

Abstract
N-syndecan (syndecan-3) is a transmembrane proteoglycan that is abundantly expressed in the major axonal pathways and in the migratory routes of the developing brain. When ligated by heparin-binding (HB) growth-associated molecule (GAM; pleiotrophin), N-syndecan mediates cortactin–Src kinase-dependent neurite outgrowth. However, the functional role of N-syndecan in brain development remains unexplored. In this study, we show that N-syndecan deficiency perturbs the laminar structure of the cerebral cortex as a result of impaired radial migration. In addition, neural migration in the rostral migratory stream is impaired in the N-syndecan–null mice. We suggest that the migration defect depends on impaired HB-GAM–induced Src kinase activation and haptotactic migration. Furthermore, we show that N-syndecan interacts with EGF receptor (EGFR) at the plasma membrane and is required in EGFR-induced neuronal migration.
Introduction
The expression of heparan sulfate (HS) in the developing brain is crucial for the correct formation of several brain structures. This was recently demonstrated with a knockout of the enzyme responsible for HS polymerization in proteoglycans (PGs; Inatani et al., 2003). The described phenotype indicates a general organizing role for HS and HS-binding growth factors in the brain. Because of the dramatic phenotype, it is difficult to tell how the individual HSPGs contribute to the brain morphology and what mechanisms are responsible for the phenotypic changes.
The syndecan family forms a rather diverse group of transmembrane HSPGs with a wide array of ligands and distinct cell signaling capabilities (Bernfield et al., 1992; Inatani et al., 2003). Although in some cases the family members have overlapping functions, syndecans and their HSs can bind distinct ligands and produce cellular responses unique to the particular syndecan in question (Bernfield et al., 1992; Lindahl et al., 1998). In addition, the in vivo expression pattern of each syndecan can differ greatly from the others. Thus, individual HSPGs are suspected to have unique functions in brain development.
N-syndecan and heparin-binding (HB) growth-associated molecule (GAM; pleiotrophin) act as a receptor–ligand pair in neurite outgrowth (Kinnunen et al., 1996, 1998a; for reviews see Rauvala and Peng, 1997; Rauvala et al., 2000). ECM-associated HB-GAM specifically binds the HS glycosaminoglycans present in the perinatal brain N-syndecan. Ligation of N-syndecan by HB-GAM triggers a signaling cascade involving the phosphorylation of c-Src and cortactin, thus affecting the actin assembly in the growth cones of neurites (for review see Rauvala and Peng, 1997; Kinnunen et al., 1998b). To date, their cofunction in neural migration has not been reported, although HB-GAM is known to mediate haptotactic migration of perinatal rodent forebrain cells via the receptor-type tyrosine phosphatase β/ζ, which is another transmembrane receptor of HB-GAM (Maeda and Noda, 1998). In addition, HB-GAM promotes osteoblast migration, which may be at least partially caused by its binding to N-syndecan (Imai et al., 1998).
In this study, we present findings suggesting an important role for N-syndecan in radial neural migration and in the rostral migratory stream (RMS) of the brain. Haptotactic migration caused by the binding of N-syndecan to its ligand HB-GAM offers a mechanistic explanation for the role of N-syndecan in neural migration. Furthermore, N-syndecan cooperates with the EGF receptor (EGFR) to regulate neural migration.
Results
N-syndecan knockout mice show a decreased accumulation of cells to the cortical plate
In the initial characterization of the N-syndecan knockout mice (Kaksonen et al., 2002), no changes were found in the gross anatomy of the brain. However, stereological analysis revealed an increased cell density in deep cortical areas and a decreased cell density in superficial layers of the cortex (Fig. 1 A).
Figure 1.

N-syndecan knockout cerebral cortex has altered morphology in terms of cell density. (A) In the adult cerebral cortex, the N-syndecan (NS) knockouts have an increased cell density in lamina VI and a decreased density in laminae II and III. (B) Cells labeled with BrdU at E15 do not ascend normally to the CP in N-syndecan knockout embryos. At age E18, the number of BrdU+ cells in the CP of the N-syndecan knockout (ko) is lower and in the SVZ/VZ is higher when compared with the wild-type (wt) littermates. (C) In the wild-type mice, the majority of the cells labeled at E15 migrate to the superficial parts of the CP, and the difference in BrdU+ cell density between VZ and CP is clear at age E18. In the N-syndecan knockout, the density difference between VZ and CP is small, illustrating the deficient accumulation of cells to the CP in the knockouts. (D) At P1, the distribution of BrdU+ cells in VZ and CP shifts slightly toward normal but is still clearly different from wild type. (E) At P10, all BrdU+ cells have shifted from VZ to CP in both wild types and knockouts. However, when P10 CP is divided to layers related to adult laminae, the knockout pups show similar skewing in the cell density as the adult knockouts. (F) The schematic drawing shows the cortical structures present at E18, and the gray arrow marks the radial migratory route examined in this experiment. IZ, intermediate zone; CC, corpus callosum. (G) In living brain slices, EGFR activation with EGF induces more efficient migration of BrdU-labeled cells from VZ to CP in wild-type embryos. The relative increase in BrdU+ cell density when compared with nonstimulated slices is ∼9% during 16 h. BrdU-labeled cells in the N-syndecan knockout slices do not show a similar increase in their migration upon stimulation. Error bars represent the SD of independent experiments; p-values were estimated with the t test.
Defective radial migration of neurons in the N-syndecan knockout brain is one possible mechanism to explain the phenotypic finding. This explanation appears plausible because N-syndecan is expressed in the neurites of embryonic brain neurons (Toba et al., 2002), and the major ligand of N-syndecan that enhances cell migration, HB-GAM, forms radially oriented streaks during and after the midgestation period in the rodent cortex (for review see Rauvala and Peng, 1997). Thus, we examined the migration of newly born neural cells in vivo in the N-syndecan knockout cortex by labeling the cells with BrdU at embryonic day (E) 15 and tracing the BrdU+ cells from cortical sections at E18, postnatal day (P) 1, and P10. The population that is labeled by BrdU in the ventricular zone (VZ) and sub-VZ (SVZ) of the dorsal cerebral cortex at E15 contains neurons that will end up in the most superficial laminae of the adult cerebral cortex. We determined the relative scattering of the BrdU+ cells in the cortical layers to reveal the possible differences in cell accumulation to the cortical plate (CP). At E18 in the N-syndecan knockout pups, the proportion of BrdU+ cells in the VZ/SVZ was higher than in the wild-type samples. Correspondingly, the amount of BrdU+ cells was lower in the CP in the knockouts (Fig. 1 B). The relative shift of BrdU+ cells from VZ/SVZ to the CP was clearly smaller in the knockouts than in the wild types (Fig. 1 C).
We continued to follow the fate of the labeled cell population, and at P1 in the N-syndecan knockout brains, the relative distribution of BrdU+ cells in layers in the VZ/SVZ and CP was still clearly different from the wild type but had normalized somewhat (Fig. 1 D). At P10 both in wild types and knockouts, all BrdU-labeled cells had shifted from below the subplate to the CP. The mature layer structure of cortex is already quite recognizable at P10, and we estimated the number of BrdU+ cells in the superficial layers II/III and in the deep layer VI. N-syndecan knockout pups displayed a similar phenotype as the adult knockouts, namely that layers II/III had less BrdU+ cells than the wild-type layers II/III and that layer VI in knockouts was denser with BrdU+ cells than in the wild types (Fig. 1 E).
N-syndecan knockout mice display normal neural proliferation and differentiation
N-syndecan has been proposed to act as a coreceptor in FGF-2 binding to high affinity FGF receptor (Dealy et al., 1997). Neural stem cells/progenitor cells are highly responsive to FGF-2, and N-syndecan deficiency could thus lead to decreased cell proliferation and neuron numbers in the cortex. We compared the cortical neural proliferation and differentiation between N-syndecan knockout and wild-type mice and found no differences. For the results and methods used, see supplemental material (available at http://www.jcb.org/cgi/content/full/jcb.200602043/DC1).
Radial neuronal migration is defective in N-syndecan knockout slices
Radial neuronal migration can be recapitulated in brain slices and is stimulated by EGF (Fricker-Gates et al., 2000). We followed this protocol and labeled the cells with BrdU in E15 living brain slices. In this assay, the scattering of BrdU+ cells in the different cortical layers was examined after 24 h of culture. We could detect a relative increase in the number of BrdU+ cells in the CP of the wild-type slices and a decrease in the number of cells in the VZ/SVZ, when EGF was present in the culture medium. The relative shift of cells from the VZ/SVZ to the CP was ∼10% higher in the EGF-treated slices than in nontreated slices. Interestingly, in the N-syndecan knockout slices, we could not see an EGF-induced shift of BrdU+ cells to the CP (Fig. 1 G). Results of this semi–in vivo experiment support the view that the cortical structure changes in the N-syndecan knockout mice depend on radial migration defect and, furthermore, suggest that the role of N-syndecan in neuronal migration may be coupled to EGFR activation.
Cell migration is defective in the RMS of N-syndecan knockout slices
A major neuronal migration route, the RMS, feeds the olfactory bulb (OB) with interneurons throughout life. A previous study has localized both N-syndecan mRNA and protein to migrating neuron clusters of the olfactory region and to the embryonic and adult OB (Toba et al., 2002), but expression in the RMS has not been studied. In addition, no functional evidence has been presented for a role of N-syndecan in the migrating neurons. Therefore, we used P3 living slices of RMS, in which N-syndecan is detected by immunohistochemistry in the migratory stream of cells (Fig. 2 A). For functional studies, cells in the rostral part of the RMS were labeled with the lipophilic dye DiI (Fig. 2 B). This method effectively labels only cells migrating along the RMS and not the cells that originate from the VZ of the OB itself (Liu and Rao, 2003). We cultured the injected slices for 24 h. During this time period, N-syndecan wild-type slices accumulated twice as many DiI-labeled cells in the volume of the OB as the N-syndecan knockout slices (Fig. 2, C and D). The volume of the N-syndecan knockout OB was not different from the wild type (unpublished data).
Figure 2.

The N-syndecan knockouts show diminished cell migration in the RMS to the OB at P3. (A) N-syndecan is expressed in the RMS at P3. Staining specificity was tested with an antibody-blocking peptide (CT, cortex). (B) RMS cells in living P3 parasagittal slices were labeled by injecting DiI to the rostral part of the RMS. After 24 h of incubation, the number of DiI-labeled cells in the volume of the OB was counted. (C) In the N-syndecan knockouts (ko), the number of migrated cells was half that of the wild-type (wt) slices. (D) A fluorescence microscope photograph showing the OBs from DiI-injected slices. The photographs show the pattern of labeled cells in the RMS after 24 h, and the dashed line determines the cell counting area. Insets show higher magnifications of the glomerular and periglomerular layers of N-syndecan knockout and wild-type OBs in which the knockout OB shows a lower number of DiI-labeled cells (arrows). Error bars represent the SEM of n embryos; p-values were estimated with the t test.
N-syndecan is required for HB-GAM–induced haptotactic cell migration
HB-GAM occurs in developing tissues as an ECM-associated ligand and is spatiotemporally coexpressed with N-syndecan (Nolo et al., 1995; Kinnunen et al., 1998a). Thus, N-syndecan may be a receptor of matrix-associated HB-GAM in migration. We tested this hypothesis in a transfilter migration assay using E15 forebrain cells, the most relevant cell type from the viewpoint of the knockout phenotype (see first section of Results). N-syndecan–deficient forebrain cells migrated very poorly to HB-GAM in this assay (Fig. 3 A). However, they did display elevated migration at higher HB-GAM concentrations, suggesting the presence of another migration-associated receptor for HB-GAM. The other receptor could very well be the receptor-type protein tyrosine phosphatase β/ζ (Maeda and Noda, 1998) because we could not completely abolish this migration in the knockout cells by degrading HS chains with heparitinase (unpublished data).
Figure 3.

N-syndecan is required for HB-GAM–guided haptotactic migration. (A) HB-GAM behaves as an N-syndecan–dependent guidance molecule in the haptotactic migration of mouse E15 forebrain cells. HB-GAM is a specific guidance molecule in a migration chamber assay, promoting migration already at a 0.3-μM coating concentration. At 3 μM, a fourfold increase in the number of migrating cells is observed when compared with polylysine (unstimulated). N-syndecan–deficient forebrain cells fail to migrate to HB-GAM–coated membranes at coating concentrations under 1 μM but show an increase at a 3-μM coating concentration. (B) HB-GAM–guided migration is enhanced when EGFR is activated by EGF in the migration chamber assay. The enhancement is dose dependent in normal forebrain cells. The N-syndecan knockout (ko) cells fail to respond to EGFR activation. wt, wild type. (C) The enhancement in migration can be blocked with the EGFR inhibitor AG1478. Src activity is also required for the enhancement and can be blocked with PP2. Blocking the MAPK pathway of EGFR with U0126 does not have any effect on migration. Error bars represent the SEM of independent experiments; p-values were estimated with the t test.
To rule out possible deficiency in the general migration ability of the knockout cells, we used EGFR activation to induce migratory phenotype in the cells. EGFR functions as a scatter factor in neural cell migration, and EGFR activation induces the migratory behavior of all neural cell types and, in addition, mediates chemotactic migration in the brain (Burrows et al., 2000; Caric et al., 2001; Aguirre et al., 2005). EGFR is also known to activate the c-Src–cortactin pathway in a ligand-specific manner, which links EGFR signaling to the modulation of the actin cytoskeleton (Goi et al., 2000).
First, we wanted to verify that EGFR activation alone is enough to potentiate migration in vitro. In the haptotactic cell migration assay, exogenous EGF enhanced the migration of wild-type mouse E15 forebrain cells to HB-GAM (Fig. 3 B) and to other haptotactic guidance molecules like midkine (Qi et al., 2001; and unpublished data). The increase in the number of migrating cells was not the result of EGF chemotaxis because the exogenous ligand was homogeneously available to the cells in the assay medium. Unexpectedly, N-syndecan knockout cells completely failed to respond to EGF stimulus and did not show any enhancement in cell migration (Fig. 3 B), suggesting that N-syndecan may interact with EGFR or some component required for EGFR-dependent migration.
The receptor-mediated induction in the wild-type cells was blocked with AG1478, a specific blocker for EGFR. The effect was also highly dependent on Src activity, which was revealed by the Src-blocking agent PP2, whereas blocking the MAPK pathway with U0126 did not affect the EGF-induced migration enhancement (Fig. 3 C).
N-syndecan is required for HB-GAM–induced Src activation
HB-GAM–induced activation of c-Src through N-syndecan has been demonstrated with N18 cells transfected with N-syndecan and adhered on HB-GAM matrix (Kinnunen et al., 1998b). We followed the same experimental procedure with E15 forebrain primary neurons. The cells were plated on HB-GAM–coated plates and lysed after defined time intervals. An increase in c-Src phosphorylation was clear in the wild-type cells within the first 60 min (Fig. 4). c-Src phosphorylation was not induced in the N-syndecan knockout forebrain cells in a similar manner (Fig. 4). The level of phosphorylation in the knockout cells gradually increased toward the 60-min time point but never reached the levels observed in the wild-type cells. HB-GAM has other signaling receptors but seems to induce c-Src phosphorylation specifically via N-syndecan. The slight, gradual increase in phosphorylation in knockout samples could be explained by some other weak receptor activity.
Figure 4.

HB-GAM–induced c-Src phosphorylation is deficient in N-syndecan knockout forebrain cells. Plating E15 forebrain neurons on HB-GAM matrix triggers c-Src phosphorylation in the wild-type (wt) cells. This increase in activity stays up during the first 60 min and then gradually declines. In N-syndecan knockout (ko) cells, this activation of c-Src does not occur. The level of phosphorylation gradually increases during 60 min but does not reach the same level as in the wild-type cells. Immunoblot of total Src was used as a loading control. Error bars represent the SEM of separate experiments.
EGFR-mediated chemotaxis is impaired in N-syndecan–deficient neurons
Interactions of N-syndecan with the migration-enhancing matrix ligand HB-GAM and the ensuing c-Src activation are likely to explain, at least in part, the role of N-syndecan in neural migration in the brain. However, we suspected that N-syndecan might additionally modulate EGFR functions because the induction of the migratory phenotype through EGFR ligation completely failed to enhance migration in HB-GAM–dependent haptotaxis assays (see N-syndecan is required for…cell migration). Therefore, we looked for an alternative assay to specifically monitor EGFR-dependent migration of cortical neurons.
HB EGF-like growth factor (HB-EGF) is suggested to specifically induce the migration of cortical neurons in a chemotactic manner (Caric et al., 2001), and its binding to EGFR is heparin dependent (Aviezer and Yayon, 1994). We prepared living brain slice cultures from E18 N-syndecan knockout and wild-type brains to estimate the chemotactic response of N-syndecan–deficient cells in semi–in vivo conditions. 1 d before slice preparation (E17), the mothers received BrdU injections, which should label neuronal cells from the last dividing stem cell population. These cells then migrate to the superficial cortical layers (Takahashi et al., 1995). A 1-d gap between the injections and slice preparations prevents false positive results, as there is no free BrdU left to label possibly damaged or late-proliferating cells in the brain. We placed HB-EGF–soaked agarose beads unilaterally in layer I of the cortical slices. The other cortical hemisphere in each slice was used as an internal control with a BSA-soaked bead. Using the density of BrdU-positive cells accumulating around the bead in 24 h, we estimated the relative chemotactic activity of HB-EGF in normal and N-syndecan–deficient brain. In the wild-type slices, HB-EGF induced a clear accumulation of BrdU-labeled cells around the beads (Fig. 5 A). The density around the BSA beads stayed the same as the density elsewhere in the same layer. In the N-syndecan knockout slices, the accumulation of cells was much weaker around the HB-EGF beads (only 30% of the wild-type accumulation; Fig. 5, B and C). These results suggest a physiologically significant role for N-syndecan in HB-EGF chemotaxis.
Figure 5.

To evaluate the biological significance of HB-EGF and N-syndecan interaction, we used a semi–in vivo approach involving living brain slices. Agarose beads soaked either in BSA or HB-EGF were placed to the marginal layer of E18 N-syndecan knockout and wild-type brain slices. (A) BrdU-labeled cells gathered around HB-EGF–soaked beads in the wild-type cortex (arrowhead), which was measured by estimating the relative BrdU-positive cell density around the beads. (B) Migrated cells were also observed around HB-EGF beads in the N-syndecan knockout slices, but the final cell density around the beads was much lower when compared with migration in wild-type samples. (C) To compare migration between genotypes, BSA beads in wild-type (wt) slices represent the baseline (0%), whereas HB-EGF beads represent 100% induction. ko, knockout. Bars, 100 μm. Error bars represent the SEM of n embryos.
We modified our migration chamber assay to measure more accurately the strength of chemotaxis in N-syndecan–deficient neurons. An insulating layer of matrigel was added to separate the compartment with the chemoguidance protein from the compartment containing the neural cells. Within 24 h, HB-EGF induced strong migration through the insulating layer in normal embryonic forebrain cells but not in N-syndecan–deficient cells (Fig. 6 A). If the binding of HB-EGF to HS chains is required for EGFR stimulation in this assay, HB-GAM, which binds HS chains at the neuron surface (Raulo et al., 2005) but is not chemotactic itself, might interfere in HB-EGF–induced chemotactic migration. In fact, HB-GAM in the assay medium clearly inhibited HB-EGF–induced chemotaxis, with a half-maximal inhibition at about threefold molar excess compared with HB-EGF (Fig. 6 B).
Figure 6.

HB-EGF chemotaxis requires N-syndecan in a modified migration chamber assay. (A) HB-EGF acts as a strong chemotactic protein for normal E15 forebrain cells, but N-syndecan knockout cells do not migrate toward HB-EGF. (B) In normal forebrain cells, soluble HB-GAM can inhibit HB-EGF chemotaxis in a dose-dependent manner. (C) Phosphotyrosine immunoprecipitation shows increased EGFR phosphorylation upon HB-EGF stimulation in A431 cells. HB-EGF–induced phosphorylation is inhibited at a 10-fold molar excess of HB-GAM. In C, the graph shows the mean of relative optical density in two experiments. Error bars represent the SEM of separate experiments.
EGFR is normally expressed in the N-syndecan knockout forebrain at E15 (unpublished data). However, the low amount of EGFR prevents the detection of differences in receptor activation between knockout and wild-type brains. Therefore, we used the A431 cell line with abundant EGFR expression, induced EGFR phosphorylation in the cells with HB-EGF, and examined the effect of soluble HB-GAM on EGFR activation. HB-GAM inhibited HB-EGF–induced EGFR phosphorylation at a 10-fold molar concentration (Fig. 6 C).
N-syndecan colocalizes with EGFR at the plasma membrane of primary forebrain neurons and in lipid raft fractions prepared from developing brain
If N-syndecan were able to sensitize EGFR signaling in the forebrain cells by direct interaction, it should be found in the same cellular compartments. We double immunostained forebrain cells plated on HB-GAM with N-syndecan and EGFR antibodies. Cells grown on HB-GAM showed limited colocalization of N-syndecan and EGFR initially, but N-syndecan was found more in the base of the growing neurite and filopodia, whereas EGFR was more diffusely distributed near the cell soma (unpublished data). Cells treated with EGF started spreading rapidly, and EGFR relocalized closer to the edges of the growing cells and away from the cell soma (Fig. 7 A). N-syndecan and EGFR clearly colocalized in the growing edges and neurite base of the EGF-treated cells.
Figure 7.

N-syndecan interacts with EGFR in embryonic forebrain neurons and in transfected HEK293 cells. (A) In cells plated on HB-GAM, N-syndecan is found in the growing processes and at their base. When the cells start spreading upon EGF stimulation, the colocalization of EGFR and N-syndecan in the neurite base and spreading lamellipodia is very strong (arrowheads). Inset is a magnification of the boxed area, which shows the base of the growing neurite. (B) N-syndecan and EGFR are both found in the sucrose gradient–separated lipid raft fraction (labeled with an asterisk) of embryonic forebrain lysate together with the lipid raft resident protein Fyn. In addition, although cortactin is diffusely present in all of the gradient fractions, its phosphorylated form is clearly enriched in the raft fraction. Furthermore, both HB-GAM and HB-EGF are present in the raft fraction, but only HB-GAM is clearly enriched there. HB-EGF antibody detects two major bands of 21 and 9.5 kD (membrane-bound and soluble protein, respectively; arrowheads) and several diffuse bands, possibly because of protein glycosylation or oligomerization. (C) In FRET analysis, a fusion vector of CFP and YFP with a 23–amino acid linker sequence was used as a positive FRET signal control, whereas cotransfected, unmodified pECFP and pEYFP vectors served as a negative control. (D) Transfected N-syndecan–YFP and EGFR-CFP cluster together in HEK293T cells plated on HB-GAM. Adding HB-EGF to the cell culture enhances the receptor clustering significantly, especially in the base of the growing cell process (arrowhead). (E) When HEK293T cells are plated on laminin, the initial clustering of N-syndecan–YFP and EGFR-CFP is clearly weaker than in cells plated on HB-GAM. The addition of HB-EGF increases the clustering, but only temporarily, and not as strongly as in HB-GAM–plated cells. (F) Primary hippocampal neurons were transfected with a similar construct using the same FRET fluorophore pair. Control transfections were made as in HEK293 cells. (G) Despite the low expression of N-syndecan and EGFR constructs in primary neurons, the HB-EGF–induced clustering is clearly visible after 15 min of induction. Clusters localize to the base of the neurite and along it (arrowheads). Bars (C–E), 10 μm; (F and G), 20 μm.
At least some of the EGFR and c-Src signaling occurs in detergent-resistant membrane domains (Hur et al., 2004), which are also known as lipid rafts (Simons and Ikonen, 1997). To determine whether N-syndecan and its signaling partners are targeted into the same membrane fractions, we used flotation sucrose gradients to isolate lipid rafts from prenatal rat forebrains and detected the proteins of interest with specific antibodies. In agreement with previous studies (for review see Waugh et al., 2001; Hur et al., 2004), EGFR was present in the lipid raft fraction. In addition, a limited amount of the EGFR ligand HB-EGF was detected in the same fraction, although the majority of differentially processed HB-EGF forms was distributed in soluble fractions and pelleted material (Fig. 7 B). N-syndecan and its ligand HB-GAM were enriched in the lipid raft fraction as well as Fyn kinase, which was also used as a raft fraction marker. We also examined the localization of cortactin, which is specifically phosphorylated by Src family kinases to regulate cell motility (Huang et al., 1998). Furthermore, cortactin is involved in both the EGFR and N-syndecan signaling pathways. To some extent, cortactin was localized in the lipid raft fraction, although the majority was diffusely present in soluble fractions. More significantly, cortactin phosphorylated at tyrosine residue 421 was almost exclusively present in the lipid rafts. Thus, all of the key molecules appear to be localized in the lipid raft membrane regions, greatly increasing the likelihood of their association.
FRET analysis suggests the interaction of N-syndecan with EGFR at the plasma membrane
Fluorescence resonance energy transfer (FRET) analysis reveals the physical proximity of two different fluorochromes (Youvan et al., 1997; Gordon et al., 1998). This method is successfully used to demonstrate physical coupling and oligomerization of receptors and receptor complexes by tagging the receptors with selected fluorochromes, such as CFP (donor) and YFP (acceptor), and quantifying donor and acceptor fluorescence changes upon their close encounter. We used this method to examine in more detail the possible coupling of N-syndecan and EGFR at the cell surface. A CFP–YFP fusion vector was used as a positive FRET control, whereas cotransfection of unmodified CFP and YFP vectors was used as a negative control (Fig. 7 C).
When HEK293T cells transfected with the fluorescent fusion receptors were plated on HB-GAM–coated coverglass, FRET quantification clearly showed the clustering of N-syndecan and EGFR at the sites of matrix (HB-GAM) contact (Fig. 7 D). To examine whether HB-EGF was capable of inducing further receptor association, we added HB-EGF to the culture medium and measured FRET after 20 min. There was a visible increase in the FRET signal, indicating that HB-EGF binding induces the close coupling of N-syndecan and EGFR (Fig. 7 D). Interestingly, the most intense FRET signal is seen at the base of the processes of 293T cells; this area may be analogous to the base of the neurite, where the most intense colocalization of N-syndecan and EGFR is observed in neurons (Fig. 7, A and D). That clustering should occur to some extent even in the absence of EGFR ligand, which speaks for the receptor coupling via the common signaling complexes of N-syndecan and EGFR (see Discussion). When the cells were plated on laminin, a weaker FRET signal was visible for a short period of time in the presence of HB-EGF (Fig. 7 E). This indicates that laminin does not induce such a strong clustering of N-syndecan and EGFR and that the HB-EGF–induced complexes are not very stable on laminin.
HEK293 cells may handle their membrane traffic in a manner not comparable with neurons, and we repeated this FRET experiment in hippocampal primary neurons. With electroporation, we could transfect the neurons with high enough efficiency to measure FRET, although the expression level of either N-syndecan or EGFR construct was not very high and declined rapidly in culture. Nevertheless, we could essentially repeat our results obtained with HEK293 cells, as we saw the clustering of N-syndecan and EGFR after HB-EGF stimulus (Fig. 7 G). Interestingly, the clustering took place in almost exactly the same locations where we observed the colocalization of these receptors with immunostaining, namely in the base of the growing neurite and in the neurite itself.
HB-EGF and HB-GAM bind to the N-syndecan ectodomain in a competitive manner
Migration assays and FRET analysis suggest that interactions of N-syndecan and EGFR depend, at least in part, on a shared ligand such as HB-EGF. We have previously used N-syndecan ectodomain to determine the detailed interactions between the N-syndecan and HB-GAM (Raulo et al., 2005). In this study, we have used the same recombinant PG fragment to elucidate the binding of HB-EGF to the N-syndecan ectodomain in an ELISA-type assay (Fig. 8, A and B). HB-EGF indeed binds the N-syndecan ectodomain with a similar affinity as HB-GAM. We estimated the dissociation constant to be 12 nM for HB-GAM and 15 nM for HB-EGF. The binding was found to be competitive (Fig. 8, C and D), indicating an interaction with the same or overlapping HS sequences. These findings provide the apparent explanation for why N-syndecan knockout cells fail to migrate in HB-EGF–induced chemotaxis assays and why soluble HB-GAM inhibits HB-EGF–induced chemotaxis and EGFR phosphorylation.
Figure 8.

HB-EGF binds the N-syndecan ectodomain with a similar affinity as HB-GAM. (A and B) In an ELISA assay, different concentrations of the IgG-tagged N-syndecan ectodomain (ENS) were added on HB-EGF– or HB-GAM–precoated 96-well plates, and the amount of the specific binding of N-syndecan ectodomain after incubation was estimated for both proteins. HB-GAM and HB-EGF bind the N-syndecan ectodomain with similar affinity. (C and D) In the competition assay, biotinylated HB-GAM or HB-EGF were bound to the immobilized N-syndecan ectodomain. A gradient of unbiotinylated HB-GAM or HB-EGF was used to compete with biotinylated protein in N-syndecan ectodomain binding. HB-GAM inhibits HB-EGF binding strongly. HB-EGF also competes with HB-GAM binding to the N-syndecan ectodomain, although with somewhat lesser efficacy. Error bars represent the SEM of independent experiments.
Discussion
Attempting to separate the impact of the individual HSPGs on brain development from the function of the total pool of HS molecules is not straightforward. In spite of the detrimental effects observed in HS-lacking mice, available syndecan knockout or misexpressing mice do not show very dramatic phenotypes in the early embryonic development but rather show this in the regulation of adult physiology (Reizes et al., 2001; Kaksonen et al., 2002). To our knowledge, this is the first study dealing with the individual role of any syndecan in the in vivo neural migration during brain development, although indirect evidence from cell adhesion and neurite motility studies has been published previously (for review see Rauvala et al., 2000; Toba et al., 2002).
N-syndecan knockout brain phenotype
The absence of N-syndecan causes structural changes in the adult mouse cerebral cortex. Although the total number of neurons and glia does not differ between N-syndecan knockout and wild-type mice, the localization of the cells in radially aligned cortical columns is very different. Consistently, the deep laminae of the cortex have a higher cell density in the knockout mice than in the wild types, and the superficial laminae have fewer neurons in the knockouts. BrdU labeling of the embryonic neural cells and their follow-up further suggest that the density changes are caused by delayed radial migration. The delay is partially caught up in the knockouts before or ∼10 d after birth, and the neurons reach the CP. No cells are left in the white matter or below subplate. Inside the CP, however, the late-born neurons seem to have a migration defect, and they stay in the deep layers.
The delayed migration also manifests in the RMS of the mutant animals, although no significant changes in the OB volume could be detected. This is probably because the bulb has a constant renewal of interneurons during the adult life, and the deficiencies in cell migration could be compensated, making it difficult to see any structural changes in the bulb.
Interestingly, the proliferation of neural progenitors in the N-syndecan knockouts appears normal, indicating an insignificant or redundant role for N-syndecan in the mitotic regulation of neural progenitors or stem cells. Immunostaining of neural stem cells with N-syndecan antibodies suggests that N-syndecan is not present in very primitive stem cells but is present in more mature progenitors (unpublished data) and, thus, probably does not act as a coreceptor for FGFs in neural stem cell proliferation, as it evidently does in the limb mesenchyme (Dealy et al., 1997).
Within the limits of the experiments detailed in this study, we cannot find any differentiation defects in the N-syndecan knockout cortical cells in vivo or in vitro. BrdU–β-III–tubulin double immunostaining states that newly born N-syndecan knockout neurons adopt the neuronal phenotype at the same time and approximately at the same frequency as the wild-type neurons. The knockout neurons do not show displaced layer phenotypes.
N-syndecan in neuronal migration
Previous studies have suggested that N-syndecan regulates cell motility by binding to ECM-associated ligands like HB-GAM (for review see Rauvala et al., 2000). HB-GAM strongly promotes the haptotactic migration of forebrain cells, and the N-syndecan knockout cells fail completely to follow HB-GAM in vitro. The defective c-Src phosphorylation in the knockout cells is very likely associated with this failure. This interpretation is compatible with the finding that the cytosolic tail of N-syndecan binds a protein complex containing a PDZ domain protein calcium/CaM-dependent serine protein kinase (CASK), cortactin, and Fyn and Src kinases (Kinnunen et al., 1998b; Hsueh and Sheng, 1999; Lauri et al., 1999).
According to our findings, N-syndecan is important for the general migratory behavior of the forebrain cells. We demonstrate this by using EGFR-induced scatter behavior in forebrain cells as a test tool. Increasing EGFR activity in neural progenitor cells induces and promotes neural differentiation and migration even postnatally (Burrows et al., 1997; Caric et al., 2001; Aguirre et al., 2005). EGFR acts as a scatter factor for these cells and drives them to the major migratory pathways in the telencephalon, namely to the radial migration route and to the RMS (Burrows et al., 1997). In our experiments, EGFR activation enhances haptotactic migration toward HB-GAM in a Src-dependent manner in wild-type cells but not in N-syndecan knockout cells. In living brain slices, the activation of EGFR induces radial migration of newly born cells from the VZ/SVZ area to the CP, but, again, cells in the N-syndecan knockout slices do not perform normally in this task.
Localization of N-syndecan, EGFR, HB-GAM, and HB-EGF in the migration routes is of interest here. Extensive studies have been published about the individual expression of each of these molecules. N-syndecan is expressed in the neural tissue by neurons and localizes in neurites (Nolo et al., 1995; Kinnunen et al., 1998a,b). It is strongly present in the RMS, as is shown in this study. During early brain development, HB-GAM is strongly expressed by neurons and neural precursors, including radial glia (for review see Rauvala and Peng, 1997). Although it is secreted, it does not diffuse far from its source. Thus, HB-GAM can create very steep gradients and provide a high local ligand concentration to its receptors that is apparently many times higher than the concentration of growth factors (Rauvala, 1989). EGFR is found in migrating neurons in all migratory routes, and its expression induces migratory behavior in all neural cell types (Caric et al., 2001). HB-EGF is expressed in the cerebral cortex, specifically at the marginal zone and in the subplate, which is in contrast to another EGFR ligand, TGF-α (Nakagawa et al., 1998). TGF-α, on the other hand, is expressed in the OB and RMS, providing a chemotactic stimulus.
The available expression data do not conflict with our results and the hypothesis about N-syndecan function in neuronal migration. However, the data do provide new ideas for future experimentation. TGF-α interaction with N-syndecan has not been studied, yet TGF-α is a major HB chemoattractant in the RMS, where N-syndecan is also strongly present.
A recent study describes the function of syndecan in neuronal migration and axon guidance in Caenorhabditis elegans (Rhiner et al., 2005). HS is involved in modulation of the axon guidance properties of Slit, a midline guidance molecule. In C. elegans, the only existing syndecan appears to play an especially important role in axon guidance and neuronal migration, and this function is mediated by Slit/Robo (Rhiner et al., 2005). In mammals, the interplay between any syndecan and Slit has not been studied at all. Interestingly, in C. elegans, another molecular link related to this study has been made. Syndecan and EGFR form a link via the intracellular PDZ domain protein CASK/LIN-2. In mammalian neurons, CASK/LIN-2 is a cytoplasmic binding partner for syndecan-3, connecting cell surface syndecan to the cytoskeleton (Hsueh and Sheng, 1999).
N-syndecan–EGFR interaction
N-syndecan and EGFR colocalize at the plasma membrane of primary forebrain neurons. When the cell membranes are fractionated, N-syndecan and EGFR are found in lipid rafts together with some of their ligands and signaling cascade molecules. Furthermore, FRET-monitored clustering indicates a close interaction between these two receptors. It is noteworthy that although binding to the HB-GAM matrix alone induces the clustering of N-syndecan and EGFR, laminin matrix promotes a very weak association. This suggests that HB-GAM matrix–induced signaling via N-syndecan is required for enhancing the receptor clustering and may be required for stabilization. HB-EGF further enhances the receptor clustering on HB-GAM but not as strongly on laminin-plated cells, which, again, speaks for HB-GAM's role in cluster stabilization. In hippocampal neurons, the clustering takes place in surprisingly similar locations, where we observe the colocalization of N-syndecan and EGFR by immunostaining. It appears that growing neurites especially concentrate on the receptor clusters.
Src activation has been recently linked to the sensitization of the EGFR signaling cascade (Hur et al., 2004), which, in turn, is associated with EGFR-induced neural migration (Boockvar et al., 2003). N-syndecan signaling triggered by ECM-associated HB-GAM is known to activate Src (Kinnunen et al., 1998b) and could therefore function in a similar manner to enhance EGFR activity in the forebrain cells. It is possible that N-syndecan can, upon ligation, cluster with other cell surface receptors via complexes formed by the PDZ domain proteins syntenin or CASK (Grootjans et al., 1997; Hsueh and Sheng, 1999). The network of N-syndecan and EGFR interactions involved in this signaling is illustrated in Fig. 9.
Figure 9.

Molecular interactions of N-syndecan in the regulation of cell motility. ECM-associated HB-GAM interaction with N-syndecan triggers Src phosphorylation, which, in turn, activates cortactin and modulates actin cytoskeleton assembly. N-syndecan is also present in lipid rafts, where Src activation could lead to the sensitization of EGFR and potentiate the receptor signaling. In addition, EGFR and N-syndecan share a HS-binding ligand, HB-EGF, and N-syndecan acts as a coreceptor in EGFR ligand binding. These interactions together lead to enhanced cell motility. GAG, glycosaminoglycan; p, phosphorylation.
In our assays, HB-EGF binds the glycosylated N-syndecan ectodomain with a similar affinity as HB-GAM and in a competitive manner. This would allow modulation of HB-EGF/EGFR contact via direct competition by HB-GAM. HB-EGF binding to N-syndecan is biologically important because N-syndecan–deficient cells cannot migrate in vitro toward HB-EGF, and, in living slices, the chemotactic activity of HB-EGF is strongly attenuated in the absence of N-syndecan. In addition, FRET analysis and binding experiments suggest that HB-EGF acts as a shared ligand between N-syndecan and EGFR, bringing together these two receptors at the cell surface. Based on these findings, we propose that both extracellular and intracellular events (Fig. 9) contribute to N-syndecan–EGFR interactions in neural cell migration.
Materials and methods
Animals
N-syndecan knockout mouse production and genotyping has been described previously (Kaksonen et al., 2002). Methods used in animal studies were approved by the board of laboratory animal experiments at the University of Helsinki. The mice were back bred to the C57/BL6 mouse strain (>10 generations) to obtain an inbred line of mutant animals.
BrdU labeling of radially migrating neurons in vivo and in slice culture
Timed pregnant females received an i.p. injection of 50 μg/g BrdU at E15. For in vivo migration assay, brain samples were collected at E18, P1, or P10, fixed with 4% PFA, and embedded in paraffin. For slice culture migration assay, the females were killed 30 min after the injection, and the embryos were collected. The brains of the embryos were prepared as slice cultures. The slices were cultured for 24 h with or without 20 ng/ml EGF. The slices were fixed and embedded in paraffin. 5-μm–thick sections were cut from the slice culture and E18 paraffin blocks, whereas 20-μm sections were cut from the P1 and P10 blocks. The sections were stained with anti-BrdU antibody. The density of BrdU+ cells in the SVZ/VZ and CP was estimated in relation to the other laminae from photographs or with Stereo Investigator (MicroBrightField).
In vitro migration assays
Haptotactic migration assays were performed on Transwell plates with a 12-μm pore size (Costar). The chamber-side membrane was coated with HB-GAM, laminin, or 0.01% poly-l-lysine. 150,000 primary neurons were plated per well in the assay medium. In the assay for EGFR-promoted migration, EGF was added to the medium (2, 20, or 50 ng/ml; Promega). The medium was freely diffusing across the membrane.
In chemotactic migration assays, a 1:1 mixture of matrigel (BD Biosystems) and collagen (rat tail; Sigma-Aldrich) was solidified on the Transwell membranes to separate the chamber compartments. Different concentrations of HB-EGF were added to the bottom chamber with or without HB-GAM, and cells were applied to the top chamber in the assay medium.
The cells were allowed to migrate for 16 h and were fixed with methanol. The wells were washed with water and stained with 1% toluidine blue for 20 min. The excess dye was removed with water, and the inner membranes were wiped clean with cotton sticks. The membranes were allowed to air dry, and the cells on the coated side were counted using an inverted light microscope (IX70; Olympus).
DiI labeling of the RMS cells
Mouse P3 brains were collected and prepared as slice cultures. 100 ng/ml DiI in dimethylformamide (Invitrogen) was injected with a glass capillary using a stereomicroscope (SZX9; Olympus) to the area of the anterior RMS (Liu and Rao, 2003). The injected slices were cultured for 24 h, rinsed with PBS, and cryopreserved with 15% sucrose in PBS. The slices were frozen and cut to 20-μm–thick sections with cryotome (Microm) and mounted with water-based mounting glue.
Preparing animal tissue and cell samples
Embryonic brain samples were collected from timed pregnant females; the day of detection of the vaginal plug was determined as E0. Females were killed with CO2 and by cervical dislocation, the uteri were dissected out, and the embryos were collected and dissected. The torsos were used for genotyping, and the heads were either further dissected for cell culture or fixed in 4% PFA/PBS overnight.
Pups under P10 were killed by decapitation, and the heads were washed in PBS. DNA from the abdominal organs was used for genotyping the pups. The brains were dissected out of the skull on a petri dish and kept on ice. The whole brain was either fixed with 4% PFA in PBS overnight or used for cell culture experiments. Adult brains were collected by killing the mice with CO2 and by cervical dislocation. The brains were rinsed in cold PBS and fixed with 4% PFA in PBS overnight.
Primary forebrain neurons were collected from E15 mouse embryos. The frontal lobes of the brain were dissociated and triturated with a 20-G needle and sterile syringe. Hippocampal cells were prepared from E18 rat brains in a similar manner. Primary cells were allowed to recover in a cell incubator with medium containing 10% FCS for 30 min before use in the assays. Assay medium was serum free with 10 mg/ml BSA. After recovery, the cells were centrifuged and suspended to the assay medium. The cell preparation was left to stand for 5 min to sediment the undissociated material. The cleared cell suspension containing 80–90% of neurons or neuronal precursors (Rauvala et al., 1988) was then collected and plated for the assays.
For slice culture preparations, the brains were cast in molten 3% agarose in PBS. 400-μm–thick sections were cut from the solidified agarose blocks with vibratome and collected on coverglasses on ice. The coverglasses were placed on parafilm sheets in petri dishes, and a drop of serum-containing medium was applied on the sections for the recovery period. After recovery, the slice cultures were kept in a 2% glucose assay medium.
Cell and tissue culture materials
DME (Invitrogen) with 10% FCS (Invitrogen) was used with added l-glutamine and penicillin-streptomycin (Invitrogen) in primary cell preparations and in cell line cultures. Cells were cultured at 37°C in 5% CO2. In migration assays and assays involving the application of recombinant proteins, FCS was omitted, and 10 mg/ml BSA (Sigma-Aldrich) was used instead in the assay medium. In the slice culture assays, 2% glucose was added to the assay medium. Primary forebrain neurons were collected from E15 mouse or E18 rat embryos. HEK293T and A431 cells were purchased from the American Type Culture Collection and cultured according to the supplier's instructions.
Recombinant proteins and primary antibodies
Human recombinant EGF was purchased from Promega; human recombinant HB-EGF was obtained from R&D Systems; poly-l-lysine was purchased from Invitrogen; and laminin was purchased from Sigma-Aldrich. HB-GAM and N-syndecan ectodomain IgG fusion protein (ENS-IgG) were produced as described previously (Raulo et al., 2005). Affinity-purified chicken anti–N-syndecan was produced against N-terminal peptide (Nolo et al., 1995) by AgriSera. Rabbit anti–HB-GAM production has been described previously (Rauvala et al., 1988). Goat anti-EGFR, rabbit anti–c-Src, rabbit anti-Fyn, goat anti–HB-EGF, and rabbit anticortactin were purchased from Santa Cruz Biotechnology, Inc. Rabbit anti–β-III–tubulin and rabbit antineurofilament were purchased from Chemicon. Mouse anti-BrdU and detection reagents were purchased from GE Healthcare. Agarose-conjugated antiphosphotyrosine antibody was obtained from Cell Signaling Technologies, and unconjugated mouse antiphosphotyrosine was purchased from Sigma-Aldrich.
Immunoprecipitation
The cell samples were lysed in 1% NP-40 PBS with protease and phosphatase inhibitors (1 μg/ml aprotinin, 1 mM PMSF, and 1 mM Na-o-vanadate). The cell lysates were homogenized by triturating with a 20-G needle. The homogenized samples were immunoprecipitated with either agarose-conjugated or soluble antibodies at 4°C overnight. Complexes formed with soluble antibodies were further collected with agarose-conjugated protein G (Sigma-Aldrich) for 2 h at RT. The precipitates were washed twice with the lysis buffer and suspended in SDS-containing loading dye. The samples were separated by denaturing PAGE, transferred to nitrocellulose filters, and blotted with antibodies of interest. HRP-conjugated secondary antibodies were detected with ECL (GE Healthcare).
Immunostaining of brain sections and cells
Paraffin sections were dewaxed and hydrated. Antibodies were diluted 1–5 μg/ml in PBS containing 2% BSA. The sections were incubated with the antibody solution overnight and biotinylated, or fluorescent secondary antibodies were used to detect the immunostaining signal. For quantifications, the required brain areas were photographed.
E15 forebrain cells from wild-type mice were plated on 50 μg/ml HB-GAM–coated object glasses and cultured for 24 h in growth factor and serum-free medium. On some glasses, 20 ng/ml EGF was added for 30 min before washing. The cells were washed and fixed with 4% PFA for 15 min and blocked with BSA and goat serum. The object glasses were incubated with 5 μg/ml anti–N-syndecan and 1 μg/ml anti-EGFR (rabbit IgG; Santa Cruz Biotechnology, Inc.) for 2 h and washed. FITC and TRITC conjugates were used as secondary antibodies. Mounted glasses were observed and photographed with a laser scanning confocal microscope (TCS SP2; Leica).
Optical analyses
All quantifications of band intensities in films or cell number estimates from photographs were made using ImageJ software (National Institutes of Health; http://rsb.info.nih.gov/ij/index.html). Thin (5 μm) paraffin sections were photographed at 63× magnification (NA 1.4) with a camera (AxioCam; Carl Zeiss MicroImaging, Inc.) mounted in a microscope (Axioplan; Carl Zeiss MicroImaging, Inc.). The relative density of immunostained or hematoxylin-eosin–stained cells in systematic random sample was estimated using the selector method, a variant of optical fractionator suitable for thin sections (Everall et al., 1997). The number of immunostained or DiI-labeled cells in thick (20 μm) sections was estimated with Stereo Investigator (MicroBrightField) and a microscope (BX51; Olympus) using an optical fractionator (Mouton, 2002).
HB-GAM–triggered c-Src phosphorylation
Petri dishes were coated with 10 μg/ml HB-GAM overnight at 4°C. The plates were washed with sterile PBS and equilibrated with the assay medium for 30 min before use. E15 forebrain neurons were prepared and plated on the HB-GAM–coated plates for 20, 40, and 60 min, after which the medium was carefully removed from the plates and the cells were washed and scraped off in the immunoprecipitation lysis buffer. The samples were immunoprecipitated with agarose-conjugated antiphosphotyrosine antibodies and blotted with anti–c-Src antibodies.
EGFR phosphorylation in N-syndecan knockout cells
E16 forebrain cells were collected from N-syndecan knockout and wild-type embryos. The cells were plated on 5-cm plates coated with poly-l-lysine (2.5 million cells per plate). The cells were allowed to attach to the plates for 2–3 h. 20 ng/ml EGF or an equal volume of PBS was added on the cells for 15 min, the plates were carefully washed with PBS, and the cells were lysed in the immunoprecipitation lysis buffer. The samples were immunoprecipitated with anti-EGFR antibody and blotted with antiphosphotyrosine antibody and anti-EGFR.
Lipid raft isolation and analysis
Rat forebrains were dissected from E17 embryos and homogenized on ice by passing through a 20-G needle in a small amount of 1% Triton X-100, TNE buffer (20 mM Tris, pH 7.4, 150 mM NaCl, and 5 mM EDTA) with complete protease inhibitors (Roche), and 1 mM Na3VO4. After 15 min on ice, the lysate volume was adjusted to 0.5 ml, mixed with 0.5 ml of 80% sucrose, and overlayed with discontinuous sucrose gradient (2 ml of 36% sucrose in TNE and 1.5 ml of 5% sucrose in TNE) in a 5-ml polyclear centrifuge tube (Beckman Coulter). The samples were centrifuged for 16–20 h at 100,000 g in a rotor (SW55; Beckman Coulter) at 4°C. Fractions of 0.5 ml were collected from the top, and preliminary identification of the lipid raft fraction was performed based on the density and presence of white scattering material. Fraction aliquots containing equal protein amounts were precipitated with 13% TCA, resuspended in SDS-PAGE sample buffer, and resolved by 4–15% PAGE. Protein content was analyzed by Western blotting using antibodies against syndecan-3 (rabbit IgG against the cytoplasmic domain), EGFR, Fyn, HB-EGF, and HB-GAM.
cDNA constructs and transfection used in FRET
Control DNA construct pECFP-YFP was produced by ligating a 0.8-kb AgeI–MfeI restriction fragment of pEYFP-C1 (CLONTECH Laboratories, Inc.) to the MfeI–XmaI fragment of pECFP-C1 via the compatible overhangs of XmaI and AgeI. The sequences for CFP and YFP were connected with a 23–amino acid linker. This expression vector was used as a positive control in FRET experiments. Cotransfected, unmodified pECFP and pEYFP vectors were used as negative controls. Rat EGFR cDNA was tagged with CFP sequence directly after the transmembrane domain of the receptor in the cytosolic part (amino acids Met668–Arg669). Rat N-syndecan cDNA was tagged with YFP in a similar manner (amino acids Tyr409–Arg410). In both constructs, a flexible amino acid linker sequence of six amino acids was left between the receptor and the fluorochrome cDNA. For transfection to hippocampal neurons, we prepared similar constructs with opposing FRET fluorophore pairs (i.e., EGFR-YFP and NSyn-CFP).
The FuGene 6 transfection kit (Roche) was used to transfect HEK293 cells. Hippocampal primary neurons were transfected by electroporation with a Nucleofector I device (Amaxa) and Rat Neuron Nucleofector Kit (Amaxa) using a standard protocol (O-03 or G-13 programs).
FRET imaging
For production and transfection of control and assay cDNA vectors, see supplemental material. HEK293T cells or hippocampal primary neurons were grown in 35-mm glass-bottom culture dishes (MatTek) in DME with 10% FCS. Petri dishes were coated by 20 μg/ml poly-l-lysine, 50 μg/ml HB-GAM, 10 μg/ml laminin, or 100 μg/ml BSA. After 24 h, cells were washed with serum-free medium and incubated overnight without serum. During image acquisition, the temperature was decreased from 37 to 22–24°C to reduce the movements of cellular organelles. Cells were incubated with or without 20 ng/ml HB-EGF for up to 30 min.
FRET signal was quantified with three filter sets (three-cube method): CFP channel (CFPF; excitation of D436/20x, emission of D480/40m, and dichromatic long-pass filter [DCLP] 455); FRET channel (FRETF; excitation of D436/20x, emission of D535/30m, and 455 DCLP); and YFP channel (YFPF; excitation of HQ500/20x, emission of HQ535/30m, and 515 DCLP; Chroma Technology Corp.). The image was recorded in live transfected cells in a platform (Cell Imaging Systems) consisting of a 60× planApo oil immersion objective (Olympus), CCD camera (C9260-905; Hamamatsu), inverted microscope (IX71; Olympus), and illumination system (MT20; Olympus). The excitation light source was a 150-W Xenon lamp (3.42% of intensity).
The images were acquired in binning 2 × 2 modes to increase the signal to noise ratio and 300–500-ms integration times. The background subtraction was made before the FRET calculations. The signals measured in the FRET channel were corrected for cross talk between CFP and YFP channels using a linear spectral unmixing algorithm (Youvan et al., 1997; Gordon et al., 1998). Corrected FRET (Fc) was calculated on a pixel by pixel basis with the following formula:
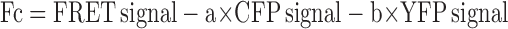 |
where a and b are correction factors for donor and aceptor. For HEK293 cells, factors a and b were found to be 0.39 ± 0.009 (n = 30) and 0.022 ± 0.002 (n = 30), respectively, and for primary neurons were found to be 0.3975 ± 0.0537 (n = 12) and 0.0281 ± 0.0053 (n = 8). EGFR-CFP and N-syndecan–YFP were expressed separately in the cells, and, for each fluorophore, the emission from the FRET channel was divided by the emission measured with either the CFP or YFP channels as follows: a = FRET/CFP, and b = FRET/YFP. FRET-corrected images are displayed in pseudocolor mode (red areas represent the high values of FRET, and blue areas represent the low values of FRET).
HB-EGF binding to the N-syndecan ectodomain
The N-syndecan ectodomain was produced as a human IgG fusion protein as previously described (Raulo et al., 2005). The concentration of the fusion protein was determined with a dot blot using human IgG protein as a standard and HRP-conjugated anti–human IgG antibody for detection. 1 μg/ml HB-GAM or HB-EGF was diluted in TBS and bound to nonimmune 96-well plates for 1 h (100 μl per well). The wells were blocked with 2% BSA in TBS, and a dilution series of N-syndecan ectodomain was pipetted to the wells starting from 10 pmol/well. After 1 h, the excess of ectodomain was washed away, and the amount of bound ectodomain was estimated using human IgG as a control protein and HRP-conjugated anti–human IgG for detection. The Kd values for both HB-GAM and HB-EGF were estimated from the binding curves using Scatchard analysis.
To further examine the binding properties of HB-EGF, 96-well plates were precoated with 1 μg/ml protein A, to which ∼1 nM of the N-syndecan ectodomain IgG fusion protein was immobilized. HB-GAM and HB-EGF were biotinylated, and their relative biotinylation level was estimated with Western blotting. HB-GAM and HB-EGF gradients were added on N-syndecan ectodomain and the excess of unbiotinylated HB-EGF or HB-GAM to assay their ability to compete for N-syndecan binding. The biotin conjugate was detected after washing using HRP-conjugated streptavidin.
HB-EGF chemotaxis in living slices
HB-EGF was diluted to 10 μg/ml in PBS with 0.1% BSA. 100 AffiGel agarose beads (Bio-Rad Laboratories) were collected and washed with micropipette in PBS. The beads were incubated with diluted HB-EGF or only dilution buffer for 1 h in 37°C. The beads were washed with PBS and placed in a drop of PBS on a glass plate. E17 timed pregnant N-syndecan heterozygote females received one 50-μg/g BrdU injection, and, 1 d later, cortical brain slice cultures were prepared from embryos. Embryos were genotyped afterward. Under a stereomicroscope (SZX9; Olympus), the HB-EGF beads were placed to the marginal zone of the cortex unilaterally, and a dilution buffer bead was inserted to the other hemisphere. After 24 h in culture, the slices were processed for BrdU immunostaining. The area for cell density estimation was defined to be within the longest radius still containing higher cell density than the layer mean.
Online supplemental material
Online supplemental material provides analysis methods for neural cell proliferation. Table S1 provides data on cortical layer differentiation. Table S2 presents data on neural cell differentiation in vitro, and Table S3 provides the corresponding results received from the N-syndecan knockout mouse strain. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200602043/DC1.
Supplementary Material
Acknowledgments
Special thanks are due to our technicians Erja Huttu, Eveliina Saarikalle, and Seija Lehto, to Dr. Eero Castrén for invaluable critical comments and suggestions, to Dr. Pertti Panula for assistance with microscopy, and to M.Sc. Juha Kuja-Panula (Neuroscience Center, University of Helsinki, Helsinki, Finland) for donating the EGFR cDNA.
This work was supported by the Sigrid Jusélius Foundation and the Academy of Finland.
Abbreviations used in this paper: CASK, calcium/CaM-dependent serine protein kinase; CP, cortical plate; EGFR, EGF receptor; FRET, fluorescence resonance energy transfer; GAM, growth-associated molecule; HB, heparin binding; HB-EGF, HB EGF-like growth factor; HS, heparan sulfate; OB, olfactory bulb; PG, proteoglycan; RMS, rostral migratory stream; SVZ, sub-VZ; VZ, ventricular zone.
References
- Aguirre, A., T.A. Rizvi, N. Ratner, and V. Gallo. 2005. Overexpression of the epidermal growth factor receptor confers migratory properties to nonmigratory postnatal neural progenitors. J. Neurosci. 25:11092–11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviezer, D., and A. Yayon. 1994. Heparin-dependent binding and autophosphorylation of epidermal growth factor (EGF) receptor by heparin-binding EGF-like growth factor but not by EGF. Proc. Natl. Acad. Sci. USA. 91:12173–12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield, M., R. Kokenyesi, M. Kato, M. Hinkes, J. Spring, R. Gallo, and E. Lose. 1992. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 8:365–393. [DOI] [PubMed] [Google Scholar]
- Boockvar, J., D. Kapitonov, G. Kapoor, J. Schouten, G. Counelis, O. Bogler, E. Snyder, T. McIntosh, and D. O'Rourke. 2003. Constitutive EGFR signalling confers a motile phenotype to neural stem cells. Mol. Cell. Neurosci. 24:1116–1130. [DOI] [PubMed] [Google Scholar]
- Burrows, R., D. Wancio, P. Levitt, and L. Lillien. 1997. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron. 19:251–267. [DOI] [PubMed] [Google Scholar]
- Burrows, R., L. Lillien, and P. Levitt. 2000. Mechanisms of progenitor maturation are conserved in the striatum and cortex. Dev. Neurosci. 22:7–15. [DOI] [PubMed] [Google Scholar]
- Caric, D., H. Raphael, J. Viti, A. Feathers, D. Wancio, and L. Lillien. 2001. EGFRs mediate chemotactic migration in the developing telencephalon. Development. 128:4203–4216. [DOI] [PubMed] [Google Scholar]
- Dealy, C., M. Seghatoleslami, D. Ferrari, and R. Kosher. 1997. FGF-stimulated outgrowth and proliferation of limb mesoderm is dependent on syndecan-3. Dev. Biol. 184:343–350. [DOI] [PubMed] [Google Scholar]
- Everall, I.P., R. DeTeresa, R. Terry, and E. Masliah. 1997. Comparison of two quantitative methods for the evaluation of neuronal number in the frontal cortex in Alzheimer disease. J. Neuropathol. Exp. Neurol. 56:1202–1206. [DOI] [PubMed] [Google Scholar]
- Fricker-Gates, R., C. Winkler, D. Kirik, C. Rosenblad, M. Carpenter, and A. Bjorklund. 2000. EGF infusion stimulates the proliferation and migration of embryonic progenitor cells transplanted in the adult rat striatum. Exp. Neurol. 165:237–247. [DOI] [PubMed] [Google Scholar]
- Goi, T., M. Shipitsin, Z. Lu, D. Foster, S. Klinz, and L. Feig. 2000. An EGF receptor/Ral-GTPase signalling cascade regulates c-Src activity and substrate specificity. EMBO J. 19:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, G.W., G. Berry, X.H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootjans, J.J., P. Zimmermann, G. Reekmans, A. Smets, G. Degeest, J. Durr, and G. David. 1997. Syntenin, a PDZ protein that binds syndecan cytoplasmic domains. Proc. Natl. Acad. Sci. USA. 94:13683–13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh, Y.P., and M. Sheng. 1999. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J. Neurosci. 19:7415–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C., J. Liu, C.C. Haudenschild, and X. Zhan. 1998. The role of tyrosine phosphorylation of cortactin in the locomotion of endothelial cells. J. Biol. Chem. 273:25770–25776. [DOI] [PubMed] [Google Scholar]
- Hur, E.-M., Y.-S. Park, B.D. Lee, I.H. Jang, H.S. Kim, T.-D. Kim, P.-G. Suh, S.H. Ryu, and K.-T. Kim. 2004. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. J. Biol. Chem. 279:5852–5860. [DOI] [PubMed] [Google Scholar]
- Imai, S., M. Kaksonen, E. Raulo, T. Kinnunen, C. Fages, X. Meng, M. Lakso, and H. Rauvala. 1998. Osteoblast recruitment and bone formation enhanced by cell matrix-associated heparin-binding growth-associated molecule (HB-GAM). J. Cell Biol. 143:1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inatani, M., F. Irie, A. Plump, M. Tessier-Lavigne, and Y. Yamaguchi. 2003. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 302:1044–1046. [DOI] [PubMed] [Google Scholar]
- Kaksonen, M., I. Pavlov, V. Voikar, S. Lauri, A. Hienola, R. Riekki, M. Lakso, T. Taira, and H. Rauvala. 2002. Syndecan-3-deficient mice exhibit enhanced LTP and impaired hippocampus-dependent memory. Mol. Cell. Neurosci. 21:158–172. [DOI] [PubMed] [Google Scholar]
- Kinnunen, T., E. Raulo, R. Nolo, M. Maccarana, U. Lindahl, and H. Rauvala. 1996. Neurite outgrowth in brain neurons induced by heparin-binding growth-associated molecule (HB-GAM) depends on the specific interaction of HB-GAM with heparan sulfate at the cell surface. J. Biol. Chem. 271:2243–2248. [DOI] [PubMed] [Google Scholar]
- Kinnunen, A., T. Kinnunen, M. Kaksonen, R. Nolo, P. Panula, and H. Rauvala. 1998. a. N-syndecan and HB-GAM (heparin-binding growth-associated molecule) associate with early axonal tracts in the rat brain. Eur. J. Neurosci. 10:635–648. [DOI] [PubMed] [Google Scholar]
- Kinnunen, T., M. Kaksonen, J. Saarinen, N. Kalkkinen, H. Peng, and H. Rauvala. 1998. b. Cortactin-Src kinase signalling pathway is involved in N-syndecan-dependent neurite outgrowth. J. Biol. Chem. 273:10702–10708. [DOI] [PubMed] [Google Scholar]
- Lauri, S.E., S. Kaukinen, T. Kinnunen, A. Ylinen, S. Imai, K. Kaila, T. Taira, and H. Rauvala. 1999. Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J. Neurosci. 19:1226–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, U., M. Kusche-Gullberg, and L. Kjellen. 1998. Regulated diversity of heparan sulfate. J. Biol. Chem. 273:24979–24982. [DOI] [PubMed] [Google Scholar]
- Liu, G., and Y. Rao. 2003. Neuronal migration from the forebrain to the olfactory bulb requires a new attractant persistent in the olfactory bulb. J. Neurosci. 23:6651–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, N., and M. Noda. 1998. Involvement of receptor-like protein tyrosine phosphatase ζ/RPTPβ and its ligand pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) in neuronal migration. J. Cell Biol. 142:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton, P.R. 2002. Principles and Practices of Unbiased Stereology: an Introduction for Bioscientists. Johns Hopkins University Press, Baltimore. 214 pp.
- Nakagawa, T., M. Sasahara, Y. Hayase, M. Haneda, H. Yasuda, R. Kikkawa, S. Higashiyama, and F. Hazama. 1998. Neuronal and glial expression of heparin-binding EGF-like growth factor in central nervous system of prenatal and early-postnatal rat. Brain Res. Dev. Brain Res. 108:263–272. [DOI] [PubMed] [Google Scholar]
- Nolo, R., M. Kaksonen, E. Raulo, and H. Rauvala. 1995. Co-expression of heparin-binding growth-associated molecule (HB-GAM) and N-syndecan (syndecan-3) in developing rat brain. Neurosci. Lett. 191:39–42. [DOI] [PubMed] [Google Scholar]
- Qi, M., S. Ikematsu, N. Maeda, K. Ichihara-Tanaka, S. Sakuma, M. Noda, T. Muramatsu, and K. Kadomatsu. 2001. Haptotactic migration induced by midkine. Involvement of protein-tyrosine phosphatase zeta. Mitogen-activated protein kinase, and phosphatidylinositol 3-kinase. J. Biol. Chem. 276:15868–15875. [DOI] [PubMed] [Google Scholar]
- Raulo, E., S. Tumova, I. Pavlov, M. Pekkanen, A. Hienola, E. Klankki, N. Kalkkinen, T. Taira, I. Kilpeläinen, and H. Rauvala. 2005. The two thrombospondin type i repeat domains of the heparin-binding growth-associated molecule bind to heparin/heparan sulfate and regulate neurite extension and plasticity in hippocampal neurons. J. Biol. Chem. 280:41576–41583. [DOI] [PubMed] [Google Scholar]
- Rauvala, H. 1989. An 18-kd heparin-binding protein of developing brain that is distinct from fibroblast growth factors. EMBO J. 8:2933–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauvala, H., and B. Peng. 1997. HB-GAM (heparin-binding growth-associated molecule) and heparin-type glycans in the development and plasticity of neuron-target contacts. Prog. Neurobiol. 52:127–144. [DOI] [PubMed] [Google Scholar]
- Rauvala, H., J. Merenmies, R. Pihlaskari, M. Korkolainen, M.L. Huhtala, and P. Panula. 1988. The adhesive and neurite-promoting molecule p30: analysis of the amino-terminal sequence and production of antipeptide antibodies that detect p30 at the surface of neuroblastoma cells and of brain neurons. J. Cell Biol. 107:2293–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauvala, H., H. Huttunen, C. Fages, M. Kaksonen, T. Kinnunen, S. Imai, E. Raulo, and I. Kilpelainen. 2000. Heparin-binding proteins HB-GAM (pleiotrophin) and amphoterin in the regulation of cell motility. Matrix Biol. 19:377–387. [DOI] [PubMed] [Google Scholar]
- Reizes, O., J. Lincecum, Z. Wang, O. Goldberger, L. Huang, M. Kaksonen, R. Ahima, M. Hinkes, G. Barsh, H. Rauvala, and M. Bernfield. 2001. Transgenic expression of syndecan-1 uncovers a physiological control of feeding behavior by syndecan-3. Cell. 106:105–116. [DOI] [PubMed] [Google Scholar]
- Rhiner, C., S. Gysi, E. Frohli, M.O. Hengartner, and A. Hajnal. 2005. Syndecan regulates cell migration and axon guidance in C. elegans. Development. 132:4621–4633. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., R.S. Nowakowski, and V.S. Caviness Jr. 1995. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J. Neurosci. 15:6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toba, Y., M. Horie, K. Sango, A. Tokashiki, F. Matsui, A. Oohira, and H. Kawano. 2002. Expression and immunohistochemical localization of heparan sulphate proteoglycan N-syndecan in the migratory pathway from the rat olfactory placode. Eur. J. Neurosci. 15:1461–1473. [DOI] [PubMed] [Google Scholar]
- Waugh, M.G., S. Minogue, J.S. Anderson, M. dos Santos, and J.J. Hsuan. 2001. Signalling and non-caveolar rafts. Biochem. Soc. Trans. 29:509–511. [DOI] [PubMed] [Google Scholar]
- Youvan, D.C., C.M. Silva, E.J. Bylina, W.J. Coleman, M.R. Dilworth, and M.M. Yang. 1997. Calibration of fluorescence resonance energy transfer in microscopy using genetically engineered GFP derivatives on nickel chelating beads. Biotechnology (NY). http://www.et-al.com/searchable/documents/document3.doc (accessed July 25, 2006).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.