were generated in the Schatz laboratory (J. Schatz, University of Basel, Basel, Switzerland) or our laboratory and have been described previously. Other antibodies used were as follows: mouse anti-Sec62p (gift of David Meyers, University of California, Los Angeles, Los Angeles, CA), anti–β-Actin (Abcam Inc.), anti-HA (Covance Research Products, Inc.), anti-Myc (strain 9E10; Evan et al., 1985; obtained from the Developmental Studies Hybridoma Bank; developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa), and anti-Myc (clone 9B11; Cell Signaling Technology) monoclonal antibodies, and horseradish peroxidase–conjugated secondary antibodies (Pierce Chemical Co.).
Immunoblot
After resolution on 12% SDS-PAGE gels under reducing conditions, proteins were transferred to nitrocellulose membranes (Schleicher and Schuell BioScience) at 1 Amp for 60–75 min at room temperature. Immunoblots were performed exactly as previously described (Claypool et al., 2002). All of the presented images were captured on film. For quantitation of immunoblots, images were captured with a VersaDoc controlling a charge-coupled device camera (Bio-Rad Laboratories), and bands from two exposures per blot were quantitated with the affiliated Quantity One software. Formulas for specific calculations are presented in the appropriate figure legends. Statistical analyses were performed using SigmaStat 3.0 (Jandel Corp.).
Subcellular fractionation
For the subcellular fractionation studies, wt yeast were grown at 30°C in YPEG medium containing 1% yeast extract, 2% tryptone, 3% glycerol, and 3% ethanol. The mitochondria isolated for all of the other experiments were derived from cultures grown at 30°C to an OD600 of ∼0.8–1 in rich lactate medium (1% yeast extract, 2% tryptone, 0.05% dextrose, and 2% lactic acid, 3.4 mM CaCl2 2H2O, 8.5 mM NaCl, 2.95 mM MgCl2 6H20, 7.35 mM KH2PO4, and 18.7 mM NH4Cl). Mitochondria were isolated as previously described (Daum et al., 1982). Subcellular fractions were collected through a series of differential centrifugations. The amount of protein in each fraction was determined using the BCA assay (Pierce Chemical Co.).
Immunogold labeling of ultrathin cryosections
Immunoelectron microscopy was performed as previously described (Rieder et al., 1996). In brief, cells were fixed in suspension for 15 min by adding an equal volume of freshly prepared 8% formaldehyde contained in 100 mM PO4 buffer, pH 7.4. The cells were pelleted, resuspended in fresh fixative (8% formaldehyde, 100 mM PO4, pH 7.4), and incubated for an additional 18–24 h at 4°C. The cells were washed briefly in PBS and resuspended in 1% low-gelling–temperature agarose. The agarose blocks were trimmed into 1-mm3 pieces, cryoprotected by infiltration with 2.3 M sucrose/30% polyvinyl pyrrolidone (10,000 mol wt)/PBS, pH 7.4, for 2 h, mounted on cryopins, and rapidly frozen in liquid nitrogen. Ultrathin cryosections were cut on an ultramicrotome (UCT; Leica) equipped with an FC-S cryoattachment and collected onto formvar/carbon-coated nickel grids. The grids were washed through several drops of 1× PBS containing 2.5% fetal calf serum and 10 mM glycine, pH 7.4, and then blocked in 10% FCS for 30 min and incubated overnight in rabbit anti-Taz1p antibody. After washing, the grids were incubated for 2 h in 5-nm gold donkey anti–rabbit conjugate (Jackson ImmunoResearch Laboratories). The grids were washed through several drops of PBS, followed by several drops of ddH2O. Grids were then embedded in an aqueous solution containing 3.2% polyvinyl alcohol (10,000 mol wt)/0.2% methyl cellulose (400 centipoises)/0.1% uranyl acetate. The sections were examined and photographed on a transmission electron microscope (EM 410; Philips) at 100 kV and images were collected with a digital camera (Megaview III; Soft Imaging System). Figures were assembled in Photoshop (Adobe), with only linear adjustment of contrast and brightness.
Submitochondrial localization
For submitochondrial localization, with the exception of the epitope-tagged Taz1p strains, all studies were performed using at least two different batches of isolated mitochondria per strain. To assay membrane association by sonication, the pellet fraction after osmotic shock of 0.5 mg mitochondria was resuspended in 0.6 M sucrose, 3 mM MgCl2, and 20 mM Hepes-KOH, pH 7.4, and sonicated for 3 × 10 s, with 30-s intervals in an ice bath, using a microtip attached to a Sonic Dismembrator 550 (Fisher Scientific) with the amplitude set at 3.5. After removal of unbroken mitoplasts by centrifugation at 20,000 g for 10 min at 4°C, the submitochondrial particles were separated from soluble matrix components with an airfuge at 27 psi for 30 min at 4°C. High-salt washes were performed for 15 min at 4°C by the addition of 0.5 M NaCl or 1 M KCl to either intact mitochondria or mitoplasts after osmotic shock. Alkali extraction was performed essentially as previously described (Fujiki et al., 1982), except that 0.2 ml of 0.1 M Na2CO3 at the indicated pH was added to 0.2 mg mitochondria, and the pellet and supernatant fractions were separated with an airfuge at 27 psi for 15 min at 4°C. For experiments using intact mitochondria, mitochondria were incubated in 0.6 M sorbitol and 20 mM Hepes-KOH, pH 7.4. Osmotic shock was performed by incubating mitochondria for 30 min on ice in 0.03 M sorbitol and 20 mM Hepes-KOH, pH 7.4. Where designated, the indicated concentration of proteinase K ±0.1% (vol/vol) Triton X-100 was included. Any proteinase K remaining associated with the pellets was inactivated, as previously described (Glick et al., 1992). The supernatants were TCA precipitated and the pellet and supernatant fractions resuspended in equal volumes of thorner buffer (10% glycerol, 8 M urea, 5% (wt/vol) SDS, 40 mM Tris, pH 6.8, 4 mg/ml bromophenol blue, and 5% β-mercaptoethanol).
The separation of sonicated membranes over linear sucrose gradients was performed essentially as previously described (Pon et al., 1989). In brief, 5 mg of mitochondria were osmotically shocked (1 mM EDTA added to swelling medium) for 30 min and then shrunk for 10 min on ice by the addition of sucrose to 0.45 M. Sonication was performed as before, except six cycles were performed. After removal of unbroken mitoplasts by centrifugation for 10 min at 20,000 g at 4°C, the sonicated vesicles were harvested with an airfuge at 27 psi for 30 min at 4°C and the membrane-containing pellet was resuspended with 0.5 ml of 0.45 M sucrose, 10 mM KCl, 1 mM EDTA, and 5 mM Hepes-KOH, pH 7.4, loaded onto a linear sucrose gradient (1.8–0.85 M sucrose, 10 mM KCl, 1 mM EDTA, and 5 mM Hepes-KOH, pH 7.4; 4 ml total volume), and centrifuged in a SW41Ti rotor (Beckman Coulter) at 100,000 g for 20 h at 4°C. Using a syringe plunger to control the flow, a hole was punched into the bottom of the tubes using an 18-gauge needle and ∼0.2-ml fractions collected in individual wells of a 96-well plate. The fractions were quantified using the Bio-Rad Protein Assay (Bio-Rad Laboratories).
Phospholipid analyses
Starter cultures were diluted to an OD600 = 0.2 in 2 ml of yeast peptone dextrose (wt or Δtaz1 yeast) or SC-Leu (all remaining tested strains) supplemented with 10 μCi/ml 32Pi and grown at 30°C for ∼24 h. After a wash with H2O, the yeast pellets were resuspended in 0.3 ml MTE buffer (0.65 M mannitol, 20 mM Tris, pH 8.0, and 1 mM EDTA) supplemented with 1 mM PMSF, 10 μM leupeptin, 2 μM pepstatin A, and 10 μM chymostatin (the latter three were obtained from Sigma-Aldrich), transferred to an Eppendorf tube containing 0.1 ml glass beads, and disintegrated by vortexing on high for ∼30 min at 4°C. A crude mitochondrial fraction was sedimented after a low-speed spin at 250 g to remove the glass beads and any remaining intact yeast by centrifugation for 5 min at 13,000 g at 4°C. Phospholipids from equal amounts of labeled crude mitochondria, as determined by liquid scintillation, were extracted with 1.5 ml 2:1 chloroform/methanol by vortexing at room temperature for 1 h. 0.3 ml of ddH2O was added, the samples were vortexed on high for 1 min, and the phases were separated by centrifugation at 1,000 rpm in a clinical centrifuge at room temperature. The upper aqueous phase was removed by aspiration and the organic phase washed with 0.25 ml 1:1 methanol/H2O. After phase separation carried out as before, the lower organic phase was transferred to a new borosilicate tube and dried down under a stream of liquid nitrogen. Chloroform-resuspended samples were loaded onto silica gel TLC plates (Analtech) and resolved in 1D twice using chloroform/ethanol/H2O/triethylamine (30:35:7:35), as previously described (Vaden et al., 2005). Labeled phospholipids were revealed using a K-screen and FX-Imager (Bio-Rad Laboratories), quantitation was performed using the affiliated Quantity One software, and statistical analyses were performed using SigmaStat 3.0 (Systat Software, Inc.).
Blue native gel electrophoresis
Detergent solubilization of mitochondria (5 mg/ml) was performed for 30 min on ice with 20 mM Hepes-KOH, pH 7.4, 10% glycerol, 50 mM NaCl, 1 mM EDTA, and 2.5 mM MgCl2 supplemented with 1.5% (wt/vol) digitonin (Biosynth International, Inc.) and protease inhibitors, as listed in phospholipid analyses; insoluble material was removed by centrifugation for 30 min at 20,000 g at 4°C. ∼100 μg of solubilized material was analyzed by blue native gel electrophoresis on a 6–16% linear polyacrylamide gradient and Taz1p was detected by immunoblot after transfer to PVDF membranes.
Online supplemental material
Fig. S1 shows that a rabbit anti-Taz1p antiserum specifically recognizes yeast Taz1p. Fig. S2 shows that Taz1p associates with mitochondrial membranes. Fig. S3 shows that the three epitope-tagged Taz1p constructs rescue growth of the Δtaz1 yeast strain on YP-galactose at 37oC. Fig. S4 shows the altered membrane association of the BTHS Taz1p mutants results in two fates; matrix mistargeting or aberrant complex assembly. Table S1 shows Taz1p transmembrane domain predictions. Online supplemental material is available at http://www.jcb.org/cgi/content/full/200605043/DC1.
Supplementary Material
Acknowledgments
We would like to thank Drs. Stephen Young, Sabeeha Merchant, Mike Teitell, Cathy Clarke, and Beth Marbois for very critical reading of the manuscript and Dr. Jeff Schatz for antibodies.
This work was supported by the Arnold and Mabel Beckman Foundation, American Heart Association grant 0640076N, and National Institutes of Health grant 1R01GM61721. S.M. Claypool is a postdoctoral fellow of the American Heart Association and C.M. Koehler is an Established Investigator of the American Heart Association.
Abbreviations used in this paper: AAC, ADP/ATP carrier; ANOVA, analysis of variance; BTHS, Barth syndrome; CL, cardiolipin; IM, inner membrane; IMS, intermembrane space; KDH, α–ketoglutarate dehydrogenase; MLCL, monolysocardiolipin; OM, outer membrane; PC, phosphatidylcholine; PE, phosphatidylethanolamine; TM, transmembrane; wt, wild type.
References
- Andersson, M.E., and P. Nordlund. 1999. A revised model of the active site of alternative oxidase. FEBS Lett. 449:17–22. [DOI] [PubMed] [Google Scholar]
- Ardail, D., J.P. Privat, M. Egret-Charlier, C. Levrat, F. Lerme, and P. Louisot. 1990. Mitochondrial contact sites. Lipid composition and dynamics. J. Biol. Chem. 265:18797–18802. [PubMed] [Google Scholar]
- Barth, P.G., H.R. Scholte, J.A. Berden, J.M. Van der Klei-Van Moorsel, I.E. Luyt-Houwen, E.T. Van 't Veer-Korthof, J.J. Van der Harten, and M.A. Sobotka-Plojhar. 1983. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62:327–355. [DOI] [PubMed] [Google Scholar]
- Barth, P.G., R.J. Wanders, P. Vreken, E.A. Janssen, J. Lam, and F. Baas. 1999. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060). J. Inherit. Metab. Dis. 22:555–567. [DOI] [PubMed] [Google Scholar]
- Barth, P.G., F. Valianpour, V.M. Bowen, J. Lam, M. Duran, F.M. Vaz, and R.J. Wanders. 2004. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am. J. Med. Genet. A. 126:349–354. [DOI] [PubMed] [Google Scholar]
- Bione, S., P. D'Adamo, E. Maestrini, A.K. Gedeon, P.A. Bolhuis, and D. Toniolo. 1996. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12:385–389. [DOI] [PubMed] [Google Scholar]
- Brandner, K., D.U. Mick, A.E. Frazier, R.D. Taylor, C. Meisinger, and P. Rehling. 2005. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol. Biol. Cell. 16:5202–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool, S.M., B.L. Dickinson, M. Yoshida, W.I. Lencer, and R.S. Blumberg. 2002. Functional reconstitution of human FcRn in Madin-Darby canine kidney cells requires co-expressed human beta 2-microglobulin. J. Biol. Chem. 277:28038–28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum, G., P. Bohni, and G. Schatz. 1982. Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem. 257:13028–13033. [PubMed] [Google Scholar]
- Evan, G.I., G.K. Lewis, G. Ramsay, and J.M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5:3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz, S., D. Rapaport, E. Klanner, W. Neupert, and B. Westermann. 2001. Connection of the mitochondrial outer and inner membranes by Fzo1 is critical for organellar fusion. J. Cell Biol. 152:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki, Y., A.L. Hubbard, S. Fowler, and P.B. Lazarow. 1982. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz, D., A. St Jean, R.A. Woods, and R.H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick, B.S., A. Brandt, K. Cunningham, S. Muller, R.L. Hallberg, and G. Schatz. 1992. Cytochromes c1 and b2 are sorted to the intermembrane space of yeast mitochondria by a stop-transfer mechanism. Cell. 69:809–822. [DOI] [PubMed] [Google Scholar]
- Gu, Z., F. Valianpour, S. Chen, F.M. Vaz, G.A. Hakkaart, R.J. Wanders, and M.L. Greenberg. 2004. Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol. Microbiol. 51:149–158. [DOI] [PubMed] [Google Scholar]
- Ho, S.N., H.D. Hunt, R.M. Horton, J.K. Pullen, and L.R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 77:51–59. [DOI] [PubMed] [Google Scholar]
- Koehler, C.M. 2004. New developments in mitochondrial assembly. Annu. Rev. Cell Dev. Biol. 20:309–335. [DOI] [PubMed] [Google Scholar]
- Ma, L., F.M. Vaz, Z. Gu, R.J. Wanders, and M.L. Greenberg. 2004. The human TAZ gene complements mitochondrial dysfunction in the yeast taz1Delta mutant. Implications for Barth syndrome. J. Biol. Chem. 279:44394–44399. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F. 1997. Barth syndrome may be due to an acyltransferase deficiency. Curr. Biol. 7:R465–R466. [DOI] [PubMed] [Google Scholar]
- Nina, M., S. Berneche, and B. Roux. 2000. Anchoring of a monotopic membrane protein: the binding of prostaglandin H2 synthase-1 to the surface of a phospholipid bilayer. Eur. Biophys. J. 29:439–454. [DOI] [PubMed] [Google Scholar]
- Pon, L., T. Moll, D. Vestweber, B. Marshallsay, and G. Schatz. 1989. Protein import into mitochondria: ATP-dependent protein translocation activity in a submitochondrial fraction enriched in membrane contact sites and specific proteins. J. Cell Biol. 109:2603–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder, S.E., L.M. Banta, K. Kohrer, J.M. McCaffery, and S.D. Emr. 1996. Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant. Mol. Biol. Cell. 7:985–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzala, M.G., L.M. Van Golde, B. De Kruyff, and L.L. Van Deenen. 1970. The intramitochondrial distribution of some enzymes involved in the biosynthesis of rat-liver phospholipids. Biochim. Biophys. Acta. 202:106–119. [DOI] [PubMed] [Google Scholar]
- Schlame, M., and B. Rustow. 1990. Lysocardiolipin formation and reacylation in isolated rat liver mitochondria. Biochem. J. 272:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame, M., R.I. Kelley, A. Feigenbaum, J.A. Towbin, P.M. Heerdt, T. Schieble, R.J. Wanders, S. DiMauro, and T.J. Blanck. 2003. Phospholipid abnormalities in children with Barth syndrome. J. Am. Coll. Cardiol. 42:1994–1999. [DOI] [PubMed] [Google Scholar]
- Sesaki, H., S.M. Southard, M.P. Yaffe, and R.E. Jensen. 2003. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol. Biol. Cell. 14:2342–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann, A., J. Reinders, Y. Wagner, C. Joppich, R. Zahedi, H.E. Meyer, B. Schonfisch, I. Perschil, A. Chacinska, B. Guiard, et al. 2003. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA. 100:13207–13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbeni, R., L. Pon, E. Zinser, F. Paltauf, and G. Daum. 1991. Mitochondrial membrane contact sites of yeast. Characterization of lipid components and possible involvement in intramitochondrial translocation of phospholipids. J. Biol. Chem. 266:10047–10049. [PubMed] [Google Scholar]
- Schmitt, S., H. Prokisch, T. Schlunck, D.G. Camp II, U. Ahting, T. Waizenegger, C. Scharfe, T. Meitinger, A. Imhof, W. Neupert, et al. 2005. Proteome analysis of mitochondrial outer membrane from Neurospora crassa. Proteomics. 6:72–80. [DOI] [PubMed] [Google Scholar]
- Testet, E., J. Laroche-Traineau, A. Noubhani, D. Coulon, O. Bunoust, N. Camougrand, S. Manon, R. Lessire, and J.J. Bessoule. 2005. Ypr140wp, ‘the yeast tafazzin’, displays a mitochondrial lysophosphatidylcholine (lyso-PC) acyltransferase activity related to triacylglycerol and mitochondrial lipid synthesis. Biochem. J. 387:617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuominen, E.K., C.J. Wallace, and P.K. Kinnunen. 2002. Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J. Biol. Chem. 277:8822–8826. [DOI] [PubMed] [Google Scholar]
- Vaden, D.L., V.M. Gohil, Z. Gu, and M.L. Greenberg. 2005. Separation of yeast phospholipids using one-dimensional thin-layer chromatography. Anal. Biochem. 338:162–164. [DOI] [PubMed] [Google Scholar]
- Vaz, F.M., R.H. Houtkooper, F. Valianpour, P.G. Barth, and R.J. Wanders. 2003. Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism. J. Biol. Chem. 278:43089–43094. [DOI] [PubMed] [Google Scholar]
- Voelker, D.R. 2004. Lipid synthesis and transport in mitochondrial biogenesis. In Mitochondrial Function and Bioenergetics. Vol. 8. C.M. Koehler and M.F. Bauer, editors. Springer-Verlag, Heidelberg. pp. 267–291.
- Vreken, P., F. Valianpour, L.G. Nijtmans, L.A. Grivell, B. Plecko, R.J. Wanders, and P.G. Barth. 2000. Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279:378–382. [DOI] [PubMed] [Google Scholar]
- Wach, A., A. Brachat, R. Pohlmann, and P. Philippsen. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 10:1793–1808. [DOI] [PubMed] [Google Scholar]
- Waite, M., P. Sisson, and E. Blackwell. 1970. Comparison of mitochondrial with microsomal acylation of monoacyl phosphoglycerides. Biochemistry. 9:746–753. [DOI] [PubMed] [Google Scholar]
- Xu, Y., R.I. Kelley, T.J. Blanck, and M. Schlame. 2003. Remodeling of cardiolipin by phospholipid transacylation. J. Biol. Chem. 278:51380–51385. [DOI] [PubMed] [Google Scholar]
- Xu, Y., J.J. Sutachan, H. Plesken, R.I. Kelley, and M. Schlame. 2005. Characterization of lymphoblast mitochondria from patients with Barth syndrome. Lab. Invest. 85:823–831. [DOI] [PubMed] [Google Scholar]
- Yaffe, M.P., and G. Schatz. 1984. Two nuclear mutations that block mitochondrial protein import in yeast. Proc. Natl. Acad. Sci. USA. 81:4819–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}