

Abstract
Phosphatidylinositol-(3,4,5)-trisphosphate (PIP3), a product of phosphatidylinositol 3-kinase, is an important second messenger implicated in signal transduction and membrane transport. In hippocampal neurons, the accumulation of PIP3 at the tip of neurite initiates the axon specification and neuronal polarity formation. We show that guanylate kinase–associated kinesin (GAKIN), a kinesin-like motor protein, directly interacts with a PIP3-interacting protein, PIP3BP, and mediates the transport of PIP3-containing vesicles. Recombinant GAKIN and PIP3BP form a complex on synthetic liposomes containing PIP3 and support the motility of the liposomes along microtubules in vitro. In PC12 cells and cultured hippocampal neurons, transport activity of GAKIN contributes to the accumulation of PIP3 at the tip of neurites. In hippocampal neurons, altered accumulation of PIP3 by overexpression of GAKIN constructs led to the loss of the axonally differentiated neurites. Together, these results suggest that, in neurons, the GAKIN–PIP3BP complex transports PIP3 to the neurite ends and regulates neuronal polarity formation.
Introduction
Phosphatidylinositol (PI) 3-kinase signaling plays an important role in neuronal development (Cantley, 2002; Higuchi et al., 2003; Menager et al., 2004; Aoki et al., 2005). It is essential for the NGF-induced neurite elongation of PC12 cells, as this process is inhibited by wortmannin, a relatively specific inhibitor of PI 3-kinase (Kimura et al., 1994). Also, overexpression or microinjection of active PI 3-kinase in PC12 cells is sufficient to induce neurite formation (Kobayashi et al., 1997; Kita et al., 1998). Recent technological developments made it possible to visualize the intracellular localization of PIP3 by the use of pleckstrin homology (PH) domain of PI-(3,4,5)-trisphosphate (PIP3) interacting proteins such as Akt (Varnai and Balla, 1998) and GRP1 (Sato et al., 2003) as PIP3-specific probes. These studies revealed that PIP3 is highly enriched at the tip of growing neurites in PC12 cells (Higuchi et al., 2003; Aoki et al., 2005). Similarly, in cultured hippocampal neurons, PIP3 accumulates at the distal end of the longest neurite and induces the single longest neurite to develop into an axon (Shi et al., 2003; Menager et al., 2004). The downstream signaling mechanism coordinating the impact of concentrated PIP3 at the tip of the neurite to form an axon has been extensively studied (Ming et al., 1999; Inagaki et al., 2001; Shi et al., 2003; Shi et al., 2004; Nishimura et al., 2005; Yoshimura et al., 2005).
However, how PIP3 accumulates at the tip of neurite remains an intriguing, yet unresolved, question. One possible explanation is that the activity of PI 3-kinase and its upstream signaling mediators are concentrated at the tip of neurites, thereby producing PIP3 locally (Shi et al., 2003). A possible positive-feedback loop in which PIP3 activates upstream signaling of PI 3-kinase has been proposed (Aoki et al., 2005). Alternatively, PIP3 might be transported from the cell body through neurites by the motor-dependent trafficking mechanism. Although historically PI 3-kinase activity and PIP3 production have been considered to occur at the plasma membrane (Cantley, 2002), significant evidence has emerged supporting the idea that PIP3 is also produced within intracellular membranes, such as the endocytosed vesicles containing activated receptors (Sato et al., 2003). Therefore, an attractive hypothesis predicts that such PIP3-containing vesicles, laden with its upstream and downstream signaling components, are transported to specialized destinations where the local signaling activity is most needed.
Kinesin-family proteins are microtubule-based motor proteins implicated in the transport of diverse cargos (Hirokawa and Takemura, 2004b). Some of these kinesins recognize phospholipids as their cargo molecules. A kinesin-3 family protein (Lawrence et al., 2004), Unc104/KIF1A has a PH domain located at the C-terminal tail, which directly interacts with PI-(4,5)-bisphosphate (PIP2) and transports PIP2-containing vesicles (Klopfenstein et al., 2002; Klopfenstein and Vale, 2004). Another kinesin-3 protein, KIF16B, transports PI-(3)-phosphate (PI3P) through direct binding, via its C-terminal PhoX homology domain to PI3P (Hoepfner et al., 2005). We propose that guanylate kinase–associated kinesin (GAKIN; Hanada et al., 2000; Asaba et al., 2003), classified as a kinesin-3 protein, KIF13B (Lawrence et al., 2004), transports PIP3-containing vesicles through the interaction with an adaptor protein PIP3BP/centaurin-α (Hammonds-Odie et al., 1996; Tanaka et al., 1997). PIP3BP has two PH domains, which specifically interact with PIP3 and has been implicated in the regulation of PIP3 signaling function (Tanaka et al., 1997, 1999; Venkateswarlu et al., 1999, 2004). The PIP3BP was originally identified in other species and named as centaurin-α (Hammonds-Odie et al.,1996). Centaurin-α/PIP3BP was recently shown to exhibit Arf GAP (ADP-ribosylating factor GTPase activating protein) activity in vivo (Venkateswarlu et al., 2004). Our results provide the first evidence that PIP3 is transported by the motor-dependent mechanism.
Results
The forkhead-associated (FHA) domain of GAKIN mediates the biochemical interaction with PIP3BP
Using full-length bovine PIP3BP as bait, we performed a yeast two-hybrid screen in HeLa cell cDNA library and isolated a partial cDNA of human GAKIN, a kinesin-like protein. Recently, in a collaborative study, human centaurin-α1 was used as a bait to isolate rat homologue of GAKIN/KIF13B from rat brain cDNA library (Venkateswarlu et al., 2005). We confirmed the interaction between PIP3BP and GAKIN and mapped the binding site using the GST pull-down assay. Defined segments of GAKIN, covering the region originally isolated by the yeast two-hybrid screen, were expressed in 293T human kidney cells, and their biochemical interactions with GST-PIP3BP immobilized on glutathione–Sepharose beads were tested. The construct spanning the motor-FHA domains (1–557) interacted specifically with GST-PIP3BP but not with the GST control (Fig. 1 C). Deletion of the FHA region within this segment completely abolished the interaction (motor 1–486), and a short construct containing only the FHA domain (FHA 455–557) showed specific binding to PIP3BP. To further test whether the observed biochemical interaction is direct, we incubated GST-PIP3BP with thioredoxin (Trx) fusion protein of the GAKIN FHA domain immobilized on agarose beads. GST-PIP3BP specifically interacted with Trx-FHA but not with the control Trx (Fig. 1 D). Together, these results demonstrate that the FHA domain of GAKIN mediates its direct interaction with PIP3BP.
Figure 1.

The FHA domain of GAKIN mediates the biochemical interaction with PIP3BP. (A) Schematic representation of GAKIN domain organization and various constructs. Kinesin family motor domain (motor), FHA, MAGUK binding stalk domain (MBS), and microtubule binding CAP-Gly domain are depicted. YTH indicates the clone obtained from the yeast two-hybrid screen. (B) Domain organization of PIP3BP. Nuclear localization signal (NLS), Arf-GAP–like zinc-finger motif (zinc finger), and PH domains are depicted. (C) GST pull-down assay. Defined segments of GAKIN were expressed as GFP fusion proteins in 293T cells, and their interactions with PIP3BP were tested by GST pull-down assay using GST-PIP3BP. GFP-GAKIN segments pulled down by GST-PIP3BP were detected by Western blotting using anti-GFP polyclonal antibody. Recombinant GST and GST-PIP3BP proteins used for the pull-down experiments were shown by Ponceau staining of the nitrocellulose membrane. (D) Direct interaction between purified GST-PIP3BP and Trx-FHA of GAKIN. Trx-FHA and control Trx proteins were immobilized on S-protein agarose beads (Novagen) and incubated with GST-PIP3BP. Beads were recovered by centrifugation, and bound proteins were analyzed by SDS-PAGE and Coomassie blue staining. (E) Coimmunoprecipitation of PIP3BP and GAKIN. COS-7 cells were transfected with plasmids encoding full-length clones of FLAG-GAKIN or -KIF13A and myc-PIP3BP. Immunoprecipitation was performed from the cell lysates using anti-myc monoclonal antibody. FLAG- and myc-tagged proteins were detected by Western blotting using anti-FLAG and anti-myc monoclonal antibodies.
To demonstrate the in vivo association between PIP3BP and GAKIN, we performed coimmunoprecipitation experiments from COS-7 cells transiently expressing PIP3BP and full-length GAKIN. The COS-7 cells were transfected with myc-PIP3BP and FLAG-GAKIN, and an anti-myc antibody was used to immunoprecipitate the complex. Western blotting with anti-FLAG antibody detected GAKIN in the myc immunoprecipitate (Fig. 1 E, second lane). We also tested KIF13A, the closest homologue of GAKIN (Nakagawa et al., 2000), for its potential interaction with PIP3BP. The FHA domains of KIF13A and GAKIN share 70% amino acid identity. Interestingly, however, we failed to detect any interaction of KIF13A with PIP3BP in the coimmunoprecipitation experiments (Fig. 1 E, third lane). This result suggests that the interaction between PIP3BP and GAKIN is highly specific. It is noteworthy that the FHA domain is a widely conserved module in the kinesin-3 family members (Lawrence et al., 2004); however, its function is poorly understood in the context of motor proteins.
GAKIN is a microtubule plus-end motor
As PIP3BP specifically interacts with PIP3 via its PH domains (Tanaka et al., 1997), we hypothesized that PIP3BP mediates the physical linkage between GAKIN and PIP3-containing vesicles, thereby facilitating the microtubule-dependent transport of the cargo vesicles containing PIP3. To test this hypothesis, we first characterized the motor activity of GAKIN by the microtubule gliding assay. Recombinant GAKIN containing motor and FHA domains (1–557) was expressed in Sf9 cells using the baculovirus expression system, and purified protein was coated on the glass surface of the motility chamber. Microtubules were polarity labeled with Alexa 488 to highlight their minus ends, and their gliding movement was visualized using time-lapse fluorescence microscopy (Fig. 2 A). The smooth movement of microtubules toward their brightly labeled minus end was reproducibly observed in multiple trials, demonstrating that GAKIN is a plus end–directed microtubule motor. A histogram showing the velocity of labeled microtubules was generated from multiple tracings and is shown in Fig. 2 B. The mean velocity of moving microtubules was estimated to be 1.66 μm/s.
Figure 2.

GAKIN is a microtubule plus-end motor. (A) Microtubules gliding on the glass surface coated with motor-FHA of GAKIN. Microtubules were polarity labeled by Alexa 488 to highlight their minus ends. Arrowheads point to the minus end of the microtubule. Motility was observed on glass coverslips coated with GAKIN motor-FHA (1–557) recombinant protein. Microtubules moved toward their minus ends with a velocity of ∼1.66 μm/s, suggesting that GAKIN is a plus-end motor. Bar, 5 μm. (B) Histogram of the velocity of microtubules gliding.
PIP3BP and GAKIN form a complex on synthetic liposomes containing PIP3
Next, we tested whether PIP3BP and GAKIN can form a macromolecular complex on the surface of PIP3-containing liposomes. Purified recombinant GST-PIP3BP and GAKIN (motor-FHA segment 1–557) were incubated with synthetic liposomes containing 10% PIP3 and 90% phosphatidylcholine (PC), and the liposome and soluble protein components were separated in the top and bottom fractions, respectively, by sucrose density gradient centrifugation. GAKIN was recovered from the top (liposome fraction) of the gradient when PIP3BP and PIP3 liposomes were present (Fig. 3, top, lanes 5 and 6). In contrast, GAKIN was recovered only from the bottom (soluble protein fraction) of the gradient when control GST was used instead of GST-PIP3BP (Fig. 3, top, lanes 3 and 4) or when 15% PIP2 liposomes were used instead of PIP3 (Fig. 3, top, lanes 7 and 8). The GST-PIP3BP was consistently recovered from the top fraction with PIP3 liposomes (Fig. 3, bottom, lanes 5 and 6) and only in the bottom fraction when 10% PIP2 liposomes were used (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1). When 15% PIP2 liposomes were used as a negative control, only a trace amount (<0.5%) of GST-PIP3BP was recovered in the top fraction, and most of the protein remained in the bottom fraction (Fig. 3, bottom, lanes 7 and 8). The lipid binding specificity of PIP3BP was documented previously (Hammonds-Odie et al., 1996; Tanaka et al., 1997; Venkateswarlu et al., 1999). Based on our Western blot results, the affinity of PIP3BP to 10% PIP3 liposomes appears to be >100 times better than 15% PIP2 liposomes. As a negative control, GST was recovered only from the bottom fraction (Fig. 3, bottom, lanes 3 and 4). Together, our data are consistent with the notion that PIP3BP is a specific binder of PIP3 and demonstrate that PIP3BP can bind both GAKIN and PIP3 simultaneously, thereby serving as a molecular linker connecting GAKIN to PIP3-containing lipid vesicles.
Figure 3.

PIP3BP and GAKIN form a complex on synthetic liposomes containing PIP3. Binding of recombinant GST-PIP3BP and GAKIN motor-FHA (1–557) to liposomes was examined using a membrane flotation assay. PIP3-containing liposomes (10% PIP3 and 90% PC) or PIP2-containing liposomes (15% PIP2 and 85% PC) were incubated with GAKIN motor-FHA (1–557) and GST-PIP3BP or control GST, and liposome and soluble protein fractions were separated by sucrose gradient centrifugation. Liposomes were recovered from the top of sucrose step gradient, whereas the unbound protein remained at the bottom. GAKIN motor-FHA (1–557) and GST-PIP3BP in the top and bottom fractions were detected by Western blotting using anti-GAKIN and anti-GST polyclonal antibodies, respectively.
GAKIN transports PIP3 liposomes via PIP3BP in vitro
We then examined whether the complex of GAKIN and PIP3BP formed on PIP3 liposomes could support actual movement of the liposomes along microtubules. To demonstrate this possibility, we reconstituted the microtubule-dependent movement of liposomes using purified recombinant PIP3BP and GAKIN. A dense microtubule lawn was prepared on the glass surface of the motility chamber using Alexa 488–labeled microtubules. To visualize liposome movement by fluorescence microscopy, liposomes were labeled using rhodamine-phosphatidylethanolamine (Rh-PE). Processive movement of PIP3 liposomes along microtubules was observed in the presence of GAKIN, PIP3BP, and ATP (Fig. 4 A and Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1). Mean velocity of liposome movement was ∼0.7 μm/s (Fig. 4 B). No processive movement was observed in the absence of GAKIN, PIP3BP, or ATP or when PIP2 was used instead of PIP3, demonstrating the specificity of the liposome movement (Fig. 4 C). This result suggests that GAKIN is capable of transporting PIP3-containing vesicles via direct interaction with PIP3BP.
Figure 4.

GAKIN transports PIP3 liposomes via PIP3BP in vitro. (A) Time-lapse visualization of a rhodamine-labeled PIP3 liposome (red) moving along microtubules (green) in the presence of GST-PIP3BP and GAKIN motor-FHA (1–557) recombinant proteins. Bar, 5 μm. (B) Histogram of the velocity of liposome motility generated by GAKIN motor-FHA (1–557) at 18 mM ATP (see Materials and methods). (C) Specificity of liposome motility by recombinant GAKIN motor-FHA (1–557) with or without PIP3 and GST-PIP3BP. Liposome motility was observed only in the presence of PIP3, recombinant GST-PIP3BP, GAKIN, and ATP.
GAKIN contributes to the accumulation of PIP3 in the neurite ends of PC12 cells
Next, we asked whether this PIP3 transport by GAKIN–PIP3BP complex plays a functional role in vivo. As endogenous GAKIN and PIP3BP can be coimmunoprecipitated from rat brain lysate (Venkateswarlu et al., 2005), we surmised that the neuronal cell might be the site where such transport is important. Therefore, we selected PC12 cells, the widely used cell line with neuronal properties, for further studies. In PC12 cells, PIP3 is highly enriched at the tip of growing neurites (Higuchi et al., 2003; Aoki et al., 2005) and its signaling is important for neurite outgrowth (Kobayashi et al., 1997; Kita et al., 1998). First, we examined whether PIP3BP and GAKIN colocalize at specific sites in PC12 cells. When PC12 cells were cotransfected with myc-PIP3BP and GFP-GAKIN and treated with NGF, both proteins significantly colocalized and accumulated at the tip of neurites (Fig. 5 A). Higher magnification images show that both proteins are found at the growth cone protrusions (Fig. 5 A). To visualize the localization of PIP3, the PH domain construct of Akt (GFP-Akt-PH), which specifically interacts with PIP3 in the cells (Varnai and Balla, 1998), was used. As reported previously (Higuchi et al., 2003), accumulation of GFP-Akt-PH was observed at the tip of growing neurites (Fig. 5 B). Endogenous GAKIN was also found concentrated at the tip of the neurites, where it colocalized with GFP-Akt-PH (Fig. 5 B). To test whether the observed colocalization of GAKIN, PIP3BP, and PIP3 is the consequence of the transport of PIP3 by the GAKIN–PIP3BP complex, we decided to use the dominant-negative (DN) approach by blocking specific interactions. The DN construct of GAKIN is a motor-deleted mutant of GAKIN, which can still bind to PIP3BP via its FHA domain (unpublished data). Unlike full-length GAKIN, which accumulates only at the tip of neurites (Fig. 5 A and Fig. 6 B), the transfected DN-GAKIN in PC12 cells distributes along neurites and the cell body. The accumulation of DN-GAKIN is still evident at the tip (Fig. 6 B); however, it is not as intense and, consequently, the cell body and the entire length of neurites are visible. This distribution suggests the inability of DN-GAKIN to translocate its cargo to its final destination. Accumulation of PIP3, as monitored by the GFP-Akt-PH detector, at the tip of neurites is significantly inhibited by the overexpression of DN-GAKIN (Fig. 6, B and C). The relative intensity of GFP-Akt-PH and anti-FLAG staining was measured and is shown in the graphical format (Fig. 6 B and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1). Because the FHA domain of GAKIN is required for its interaction with PIP3BP, two additional constructs lacking the FHA domain of GAKIN were made and used as negative controls (Fig. 6). In the Δ-FHA construct of GAKIN, the FHA domain was deleted from the DN-GAKIN mutant. Thus, the Δ-FHA construct starts with the membrane-associated guanylate kinase (MAGUK) binding stalk domain and extends up to the Cap-Gly domain. A chimera construct (FHA-chimera) of GAKIN was also made where its FHA domain was replaced with the FHA domain of KIF13A, the closest homologue of GAKIN, which does not bind to PIP3BP. Thus the FHA-chimera contains the FHA domain of KIF13A and the MAGUK binding stalk to Cap-Gly domains of GAKIN. Unlike DN-GAKIN, neither of these constructs showed any effect on PIP3 localization (Fig. 6). These results suggest that PIP3 is transported to the distal end of neurites by a GAKIN-dependent mechanism.
Figure 5.

PIP3, PIP3BP, and GAKIN colocalize at the tip of growing neurites in PC12 cells. (A) PIP3BP and GAKIN colocalize at the tip of growing neurites and growth cones of PC12 cells. PC12 cells were transfected with GFP-PIP3BP (full length) and FLAG-GAKIN (full length) or GFP-GAKIN and myc-PIP3BP. 1 d after transfection, cells were stimulated with 5.0 ng/ml NGF for 36 h and fixed for immunofluorescence analysis. The merged image shows significant colocalization at the distal end of the neurite. Higher magnification images of cells transfected with GFP-PIP3BP and FLAG-GAKIN show significant colocalization of PIP3BP and GAKIN at the tip of the growth cone. (B) PIP3 colocalizes with endogenous GAKIN at the tip of growing neurites. PC12 cells were transfected with GFP-Akt-PH to monitor PIP3 localization and stimulated with NGF as described. Endogenous GAKIN was stained with anti-GAKIN monoclonal antibody. The merged image and measured relative fluorescence intensity show significant colocalization at the tip of neurites.
Figure 6.

GAKIN contributes to the accumulation of PIP3 at the tip of growing neurites in PC12 cells. (A) Schematic diagram of domain organization of several GAKIN constructs used in these experiments. (B) PIP3 accumulation is blocked by DN-GAKIN. PC12 cells were transfected with GFP-Akt-PH and FLAG-DN-GAKIN, FLAG-GAKIN (full length), FLAG-Δ-FHA, or FLAG-FHA-chimera and stimulated with NGF. Arrowheads represent neurites with PIP3 accumulation at the tip, whereas arrows represent neurites without PIP3 accumulation. Overexpression of DN-GAKIN significantly reduced the accumulation of PIP3 at the neurite tips. Bars, 20 μm. (C) Quantification of PIP3 accumulation at the neurite tips. The numbers of the neurites with and without accumulation of PIP3 were counted. Values from three independent experiments are represented as means + SD (percentage of PIP3 accumulation: GFP-Akt-PH alone, 84 ± 0.5%, n = 1007; GAKIN + GFP-Akt-PH, 87 ± 0.7%, n = 395, P = 0.002; DN-GAKIN + GFP-Akt-PH, 60 ± 3%, n = 693, P < 0002; Δ-FHA + GFP-Akt-PH, 81 ± 7%, n = 220, P = 0.5; FHA-chimera + GFP-Akt-PH, 86 ± 4%, n = 131, P = 0.4; *, P < 0.002).
GAKIN-mediated transport of PIP3 regulates neuronal polarity
In cultured hippocampal neurons, the location of PIP3 was monitored by the GFP-Akt-PH detector and found highly enriched in the distal end of the longest neurite (Fig. 7, A and B) in contrast to the diffused fluorescence observed for GFP (Fig. 8 A and Fig. S2). This location of PIP3 is consistent with previously published studies (Shi et al., 2003; Menager et al., 2004). Endogenous PIP3BP was found colocalized with GFP-Akt-PH at the end of the longest neurite (Fig. 7 A). As was the case in the PC12 cells, overexpressed full-length GAKIN was found highly enriched at the distal ends of neurites, often at their multiple termini (Fig. 7 B, middle). Significant colocalization of GFP-Akt-PH with overexpressed GAKIN was observed, and the accumulation of GFP-Akt-PH appeared to be enhanced under these conditions (Fig. 7 B, middle). On the other hand, overexpression of DN-GAKIN caused the loss of enriched accumulation of GFP-Akt-PH at the tip of the longest neurite (Fig. 7 B, right). Similar to the pattern observed in the case of GFP-Akt-PH expression alone in control neurons, some staining of GFP-Akt-PH could be visualized at the end of short neurites in DN-GAKIN transfected cells. As before, the relative intensity was measured and is shown in Fig. 7 B (Fig. S2). We quantified the GFP-Akt-PH accumulation by scoring the cells with the relative intensity ratio between neurite tip and cell body (Fig. 7 C), and the representative images are presented (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1). The data show that upon overexpression of DN-GAKIN, a subpopulation of cells that show high accumulation at the tip (tip/cell body ratio >1) disappeared. In contrast, cells with a high accumulation ratio are increased when full-length GAKIN is overexpressed. These results suggest that the accumulation of PIP3 in hippocampal neurons is dependent on the activity of GAKIN.
Figure 7.

PIP3 and PIP3BP colocalize at the tip of the axon in hippocampal neurons, and this accumulation is modulated by the overexpression of GAKIN. (A) PIP3 colocalizes with endogenous PIP3BP at the tip of the axon in cultured mouse hippocampal neurons. Hippocampal neurons were transfected with GFP-Akt-PH. Endogenous PIP3BP was stained with anti-PIP3BP rabbit antiserum. Arrowheads point to the tip of the axon. Magnified images are shown in the bottom panels. (left) Diagram shows the outline of the cell observed. (B) Overexpression of GAKIN constructs alters the PIP3 localization. Hippocampal neurons were transfected with GFP-Akt-PH and FLAG-GAKIN or FLAG-DN-GAKIN. Overexpression of full-length GAKIN enhanced the accumulation of PIP3, often at the multiple termini of the neurites. Overexpression of DN-GAKIN caused the loss of accumulation of PIP3 at the end of the axon-like neurite. (C) The subpopulation of hippocampal neurons scored as relative fluorescence intensity of GFP-Akt-PH as (neurite tip/cell body) ratio. Fluorescence intensity at the tip was measured and normalized by the intensity at the cell body. The population of cells that show high accumulation at the tip (tip/cell body ratio >1), normal accumulation (tip/cell body ratio >0.5 to <1) or less accumulation (tip/cell body ratio <0.5) are shown in the graphical format (number of cell analyzed and value of a t test: GFP-Akt-PH, n = 23; DN-GAKIN + GFP-Akt-PH, n = 31, P < 0.01; GAKIN + GFP-Akt-PH, n = 27, P < 0.01).
Figure 8.

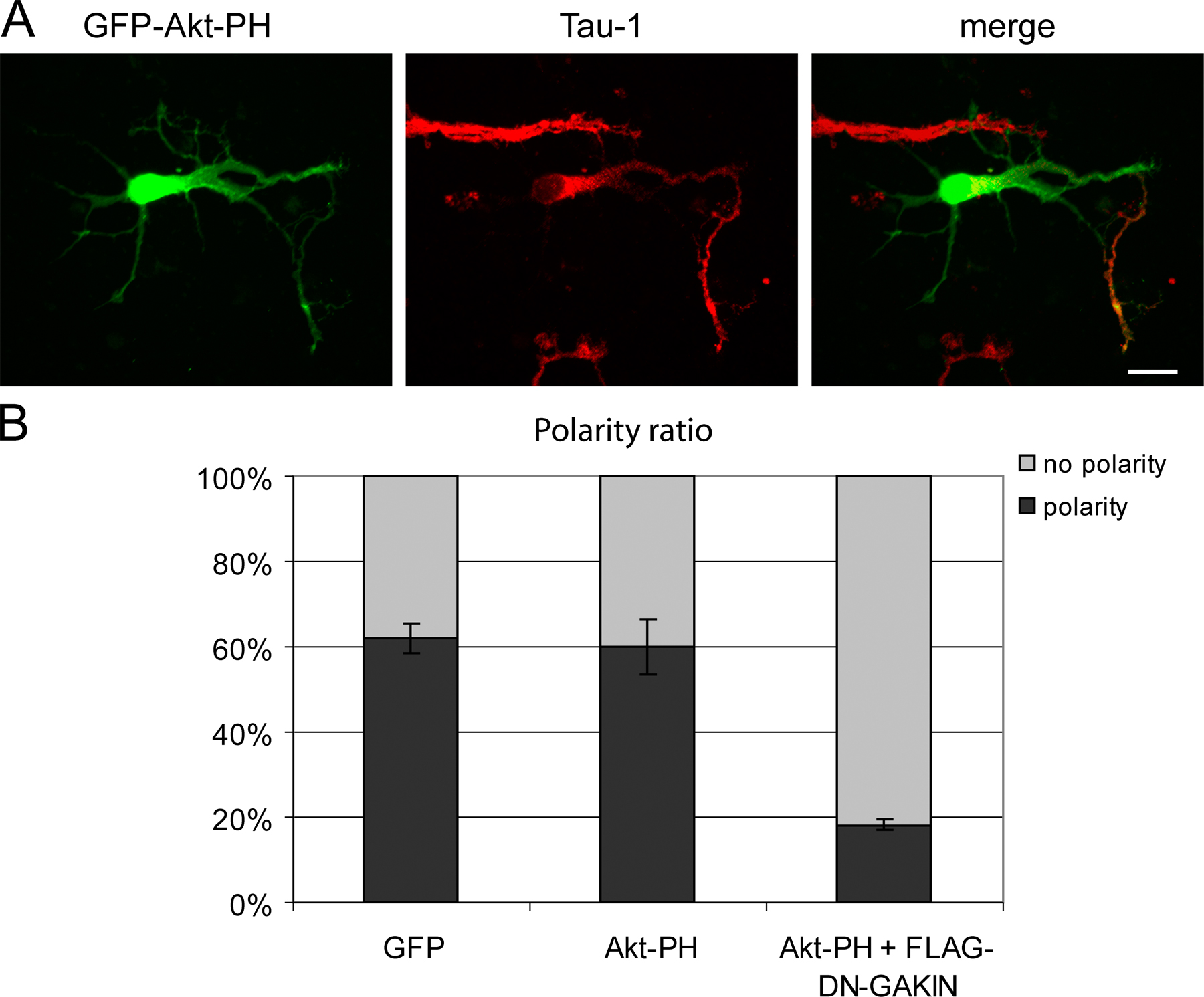
GAKIN-mediated transport of PIP3 regulates neuronal polarity of hippocampal neurons. (A) Overexpression of GAKIN constructs affects the neuronal polarity. GFP, as a morphological marker, was cotransfected with FLAG-GAKIN or FLAG-DN-GAKIN. Neuronal polarity was determined as described in Materials and methods. Overexpression of GAKIN, as well as DN-GAKIN, caused the loss of the axonally specified neurites. (B) Quantification of neuronal polarity formation as determined in A (percentage of neurons polarized: GFP, 62 ± 3%, n = 185; GFP-GAKIN, 26 ± 8%, n = 131, P < 0.002; GFP-DN-GAKIN, 19 ± 5%, n = 193, P < 0.0005; GFP-motor, 74 ± 8%, n = 55, P = 0.08; GFP-motor-FHA, 31 ± 13%, n = 43, P = 0.02; GFP-KIF13A, 74 ± 11%, n = 32, P = 0.16; FLAG-GAKIN, 19 ± 4%, n = 120, P < 0.0002: FLAG-DN-GAKIN, 6.4 ± 1%, n = 185, P < 0.00002; *, P < 0.05). (C) Loss of the Tau-1–positive axon structure by overexpression of GAKIN and DN-GAKIN. Bars, 20 μm.
Also, we noticed that the cells overexpressing GAKIN and DN-GAKIN often lost the typical morphology of the well-differentiated axon-dendrite structure, leading to the formation of multiple, highly branched neurites (Fig. 7 B). In the case of full-length GAKIN, this could be attributed to the hyperaccumulation of PIP3 at the ends of multiple neurites. Generally, PIP3 without the overexpression of GAKIN would accumulate in the longest neurite observed in normally developing neurons. In the case of DN-GAKIN, the reduction of PIP3 accumulation in the longest neurite end might inhibit proper axon-dendrite specification. Therefore, we quantified the number of cells with axon-dendrite polarity based on the morphological observation (Shi et al., 2003; Menager et al., 2004) upon overexpression of GAKIN and DN-GAKIN. It is noteworthy that a recent study examined the role of PIP3 in neuronal polarization and axon formation and established that the hippocampal neurons expressing low levels of the GFP-Akt-PH construct retained cell polarity (Menager et al., 2004). Following this experimental strategy, we used a highly purified preparation of GFP-Akt-PH plasmid and selected neurons expressing low levels of GFP-Akt-PH. Similar to the GFP transfected control neurons, the GFP-Akt-PH transfected cells maintained cell morphology as established by the observation of Tau-1–positive neurites in individual neurons (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1). However, to further eliminate the possibility of deleterious effects of the GFP-Akt-PH detector on the neuronal polarity (Shi et al., 2003), GAKIN and DN-GAKIN were cotransfected with GFP as a morphological marker. Representative images (Fig. 8 A) and the quantification data (Fig. 8 B) demonstrate that overexpression of GAKIN and DN-GAKIN caused the loss of neuronal polarity. To confirm that such an effect is mediated by the FHA–PIP3BP interaction, the effect of the motor-FHA (1–557) domain of GAKIN was tested. When expressed in primary neurons, the motor-FHA domain showed diffuse distribution throughout the cell and did not accumulate at the neurite ends (Fig. S5), suggesting that it is not capable of transporting cargo in vivo. The motor-FHA domain suppressed neuronal polarity, whereas the motor domain alone (1–486, without the FHA domain) did not (Fig. 8 B and Fig. S5). These results suggest the necessity of the FHA domain for the DN effect of GAKIN mutants on neuronal polarity. As a negative control, KIF13A had no effect on polarity.
To further confirm this effect on neuronal polarity, transfected neurons were stained for tau, a marker for axon differentiation (Shi et al., 2003; Menager et al., 2004). GFP-expressing control neuron exhibited a single long Tau-1–positive neurite, which is an axon (Fig. 8 C, left). However, neurons expressing full-length GAKIN or DN-GAKIN did not have Tau-1–positive neurites (Fig. 8 C). These results confirmed the loss of polarity observed by the morphological analysis.
Discussion
In this study, we demonstrate that a complex of PIP3BP and GAKIN provides a novel mechanism for motor-mediated transport of PIP3-containing vesicles along microtubules (Fig. 9). Previously, a model for the transport of PIP2-containing vesicles by Unc104/KIF1A, an evolutionally conserved kinesin-like motor protein, was proposed (Klopfenstein et al., 2002). In that model, a PH domain located at the C-terminal tail of Unc104 interacts with PIP2 specifically, thereby connecting the PIP2 vesicles to microtubule-dependent transport (Fig. 9, left). Our results propose a new model in which PIP3BP serves as a molecular linker connecting PIP3 vesicles to a motor protein (Fig. 9, right).
Figure 9.

A proposed model for the intracellular transport mechanism of PIP2 and PIP3 cargo vesicles by kinesins.
The PIP3BP binding site in GAKIN was localized to a small region encoded by the FHA domain (Fig. 1). The FHA domain was originally discovered in transcription factors and DNA repair proteins and is now well established as the phosphothreonine binding protein domain (Durocher and Jackson, 2002). All members of the kinesin-3 subfamily of proteins, including GAKIN, contain an FHA domain in close proximity to the N-terminal motor domain (Lawrence et al., 2004). However, it is not known whether the FHA domain of kinesins recognizes phosphothreonine residues, as do other conventional FHA domains. Our direct binding experiments used bacterially expressed GST-PIP3BP and GAKIN FHA domain, suggesting that phosphorylation was not required for the ligand recognition of FHA, although we could not rule out the possibility that the recombinant GST-PIP3BP was phosphorylated in the bacterial host. Nonetheless, our results provide the first evidence indicating that the FHA domain of kinesin-3 family protein is a cargo binding domain.
Using in vitro motility assays, previous studies have determined the velocity range of kinesins to be ∼0.1–1.5 μm/s (for review see Hirokawa and Takemura, 2004a). We measured the velocity of the recombinant motor-FHA domain of GAKIN using a microtubule gliding assay in the presence of 1.0 mM ATP and ATP regeneration system, and it was determined to be ∼1.66 μm/s. Using a liposome motility assay, the velocity was determined to be ∼0.7 μm/s, which is slightly less than the velocity measured by the microtubule gliding assay. It may be speculated that in the microtubule gliding assay, multiple motor molecules contribute to the movement of each microtubule, whereas in the vesicle motility system, fewer motor molecules or possibly a single motor could be responsible for the movement of a single liposome. Overall, these values are consistent with the microtubule-dependent ATPase activity of the GAKIN motor domain that we reported previously (Asaba et al., 2003). These results indicate that the motor and FHA segment of GAKIN shows relatively fast anterograde activity; however, the motor activity of the full-length GAKIN remains to be established.
GAKIN was originally discovered as the binding partner for human discs large tumor suppressor protein (hDlg), a member of the MAGUK superfamily (Hanada et al., 2000). As a scaffolding protein with multiple protein–protein interaction motifs, hDlg is proposed to link specific cargo vesicles to GAKIN for their transport to specialized membrane regions (Asaba et al., 2003). Its Drosophila melanogaster homologue, Dlg, is an important polarity determination factor in epithelial cells and asymmetric cell division of neuroblasts (Knust and Bossinger, 2002). It is, therefore, possible that the transport of hDlg by GAKIN also has significant effects on cell polarity in general. However, in the current setting, we believe that the effect on the neuronal polarity is primarily regulated by GAKIN–PIP3BP–mediated transport of PIP3, mainly because (1) significant enhancement of PIP3 accumulation that was observed upon overexpression of full-length GAKIN correlated with the loss of polarity and (2) the motor-FHA construct, which binds to PIP3BP but not to hDlg, exhibited DN effects on neuronal polarity. The precise relationship of two potential cargos of GAKIN, mediated by PIP3BP and hDlg, will be a subject of future studies.
Generally, PIP3 accumulates only at the tip of the longest neurite of a neuronal cell and, thus, the PIP3-positive neurite receives the exclusive differentiation cue to develop into an axon. Our images of GFP-Akt-PH localization in the control neurons (Fig. 7, A and B, left) are consistent with this model, with the signal accumulating only in the single terminus of the longest neurite. When the full-length GAKIN is overexpressed in neurons, it tends to accumulate not only to the longest neurite but also at multiple termini of every growing neurite (Fig. 8). Consequently, the GFP-Akt-PH accumulation is detected in multiple termini of several neurites where GAKIN accumulates (Fig. 7 B). Our interpretation is that the accumulation of PIP3 in the longest neurite as well as at the ends of multiple neurites disrupts or delays the axon-specification signal, thus causing the loss of tau-positive (axon-bearing) neurons under these experimental conditions (Fig. 8). In differentiated neurons, how kinesin motors recognize axon from dendrites and transport axon-specific cargo to axons and dendrite-specific cargo to dendrites remains unresolved. However, in cultured hippocampal neurons that are yet to be fully differentiated, motors may not be able to distinguish one neurite from another, which appears to be the case for GAKIN.
PIP3, by accumulating in specialized membrane sites in the cell, is an important cellular polarity determination factor (Bourne and Weiner, 2002). As is the case in hippocampal neurons, where the localized accumulation of PIP3 determines the axon-dendrite polarity, localization of PIP3 at the leading edge determines the internal polarity of neutrophil and Dictyostelium discoideum during their chemotaxis (Chung et al., 2001; Wang et al., 2002; Sadhu et al., 2003; Xu et al., 2005). How is the local gradient of PIP3 formed? One possible mechanism to initiate and maintain localized PIP3 is the positive-feedback loop formed between PIP3 and small GTPases, Rac and Cdc42 (Weiner et al., 2002; Srinivasan et al., 2003). In neutrophils, exogenously added PIP3 activates endogenous PI 3-kinase, suggesting the existence and importance of such a positive-feedback loop (Weiner et al., 2002). It was suggested that such positive-feedback pathways operate in the growth cone of neuronal cells (Aoki et al., 2005). Therefore, it has been assumed that local production of PIP3 at the tip of neurites and the enhancement of the signal by positive feedback, which activates further production of PIP3, are sufficient to explain the sustained local accumulation of PIP3 in neuronal cells (Menager et al., 2004; Aoki et al., 2005).
How, then, does our proposed model of PIP3 transport fit into the current paradigm? Our results demonstrate that full-length GAKIN overexpressed in hippocampal neurons accumulated at the distal ends of neurites and PIP3 accumulation is enhanced at sites where GAKIN is accumulated. In contrast, overexpression of a related motor protein, KIF13A, did not significantly affect PIP3 accumulation. These results suggest that GAKIN specifically transports PIP3 itself or transports the factors that stimulate the production of PIP3, to contribute to the local accumulation of PIP3. From our present data, it is not possible to differentiate between these two possibilities at this stage. In any event, these two possibilities may not be mutually exclusive, as the transported PIP3 can stimulate the local production of PIP3 by the aforementioned positive-feedback mechanism. The transport of PIP3-containing vesicles could move upstream and downstream components of the PIP3 signaling complex residing on the same vesicles as PIP3. Overall, our data suggests that GAKIN-dependent transport contributes to the local accumulation of PIP3 at the tip of neurites, thus supporting the existence of a novel mechanism for the proposed transport of PIP3 in vivo.
The conventional model of PIP3 as a second messenger describes that it is produced on the plasma membrane by a signaling complex formed by the activated receptors and is metabolized quickly by the lipid phosphatase activity such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), thus eliciting only the transient response on site (Cantley, 2002). Can PIP3 be found on the intracellular vesicles that are transportable? Significant evidence has been reported that demonstrates the production of PIP3 at the intracellular membranes (Sato et al., 2003). Also, in sympathetic neurons, locally administered NGF at the tip of axons leads to activation of PI 3-kinase at the cell body (Kuruvilla et al., 2000) by the retrograde transport of the internalized activated NGF receptor (Miller and Kaplan, 2001; Ye et al., 2003; Kuruvilla et al., 2004). Therefore, it is reasonable to assume that in developing neurons a significant amount of PIP3 is continuously produced at intracellular membrane vesicles in the cell body.
In neurons, which often extend exceptionally long processes, transport of specific cargo molecules by motor proteins is essential for the proper targeting of cellular components (Hirokawa and Takemura, 2004b). For example, signaling components downstream of PIP3 that regulate axonal specification, such as CRMP-2 (collapsin response mediated protein 2) and Par3/Par6/aPKC, are transported by kinesin-1 and -2, respectively (Nishimura et al., 2004; Kimura et al., 2005). We propose that the GAKIN–PIP3BP complex transports PIP3-containing vesicles from the cell body to the distal end of the neurites. Directed transport of PIP3 and its signaling components to the specialized membrane region contributes to the local enhancement of the signaling strength at the neurite ends, thus leading to a precipitous breakdown of the cellular symmetry.
Materials and methods
Tissue culture and transfections
HEK293T and COS-7 cells were maintained in DME (Invitrogen) supplemented with 10% FCS (Hyclone). PC12 cells were cultured in DME supplemented with 10% FCS and 5% horse serum (Invitrogen). DNA transfection in COS-7, PC12 cells, and hippocampal neurons were performed using Lipofectamine 2000 (Invitrogen). HEK293T cells were transfected using the calcium phosphate method (Tanaka et al., 1999).
Plasmids
GFP- and myc-PIP3BP (Tanaka et al., 1999), GFP-GAKIN, and GFP-DN- GAKIN (Asaba et al., 2003) were described previously. Other plasmids used to express tagged proteins were GFP and pEGFP-C1 (CLONTECH Laboratories, Inc.), Trx and pET32a (Novagen), and FLAG and pCMV-Tag2b (Stratagene). Corresponding cDNA fragments were cloned into vectors by conventional molecular biology methods. GFP-Akt-PH was a gift from T. Balla (National Institutes of Health, Bethesda, MD; Varnai and Balla, 1998).
Antibodies
Anti-GAKIN rabbit polyclonal antibody (Hanada et al., 2000) and anti-PIP3BP/centaurin-α rabbit anti-serum (Venkateswarlu et al., 2005) were described previously. Anti-GAKIN mouse monoclonal antibody (4A05) was generated against recombinant protein of GAKIN (1414–1826). Commercial antibodies used are anti-myc (9E10; Sigma-Aldrich), anti-FLAG (M2; Sigma-Aldrich), anti-GFP (Invitrogen), HRP-conjugated anti-GST (Santa Cruz Biotechnology, Inc.), and Tau-1 (Chemicon).
Recombinant protein expression
GST-PIP3BP and GST were expressed and purified as described previously (Tanaka et al., 1999). Trx-FHA protein of GAKIN, which also carries His- and S-protein tags, was expressed in BL21(DE3) cells and purified by Ni-NTA agarose (Novagen) chromatography. GAKIN His-motor-FHA (aa 1–557) was expressed in Sf9 cells using the Bac-to-Bac baculovirus expression system (Invitrogen). The protein was purified by Ni-NTA agarose.
Immunoprecipitation and GST pull-down assays
For immunoprecipitations, COS-7 cells were transfected with FLAG-GAKIN or -KIF13A along with myc-PIP3BP. Cells were lysed in the IP buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1.0 mM MgCl2, and 0.5% Triton X-100), and immunoprecipitations were performed using anti-myc monoclonal antibody (9E10). FLAG- and myc-tagged proteins were detected by Western blotting. For GST pull-down experiments, 293T cells were transfected with various GFP-GAKIN constructs and lysed in the IP buffer. GST-PIP3BP or control GST proteins immobilized on glutathione–Sepharose 4B beads were used to pull down interacting proteins from the lysate. GFP-GAKIN proteins bound to the beads were detected by Western blot using anti-GFP antibody.
Preparation of fluorescent microtubules
Tubulin was prepared from cow brain (Desai and Mitchison, 1998) and fluorescently labeled using Alexa 488 carboxylic acid succinimidyl ester (Invitrogen) as described previously (Nielsen et al., 2001). Microtubules were polymerized in the presence of 20 μl of unlabeled tubulin (2.5 mg/ml) and 5 μl of Alexa 488–labeled tubulin (0.5 mg/ml) using taxol (Calbiochem). Minus-end polarity-marked microtubules were prepared by a two-step polymerization protocol of labeled and unlabeled tubulins mixed at a defined ratio (Desai and Mitchison, 1998; Nielsen et al., 2001).
Microtubule gliding assay
A flow chamber with a volume of ∼10 μl was prepared using two glass coverslips (Corning) separated by two parallel strips of double-sided tape. 0.5 mg/ml of recombinant GAKIN motor-FHA (1–557) protein in BRB80 (80 mM Pipes, pH 6.8, 2.0 mM MgCl2, and 1.0 mM EGTA) supplemented with 0.2 mM Mg-ATP, 2.0 mM DTT, and 20% glycerol was incubated inside the flow cell for 10 min. After washing the flow cell with 10 μl of motility buffer (BRB80, 1.0 mM ATP, 20 μM taxol, 1.0 mg/ml casein, 50 mM glucose, 0.5 mg/ml glucose oxidase, 4.0 mM phosphoenolpyruvate, 20 μg/ml pyruvate kinase, and 0.14 mg/ml catalase), 10 μl of gliding buffer containing polarity-marked microtubules was introduced into the flow cell. The flow cell was sealed with petroleum jelly and visualized by fluorescence microscopy (TE2000E [Nikon]; Plan Apo 100× objective lens) at room temperature. Images were recorded by a camera (CoolSnapHQ; Roper Scientific) and MetaMorph software (Universal Imaging Corp.). Sequential photographs were taken every 1 or 5 s.
Preparation of liposomes
Egg yolk PC, Rh-PE, synthesized unsaturated PIP3, and synthesized unsaturated PIP2 (all from Avanti Polar Lipids, Inc.) were stored in organic solvents. 1.0 μmol PC was mixed with 1.0 or 10% (g/g lipids) PIP3 or 10 or 15% PIP2 and 0.05% Rh-PE and dried in a glass vial under the stream of dry nitrogen. The lipid film was further desiccated under high vacuum overnight, and lipids were rehydrated by the addition of BRB80 containing 5% sucrose and incubated at 70°C for 30 min with occasional mixing by vortex. Three freeze/thaw cycles were performed in the dry ice box and a 37°C water bath. A mini-extruder device with 250-μl syringes (Avanti Polar Lipids, Inc.) was used to extrude liposomes through a 100-nm pore polycarbonate filter (Whatman) at 70°C. Liposomes were stored in the dark at 4°C and used within 2 wk.
Liposome flotation assay
Liposomes containing 10 or 15% PIP2 or 10% PIP3 (5.0 nmol) were incubated on ice for 30 min with 70 pmol GST-PIP3BP and 30 pmol His-motor-FHA. BRB80 containing 2.0 M sucrose was added to the incubation reaction to bring the final sucrose concentration to 1.6 M, and this mixture was overlaid with cushions containing 1.4, 0.4, and 0.25 M sucrose in the same buffer in a centrifuge tube (7 × 20 mm). After centrifugation at 40,200 rpm at 4°C for 30 min in a rotor (Type 42.2Ti; Beckman Coulter), the 0.25/0.4 M fraction (top fraction) and the loading fraction (1.6 M sucrose; bottom fraction) was collected and analyzed by SDS-PAGE followed by Western blotting with anti-GAKIN polyclonal antibody and anti-GST antibody. For anti-GST Western blot, the loading ratio of top/bottom fraction was 5:1. For anti-GAKIN Western blot, the ratio was 25:1. GST-PIP3BP loaded in the input lane was 15 ng. The His-motor-FHA amount was 20 ng.
Liposome motility assay
A lawn of Alexa 488–labeled microtubules was prepared on the motility chamber to visualize the movement of rhodamine-labeled liposomes by fluorescence microscopy. To ensure the attachment of microtubules on the glass surface, the flow cell was first coated with recombinant His-GAKIN-CT (aa 1414–1826) that contains a microtubule binding CAP-Gly domain and binds avidly to microtubules in vitro. A similar approach was used before to immobilize microtubules on the glass surface coated with either anti-tubulin of poly-l-lysine (McGoldrick and Sheetz, 1998). After incubation of 0.125 mg/ml His-GAKIN-CT (in BRB80 containing 0.1 mg/ml BSA) in the flow cell for 10 min at room temperature, fluorescent-labeled microtubules (0.1–0.2 mg/ml in BRB80 supplemented with 10 μM taxol and 1.0 mM GTP) were introduced and incubated for 30 min at room temperature in a humidified chamber. The flow cell was washed twice with BRB80 supplemented with 5.0 mg/ml casein, 10 μM taxol, and 1.0 mM GTP and used immediately for the motility assay. Liposomes of various compositions were incubated with 0.15 nmol GST-PIP3BP on ice for 30 min, and stable liposome–PIP3BP complex was separated from unbound PIP3BP by ultracentrifugation using sucrose gradient as described (see Liposome flotation assay). The top fraction of the gradient containing the liposome–PIP3BP complex was mixed with 0.5 mg/ml of His-motor-FHA in the motility buffer and introduced into the flow cell containing a lawn of microtubules. The final concentration of liposomes was ∼50 μg/ml. Time-lapse imaging was captured as described for the microtubule gliding assay. Sequential photographs were taken every 1 s for 30 s. Imaging of the fluorescently labeled microtubules was captured for each frame and overlaid with the imaging of liposomes. The velocity of each moving liposome was measured using MetaMorph software by tracing the position of each liposome for every second. Videos were made using the MetaMorph software. Individual experiments were repeated more than three times for each sample (nine times for 1% PIP3-containing liposomes and six times for 10% PIP3-containing liposomes).
Immunofluorescence microscopy
PC12 cells were seeded onto coverslips (Fisher Scientific) coated with 0.01% poly-l-lysine (Sigma-Aldrich), stimulated with 5.0 ng/ml NGF for 36 h, and fixed with 4% PFA in PBS at room temperature for 15 min. Cells were then washed with PBS and permeabilized by PBS supplemented with 0.1% Triton X-100. Hippocampal neurons were fixed at day 3 in vitro with 4% PFA. Primary antibodies were anti-myc (1:2,000), anti-FLAG (1:2,000), anti-GAKIN monoclonal antibody (1:1,000), and anti-PIP3BP rabbit serum (1:500). Secondary antibodies were Alexa 594–conjugated anti-mouse and anti-rabbit antibodies (Invitrogen).
Analysis of GFP-Akt-PH accumulation in PC12 cells
The intensity of GFP-Akt-PH was observed by fluorescence microscopy using a 40× magnification setting. The expression of several GAKIN constructs was detected by staining with an anti-FLAG antibody. The number of neurites with and without the accumulation of GFP-Akt-PH fluorescence was manually counted. At least three independent experiments were performed for each construct (>70 cells and >300 neurites were observed for each experimental condition).
Hippocampal neuron culture
Methods for preparing the hippocampal cell cultures were essentially the same as those described previously (Banker and Goslin, 1998). Hippocampi were dissected from mice at embryonic day 16.5, and dissociated cells were plated in plating medium (DME supplemented with 10% FCS and 5% horse serum) onto glass coverslips coated with poly-l-lysine. After neurons attached to the substrate, the medium was exchanged to neuronal culture medium (MEM [Invitrogen] with B27 supplement [Invitrogen], 0.6% glucose, and 1 mM sodium pyruvate [Sigma-Aldrich]). Around 18 h after plating, neurons were transfected with various DNA constructs using Lipofectamine 2000.
Determination of neuronal polarity
Neuronal polarity was assessed by determining the percentage of neurons with a single long process that was at least twice as long as the other processes. For each construct, at least three independent experiments were performed. The t test was used for the statistical significance.
Online supplemental material
Fig. S1 shows that GAKIN and PIP3BP form a complex on PIP3 liposomes. Fig. S2 shows that relative fluorescence intensity was measured to show the accumulation of PIP3 at the tip of the neurite in PC12 cells and in neurons. Fig. S3 demonstrates that low-level expression of GFP-Akt-PH detector does not affect neuronal polarity. Fig. S4 shows the effect on PIP3 accumulation by overexpression of full-length GAKIN and DN-GAKIN in hippocampal neurons. Fig. S5 depicts the distribution of several GAKIN constructs expressed in hippocampal neurons. Videos 1 and 2 show that GAKIN transports PIP3 liposomes via PIP3BP in vitro. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200604031/DC1.
Supplementary Material
Acknowledgments
We are grateful to Drs. K. Venkateswarlu, Y. Gotoh, and T. Balla for reagents and advice. We thank Dr. H. Hashimoto for suggestions on liposome motility experiments.
This work was supported by the National Institutes of Health grants CA 94414 and HL60755. T. Hanada is the recipient of a Campus Research Board Award from the University of Illinois at Chicago (2004–2005).
Abbreviations used in this paper: DN, dominant-negative; FHA, forkhead-associated; GAKIN, guanylate kinase–associated kinesin; MAGUK, membrane-associated guanylate kinase; PC, phosphatidylcholine; PH, pleckstrin homology; PI, phosphatidylinositol; PI3P, PI-(3)-phosphate; PIP2, PI-(4,5)-bisphosphate; PIP3, PI-(3,4,5)-trisphosphate; Rh-PE, rhodamine-phosphatidylethanolamine; Trx, thioredoxin.
References
- Aoki, K., T. Nakamura, K. Fujikawa, and M. Matsuda. 2005. Local phosphatidylinositol 3,4,5-trisphosphate accumulation recruits Vav2 and Vav3 to activate Rac1/Cdc42 and initiate neurite outgrowth in nerve growth factor-stimulated PC12 cells. Mol. Biol. Cell. 16:2207–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaba, N., T. Hanada, A. Takeuchi, and A.H. Chishti. 2003. Direct interaction with a kinesin-related motor mediates transport of mammalian discs large tumor suppressor homologue in epithelial cells. J. Biol. Chem. 278:8395–8400. [DOI] [PubMed] [Google Scholar]
- Banker, G., and K. Goslin. 1998. Culturing Nerve Cells. 2nd edition. The MIT Press, Cambridge, MA. 666 pp.
- Bourne, H.R., and O. Weiner. 2002. A chemical compass. Nature. 419:21. [DOI] [PubMed] [Google Scholar]
- Cantley, L.C. 2002. The phosphoinositide 3-kinase pathway. Science. 296:1655–1657. [DOI] [PubMed] [Google Scholar]
- Chung, C.Y., S. Funamoto, and R.A. Firtel. 2001. Signaling pathways controlling cell polarity and chemotaxis. Trends Biochem. Sci. 26:557–566. [DOI] [PubMed] [Google Scholar]
- Desai, A., and T.J. Mitchison. 1998. Preparation and characterization of caged fluorescein tubulin. Methods Enzymol. 298:125–132. [DOI] [PubMed] [Google Scholar]
- Durocher, D., and S.P. Jackson. 2002. The FHA domain. FEBS Lett. 513:58–66. [DOI] [PubMed] [Google Scholar]
- Hammonds-Odie, L.P., T.R. Jackson, A.A. Profit, I.J. Blader, C.W. Turck, G.D. Prestwich, and A.B. Theibert. 1996. Identification and cloning of centaurin-alpha. A novel phosphatidylinositol 3,4,5-trisphosphate-binding protein from rat brain. J. Biol. Chem. 271:18859–18868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, T., L. Lin, E.V. Tibaldi, E.L. Reinherz, and A.H. Chishti. 2000. GAKIN, a novel kinesin-like protein associates with the human homologue of the Drosophila discs large tumor suppressor in T lymphocytes. J. Biol. Chem. 275:28774–28784. [DOI] [PubMed] [Google Scholar]
- Higuchi, M., K. Onishi, N. Masuyama, and Y. Gotoh. 2003. The phosphatidylinositol-3 kinase (PI3K)-Akt pathway suppresses neurite branch formation in NGF-treated PC12 cells. Genes Cells. 8:657–669. [DOI] [PubMed] [Google Scholar]
- Hirokawa, N., and R. Takemura. 2004. a. Kinesin superfamily proteins and their various functions and dynamics. Exp. Cell Res. 301:50–59. [DOI] [PubMed] [Google Scholar]
- Hirokawa, N., and R. Takemura. 2004. b. Molecular motors in neuronal development, intracellular transport and diseases. Curr. Opin. Neurobiol. 14:564–573. [DOI] [PubMed] [Google Scholar]
- Hoepfner, S., F. Severin, A. Cabezas, B. Habermann, A. Runge, D. Gillooly, H. Stenmark, and M. Zerial. 2005. Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell. 121:437–450. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., K. Chihara, N. Arimura, C. Menager, Y. Kawano, N. Matsuo, T. Nishimura, M. Amano, and K. Kaibuchi. 2001. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 4:781–782. [DOI] [PubMed] [Google Scholar]
- Kimura, K., S. Hattori, Y. Kabuyama, Y. Shizawa, J. Takayanagi, S. Nakamura, S. Toki, Y. Matsuda, K. Onodera, and Y. Fukui. 1994. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 269:18961–18967. [PubMed] [Google Scholar]
- Kimura, T., H. Watanabe, A. Iwamatsu, and K. Kaibuchi. 2005. Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 93:1371–1382. [DOI] [PubMed] [Google Scholar]
- Kita, Y., K.D. Kimura, M. Kobayashi, S. Ihara, K. Kaibuchi, S. Kuroda, M. Ui, H. Iba, H. Konishi, U. Kikkawa, et al. 1998. Microinjection of activated phosphatidylinositol-3 kinase induces process outgrowth in rat PC12 cells through the Rac-JNK signal transduction pathway. J. Cell Sci. 111:907–915. [DOI] [PubMed] [Google Scholar]
- Klopfenstein, D.R., and R.D. Vale. 2004. The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell. 15:3729–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein, D.R., M. Tomishige, N. Stuurman, and R.D. Vale. 2002. Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell. 109:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust, E., and O. Bossinger. 2002. Composition and formation of intercellular junctions in epithelial cells. Science. 298:1955–1959. [DOI] [PubMed] [Google Scholar]
- Kobayashi, M., S. Nagata, Y. Kita, N. Nakatsu, S. Ihara, K. Kaibuchi, S. Kuroda, M. Ui, H. Iba, H. Konishi, et al. 1997. Expression of a constitutively active phosphatidylinositol 3-kinase induces process formation in rat PC12 cells. Use of Cre/loxP recombination system. J. Biol. Chem. 272:16089–16092. [DOI] [PubMed] [Google Scholar]
- Kuruvilla, R., H. Ye, and D.D. Ginty. 2000. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 27:499–512. [DOI] [PubMed] [Google Scholar]
- Kuruvilla, R., L.S. Zweifel, N.O. Glebova, B.E. Lonze, G. Valdez, H. Ye, and D.D. Ginty. 2004. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 118:243–255. [DOI] [PubMed] [Google Scholar]
- Lawrence, C.J., R.K. Dawe, K.R. Christie, D.W. Cleveland, S.C. Dawson, S.A. Endow, L.S. Goldstein, H.V. Goodson, N. Hirokawa, J. Howard, et al. 2004. A standardized kinesin nomenclature. J. Cell Biol. 167:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoldrick, C., and M. Sheetz. 1998. Organelle motility and membrane network formation from cultured mammalian cells. Methods Enzymol. 298:353–360. [DOI] [PubMed] [Google Scholar]
- Menager, C., N. Arimura, Y. Fukata, and K. Kaibuchi. 2004. PIP3 is involved in neuronal polarization and axon formation. J. Neurochem. 89:109–118. [DOI] [PubMed] [Google Scholar]
- Miller, F.D., and D.R. Kaplan. 2001. On Trk for retrograde signaling. Neuron. 32:767–770. [DOI] [PubMed] [Google Scholar]
- Ming, G., H. Song, B. Berninger, N. Inagaki, M. Tessier-Lavigne, and M. Poo. 1999. Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron. 23:139–148. [DOI] [PubMed] [Google Scholar]
- Nakagawa, T., M. Setou, D. Seog, K. Ogasawara, N. Dohmae, K. Takio, and N. Hirokawa. 2000. A novel motor, KIF13A, transports mannose-6-phosphate receptor to plasma membrane through direct interaction with AP-1 complex. Cell. 103:569–581. [DOI] [PubMed] [Google Scholar]
- Nielsen, E., F. Severin, A.A. Hyman, and M. Zerial. 2001. In vitro reconstitution of endosome motility along microtubules. Methods Mol. Biol. 164:133–146. [DOI] [PubMed] [Google Scholar]
- Nishimura, T., K. Kato, T. Yamaguchi, Y. Fukata, S. Ohno, and K. Kaibuchi. 2004. Role of the PAR-3-KIF3 complex in the establishment of neuronal polarity. Nat. Cell Biol. 6:328–334. [DOI] [PubMed] [Google Scholar]
- Nishimura, T., T. Yamaguchi, K. Kato, M. Yoshizawa, Y. Nabeshima, S. Ohno, M. Hoshino, and K. Kaibuchi. 2005. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat. Cell Biol. 7:270–277. [DOI] [PubMed] [Google Scholar]
- Sadhu, C., B. Masinovsky, K. Dick, C.G. Sowell, and D.E. Staunton. 2003. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J. Immunol. 170:2647–2654. [DOI] [PubMed] [Google Scholar]
- Sato, M., Y. Ueda, T. Takagi, and Y. Umezawa. 2003. Production of PtdInsP3 at endomembranes is triggered by receptor endocytosis. Nat. Cell Biol. 5:1016–1022. [DOI] [PubMed] [Google Scholar]
- Shi, S.H., L.Y. Jan, and Y.N. Jan. 2003. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 112:63–75. [DOI] [PubMed] [Google Scholar]
- Shi, S.H., T. Cheng, L.Y. Jan, and Y.N. Jan. 2004. APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 14:2025–2032. [DOI] [PubMed] [Google Scholar]
- Srinivasan, S., F. Wang, S. Glavas, A. Ott, F. Hofmann, K. Aktories, D. Kalman, and H.R. Bourne. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K., S. Imajoh-Ohmi, T. Sawada, R. Shirai, Y. Hashimoto, S. Iwasaki, K. Kaibuchi, Y. Kanaho, T. Shirai, Y. Terada, et al. 1997. A target of phosphatidylinositol 3,4,5-trisphosphate with a zinc finger motif similar to that of the ADP-ribosylation-factor GTPase-activating protein and two pleckstrin homology domains. Eur. J. Biochem. 245:512–519. [DOI] [PubMed] [Google Scholar]
- Tanaka, K., K. Horiguchi, T. Yoshida, M. Takeda, H. Fujisawa, K. Takeuchi, M. Umeda, S. Kato, S. Ihara, S. Nagata, and Y. Fukui. 1999. Evidence that a phosphatidylinositol 3,4,5-trisphosphate-binding protein can function in nucleus. J. Biol. Chem. 274:3919–3922. [DOI] [PubMed] [Google Scholar]
- Varnai, P., and T. Balla. 1998. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol. 143:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswarlu, K., P.B. Oatey, J.M. Tavare, T.R. Jackson, and P.J. Cullen. 1999. Identification of centaurin-alpha1 as a potential in vivo phosphatidylinositol 3,4,5-trisphosphate-binding protein that is functionally homologous to the yeast ADP-ribosylation factor (ARF) GTPase-activating protein, Gcs1. Biochem. J. 340:359–363. [PMC free article] [PubMed] [Google Scholar]
- Venkateswarlu, K., K.G. Brandom, and J.L. Lawrence. 2004. Centaurin-alpha1 is an in vivo phosphatidylinositol 3,4,5-trisphosphate-dependent GTPase-activating protein for ARF6 that is involved in actin cytoskeleton organization. J. Biol. Chem. 279:6205–6208. [DOI] [PubMed] [Google Scholar]
- Venkateswarlu, K., T. Hanada, and A.H. Chishti. 2005. Centaurin-alpha1 interacts directly with kinesin motor protein KIF13B. J. Cell Sci. 118:2471–2484. [DOI] [PubMed] [Google Scholar]
- Wang, F., P. Herzmark, O.D. Weiner, S. Srinivasan, G. Servant, and H.R. Bourne. 2002. Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat. Cell Biol. 4:513–518. [DOI] [PubMed] [Google Scholar]
- Weiner, O.D., P.O. Neilsen, G.D. Prestwich, M.W. Kirschner, L.C. Cantley, and H.R. Bourne. 2002. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X., M. Meier-Schellersheim, X. Jiao, L.E. Nelson, and T. Jin. 2005. Quantitative imaging of single live cells reveals spatiotemporal dynamics of multistep signaling events of chemoattractant gradient sensing in Dictyostelium. Mol. Biol. Cell. 16:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, H., R. Kuruvilla, L.S. Zweifel, and D.D. Ginty. 2003. Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron. 39:57–68. [DOI] [PubMed] [Google Scholar]
- Yoshimura, T., Y. Kawano, N. Arimura, S. Kawabata, A. Kikuchi, and K. Kaibuchi. 2005. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 120:137–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}