

Abstract
Receptor endocytosis is a fundamental step in controlling the magnitude, duration, and nature of cell signaling events. Confluent endothelial cells are contact inhibited in their growth and respond poorly to the proliferative signals of vascular endothelial growth factor (VEGF). In a previous study, we found that the association of vascular endothelial cadherin (VEC) with VEGF receptor (VEGFR) type 2 contributes to density-dependent growth inhibition (Lampugnani, G.M., A. Zanetti, M. Corada, T. Takahashi, G. Balconi, F. Breviario, F. Orsenigo, A. Cattelino, R. Kemler, T.O. Daniel, and E. Dejana. 2003. J. Cell Biol. 161:793–804). In the present study, we describe the mechanism through which VEC reduces VEGFR-2 signaling. We found that VEGF induces the clathrin-dependent internalization of VEGFR-2. When VEC is absent or not engaged at junctions, VEGFR-2 is internalized more rapidly and remains in endosomal compartments for a longer time. Internalization does not terminate its signaling; instead, the internalized receptor is phosphorylated, codistributes with active phospholipase C–γ, and activates p44/42 mitogen-activated protein kinase phosphorylation and cell proliferation. Inhibition of VEGFR-2 internalization reestablishes the contact inhibition of cell growth, whereas silencing the junction-associated density-enhanced phosphatase-1/CD148 phosphatase restores VEGFR-2 internalization and signaling. Thus, VEC limits cell proliferation by retaining VEGFR-2 at the membrane and preventing its internalization into signaling compartments.
Introduction
Endothelial cells are contact inhibited in their growth and lose the capacity to respond to growth factors when they reach confluence. This phenomenon is mediated by different concurrent mechanisms. Molecules at cell to cell junctions, such as cadherins, may transfer signals that reduce the capacity of the cells to respond to proliferative stimuli (Dejana, 2004; Gumbiner, 2005). Cadherins are located at intercellular adherens junctions and are linked to different intracellular partners that include β-catenin, plakoglobin, p120, Src (Gumbiner, 2005), csk (Baumeister et al., 2005), and density-enhanced phosphatase-1 (DEP-1)/CD148. β-catenin, plakoglobin, and p120 can also translocate to the nucleus and modulate cell transcription. In tumor cells, the negative effect of epithelial cadherin (E-cadherin) on cell growth is a result of its capacity to bind β-catenin and inhibit its translocation to the nucleus. This effect is detected in tumor cell lines in which cytosolic β-catenin ubiquitination and destruction is impaired (St Croix et al., 1998; Mueller et al., 2000; Gottardi et al., 2001; Stockinger et al., 2001; Bryant and Stow, 2005).
Endothelial cells express a cell-specific cadherin called vascular endothelial cadherin (VEC). This protein exerts a negative effect on cell growth by binding VEGF receptor (VEGFR) type 2 and inhibiting its signaling activity (Carmeliet et al., 1999; Shay-Salit et al., 2002; Lampugnani et al., 2003; Dejana, 2004). VEGF is a major growth factor for endothelial cells and plays an important role in the formation of new vessels during embryogenesis and in proliferative diseases (Alitalo et al., 2005; Carmeliet, 2005; Ferrara and Kerbel, 2005). In blood endothelium, the activities of VEGF are mediated by its interaction with two tyrosine kinase receptors, VEGFR-1 (flt-1) and -2 (flk/KDR), as well as neuropilins. The growth signals are transferred, to a large extent, through the activation of PLC-γ, PKC, and subsequently p44/42 MAPK (Takahashi et al., 1999, 2001; Matsumoto et al., 2005; Singh et al., 2005).
We found that in contact-inhibited endothelial cells, VEGFR-2 forms a complex with VEC that results in the inhibition of its tyrosine phosphorylation and, consequently, in the attenuation of MAPK activation. This effect was attributed to the phosphatase DEP-1/CD148 that, by binding β-catenin and p120, may associate with the cadherin–receptor complex and dephosphorylate the receptor (Lampugnani et al., 2003). In this study, we go further by describing another aspect of this phenomenon.
Upon activation with specific ligands, growth factor receptors are internalized via clathrin-dependent and -independent pathways. In many cases, this process leads to signaling termination via degradation of the activated receptor complex. Therefore, internalization is considered an important mechanism through which cells may control the intensity and duration of signal transduction.
However, more recent findings indicate that internalization is not just a sink through which receptors are degraded (Di Fiore and De Camilli, 2001; Miaczynska et al., 2004). On the contrary, some receptors, such as TGF-β, EGF, or NGF receptors, can maintain their signaling activity from within intracellular compartments (Suyama et al., 2002; Di Guglielmo et al., 2003; Bryant et al., 2005; Sigismund et al., 2005).
Little is known about the internalization pathways followed by VEGFR-2 or their functional significance (Labrecque et al., 2003; Bhattacharya et al., 2005; Mitola et al., 2006). It has been reported that cadherins may influence growth factor receptor internalization, but the extent to which they do depends on the cadherin or growth factor receptor. In tumor cell lines, N-cadherin forms a complex with FGF receptor 1 that inhibits its internalization and degradation. This causes a sustained FGF signaling and abnormal cell growth (Suyama et al., 2002). In contrast, E-cadherin cointernalizes with FGF receptor 1, which facilitates its nuclear translocation and signaling activity (Bryant and Stow, 2005; Bryant et al., 2005).
In this study, we analyzed the role of VEC on VEGFR-2 internalization and signaling in endothelial cells. We found that the receptor is internalized more rapidly and efficiently when VEC is absent or not clustered at intercellular contacts. Strikingly, internalization does not terminate receptor signaling, which instead continues in endosomes. This may explain why VEC-null cells present increased and uncontrolled growth.
Results
VEC expression inhibits VEGFR-2 internalization
We first investigated whether the establishment of cell to cell contact modulates VEGFR-2 internalization. Using freshly isolated human umbilical vein endothelial cells (HUVECs) stimulated with VEGF, we observed that VEGFR-2 endocytosis, which was evaluated by immunofluorescence labeling of intracellular vesicular compartments, was significantly reduced by cell density (Fig. 1 A). Time course analysis revealed that in sparse cells, the number of receptor-positive vesicles increased more rapidly and to a larger extent than in confluent cells (Fig. 1 A, bottom).
Figure 1.

VEC clustering at cell–cell contacts inhibits VEGFR-2 endocytosis. (A) The internalization of VEGFR-2 from the plasma membrane was analyzed in sparse and confluent HUVECs treated with VEGF for 5 min. To detect the internalized receptor, cells were treated with a recombinant single chain antibody to human VEGFR-2, scFvA7, and acid washed before fixation and processing for immunofluorescence microscopy. Internalized VEGFR-2 appears in a vesicular pattern that is more abundant in sparse than in confluent cultures. (bottom) The granular staining after different incubation lengths with VEGF was quantified using the ImageJ program (see Materials and methods). The results (referred to as events per cell) reported in the graph are means ± SD (error bars) of three independent experiments. At least seven random fields were analyzed for each time point in each experiment. (B) VEC-null and -positive confluent cultures were treated as described in A, but the anti–mouse VEGFR-2 clone Avas12α1 was used. The micrographs show a typical vesicular labeling pattern after a 10-min treatment with VEGF. The staining appears more abundant in VEC-null than in VEC-positive cells. (bottom) The time course of vesicular labeling in response to VEGF was analyzed as described in A. The binding of the antibody does not activate VEGFR-2 nor does it induce its internalization (for details see Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1). In A and B, nuclei stained with DAPI appear blue. *, P ≤ 0.05; **, P ≤ 0.01 by comparing sparse versus confluent (A, bottom) and VEC-null versus -positive (B, bottom) cells by analysis of variance and the Duncan test. Bars (A), 15 μm; (B) 20 μm.
This first observation suggested that the establishment of cell to cell contact reduced VEGFR-2 internalization. Because VEC plays a role in VEGFR-2 signaling, we investigated whether VEC could be involved inVEGFR-2 internalization. We compared syngenic endothelial cell lines differing for the expression of VEC. These cells had been characterized previously in detail and presented superimposable levels of VEGFR-2 (see Fig. 8; Lampugnani et al., 2002, 2003). As shown in Fig. 1 B, after the addition of VEGF, the number of VEGFR-2–containing vesicular compartments is markedly higher in the absence of VEC. Quantification of the amount of biotinylated receptor that was internalized, degraded, or recycled back to the plasma membrane is reported in Fig. 2. In VEC-null endothelium, the receptor is internalized more quickly and to a higher extent than in VEC-positive cells (Fig. 2, A and B).
Figure 8.

Silencing clathrin expression inhibits VEGFR-2 phosphorylation and signaling in VEC-null cells. VEC-null and -positive cells were transfected with Stealth siRNA-targeting mouse clathrin heavy chain. Two oligonucleotides (a and b; respective sequences are reported in Materials and methods) were used that target two independent sequences of the clathrin heavy chain mRNA. Negative Stealth siRNA duplex was used as a control. After 72 h, cells were treated with VEGF or control medium for 10 min. They were then extracted and processed for Western blotting. Clathrin siRNA reduced clathrin heavy chain levels by ∼75 (oligonucleotide a) and 90% (oligonucleotide b) in both cell types. VEGFR-2 phosphorylation at tyrosine 1214 and p44/42 MAPK phosphorylation in response to VEGF were strongly inhibited in VEC-null cells, whereas these parameters were only barely affected in VEC- positive cells. Comparable results were obtained for PY1054/59–VEGFR-2 (not depicted). The graphs show the quantification of these effects as means ± SD (error bars) of five independent experiments. Fold increase after VEGF is shown for PY1214–VEGFR-2 and phospho-p44/42 MAPK. *, P ≤ 0.01 versus negative siRNA by variant analysis and the Dunnet test.
Figure 2.

Internalization, degradation, and recycling of biotinylated VEGFR-2 are inhibited in VEC-positive cells. (A) Internalization and recycling of VEGFR-2 was measured after cell surface biotinylation with thiol-cleavable Sulfo-NHS-SS-Biotin. At selected time points, biotin was cleaved by GSH followed by immunoprecipitation of VEGFR-2 and probing with HRP-streptavidin as described in Materials and methods. Recycling was measured as the amount of biotinylated receptor reappearing on the cell surface (and therefore not protected by GSH cleavage) at the indicated time points after 10 min of internalization in the presence of VEGF (indicated as 0 min in recycling panel). Total, total amount of labeled receptor after Sulfo-NHS-SS-Biotin labeling at 4°C (without GSH treatment). GSH, treatment with reducing GSH to remove any labeling on the residual surface-exposed receptor. (B) Densitometric analysis of internalization expressed as a percentage of total labeling. (C and D) Quantitation of receptor degradation and recycling as a percentage of the amount of receptor internalized after 10 min with VEGF. Although A presents a representative experiment, the values in B–D are the means of four independent experiments ± SD (error bars). **, P ≤ 0.01; *, P ≤0.05 comparing VEC-null with -positive cells, respectively, by analysis of variance and the Duncan test.
In VEC-null cells, receptor internalization kinetics appear faster in biotinylation than in immunofluorescence experiments. This apparent discrepancy may be caused by internalization compartments, which can be measured in biotinylation experiments because they are protected from glutathione (GSH) reduction but are not yet clustered in structures resolvable in immunofluorescence microscopy.
The overall amount of internalized receptor for the duration of the experiment is about fourfold more in VEC-null than -positive cells. Receptor degradation exceeds recycling by about fivefold in both cell types, but both parameters are significantly increased in the absence of VEC (Fig. 2, C and D). These data indicate that a higher amount of VEGFR-2 is internalized, degraded, and recycled in the absence of VEC.
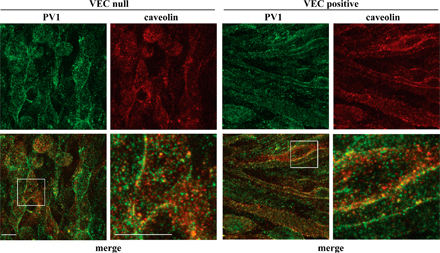
Internalization of growth factor receptors may follow clathrin-dependent or -independent pathways. Among the latter, caveolae have been shown to regulate receptor internalization directed toward degradation (Di Guglielmo et al., 2003; Sigismund et al., 2005). Our codistribution experiments of VEGFR-2 with early endosomal antigen-1 (EEA-1) and caveolin-1 show that VEGFR-2 internalizes mostly in EEA-1–positive early endosomes (Fig. 3) and to a very low extent in caveolae. Colocalization of VEGFR-2 and the caveolar component PV-1 (Stan et al., 2004; Stan, 2005) was also negligible (unpublished data). To control whether caveolae were expressed correctly and to a comparable extent in both VEC-null and -positive cells, we costained these structures with PV-1 and caveolin antibodies. As shown in Fig. S1 (available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1), the extensive and comparable colocalization of caveolin and PV-1 could be observed in both cell types, suggesting structural integrity of the caveolar compartment.
Figure 3.

Internalized VEGFR-2 colocalizes with EEA-1–positive compartments by immunofluorescence analysis. HUVECs, VEC-null, and VEC-positive cells were double labeled for VEGFR-2 and either EEA-1 or caveolin-1. Cells were activated with VEGF for 10 min. (A) Representative examples of confocal images for each antigen and their respective merges (boxed areas; 3.5-fold magnification) are shown for confluent HUVECs after treatment with VEGF. VEGFR-2 was revealed with an AlexaFluor488-conjugated secondary antibody and is shown in green. EEA-1 and caveolin revealed with AlexaFluor647-conjugated secondary antibodies are shown in red. Nuclei appear blue after DAPI staining. Bars, 10 μm. (B) To quantify colocalization events, images were analyzed using the ImageJ colocalization plugin (as described in Materials and methods). The graphs present the number of colocalization events normalized for the number of VEGFR-2– positive compartments. After VEGF treatment, ∼45–55% of VEGFR-2–positive compartments showed colocalization with EEA-1 in all of the situations examined. Colocalization of internalized VEGFR-2 with caveolin-1 was negligible. Values are the mean of at least three experiments ± SD (error bars). In each experiment, at least five random fields were analyzed for each point. *, P ≤ 0.01 by t test. (C, a and b) Immunogold labeling of EEA-1 (10 nm gold; arrows) and VEGFR-2 (15 nm gold; arrowheads) on an ultrathin cryosection of VEC-null and -positive cells treated with VEGF for 10 min. The panels display VEGFR-2 labeling of EEA-1–positive endosomes in VEC-null rather than in VEC-positive cells. (c and d) Under the same conditions, morphologically identified caveolae are devoid of VEGFR-2. Bar (a), 227 nm; (b) 222 nm; (c) 350 nm; (d) 370 nm.
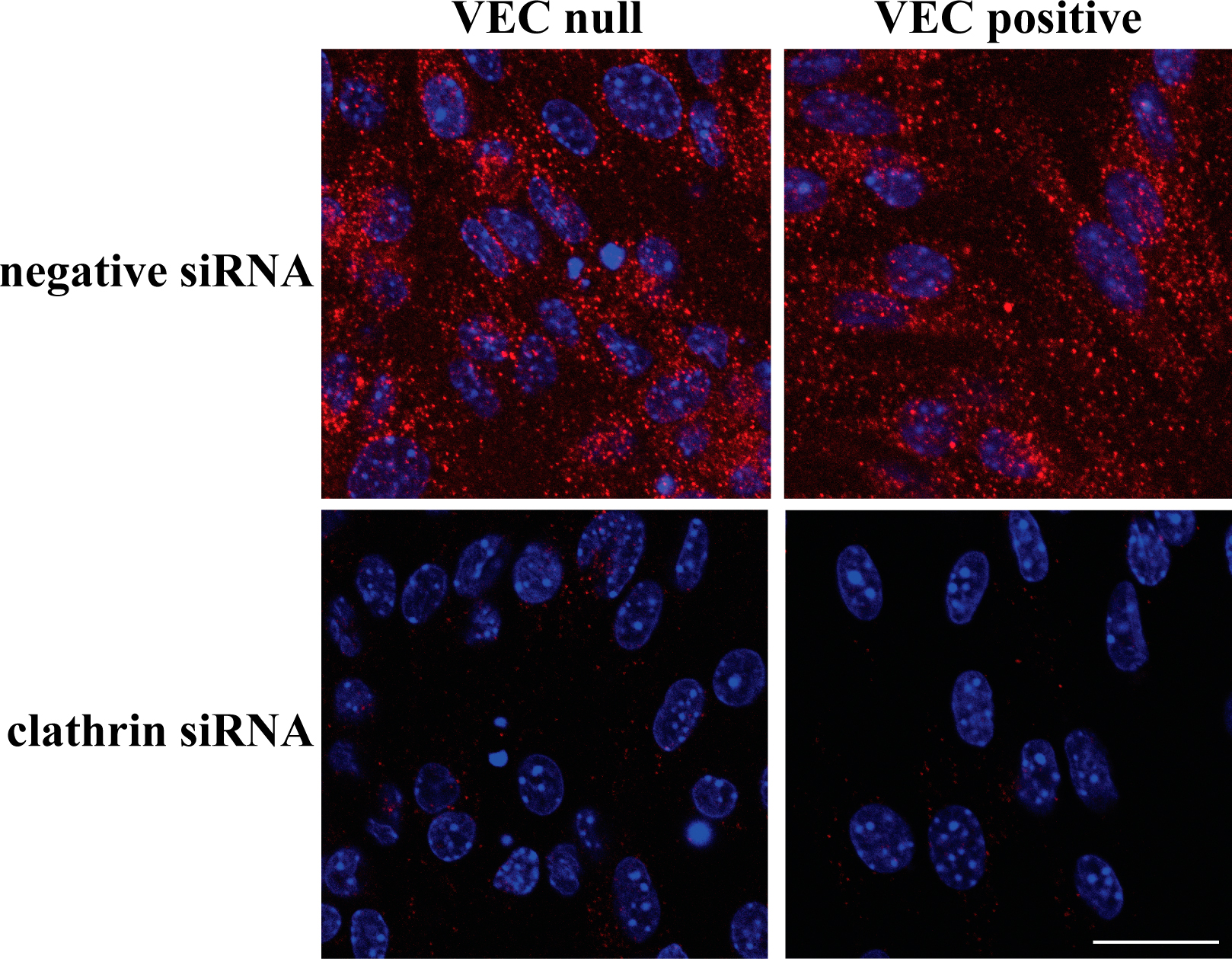
The preferential distribution of VEGFR-2 in EEA-1–positive endosomes was further confirmed using immuno-EM (Fig. 3 C). In addition, silencing clathrin heavy chain expression either by siRNA (see Fig. 8 and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1) or disrupting clathrin-coated pits by hypertonic medium blocked receptor internalization both in VEC-positive and -null cells (Fig. 4). In contrast, after incubation with filipin at a concentration able to fully disrupt lipid rafts and caveolae (Schnitzer et al., 1994; Andriopoulou et al., 1999), receptor internalization did not significantly change (Fig. 4).
Figure 4.

VEGFR-2 is internalized through a clathrin-dependent pathway. (A) VEC-null and -positive cells were transfected with either Stealth siRNA targeting the mouse clathrin heavy chain or negative Stealth siRNA control duplex (and used 72 h later), or the cells were treated with hypertonic medium (0.45 M sucrose for 30 min) or 1 μg/ml filipin for 1 h. The micrographs show VEGFR-2 immunofluorescence staining after VEGF treatment for 10 min. Both clathrin heavy chain siRNA and hypertonic medium strongly reduced VEGFR-2 vesicular patterning both in VEC-null and -positive cells. Using filipin to interfere with the caveolar compartment had no effect on either cell type. The negative Stealth siRNA control duplex produced results that were indistinguishable from untreated cells (not depicted). Nuclei appear blue after DAPI staining. Bar, 20 μm. (B) Column graphs represent VEGFR-2 vesicular labeling in VEGF-treated cells quantified by ImageJ. Ctr, control; HM, hypertonic medium. Values normalized per cell are the mean of three independent experiments ± SD (error bars). In each experiment, at least five independent fields were analyzed for each point. *, P ≤ 0.01 versus control values by analysis of variance and the Dunnet test.
We also performed experiments at different time points (5, 7, 10, 20, and 30 min) after the addition of VEGF in VEC-null and -positive cells using caveolin and PV-1 as markers of caveolae and EEA-1 and Rab-5 as markers of early endosomes. At all time points considered, we could not see internalized VEGFR-2 in vesicles positive to either caveolin or PV-1 but only in bona fide early endosomes (unpublished data). These data strongly suggest that, at least within the experimental conditions used, VEGFR-2 endocytosis in endothelial cells is mostly clathrin dependent.
Internalized VEGFR-2 retains signaling activity
In the absence of VEC, endothelial cells respond more effectively to growth signals transferred by VEGFR-2. From the aforementioned observations, we concluded that VEGFR-2 is internalized more quickly and to a larger extent in VEC-null cells than in VEC-positive cells. Therefore, we asked whether the receptor retains its signaling activity when sequestered in intracellular compartments.
We first tested whether the internalized receptor retained tyrosine phosphorylation by cell fractionation on an iodixanol gradient (Yeaman et al., 2001). Antibodies recognizing phosphotyrosine (PY) 1214– and PY1054/59–VEGFR-2 were used. As shown in Fig. 5, in the absence of VEC, a higher amount of phosphorylated VEGFR-2 is detected in intracellular fractions, whereas in the presence of VEC, the phosphorylated receptor remains preferentially in fractions corresponding to peripheral plasma membranes.
Figure 5.

VEC expression reduces the amount of PY–VEGFR-2 in internal compartments. Extracts of VEGF-treated VEC-null and -positive cells were fractionated on an iodixanol gradient as described in Materials and methods. Samples representative of the total protein content of each fraction were analyzed by SDS-PAGE and Western blotting. As expected, VEC concentrates in lower density fractions corresponding to plasmatic membranes, whereas clathrin and EEA-1 are enriched in higher density fractions corresponding to internal membranes (Yeaman et al., 2001). Upon stimulation with VEGF, PY1214–VEGFR-2 is enriched in fractions corresponding to the internal membranes in VEC-null cells. The graph shows the ratio between the phosphorylated receptor and the total receptor present in each fraction. For quantification, in each fraction, we considered the band with the molecular mass of the mature form of the receptor at the plasma membrane (∼210 kD; see fraction 1 and arrowheads; Takahashi and Shibuya, 1997). We chose this band by making the assumption that full-length VEGFR-2 represents the signaling form of the receptor. At increasing density, a lighter band appears both in VEC-null and -positive cells (asterisks). It is likely that this band derives from VEGFR-2 processing in internal compartments (for instance, proteolytic or intermediate synthesis products) and is indeed not present in the peripheral membrane fractions. Comparable results were obtained using an antibody to PY1054/59–VEGFR-2 (not depicted).
Phosphorylation of VEGFR-2 tyrosine 1175 is required for binding and activation of PLC-γ, which is the major effector of VEGF-mediated cell proliferation (Takahashi et al., 2001). By using antibodies specific for PY1175–VEGFR-2, we observed that in the absence of VEC, a higher amount of internalized receptor was phosphorylated at this specific tyrosine (Fig. 6, A and B). Consistently, more internalized PY1175–VEGFR-2 was found in sparse than in confluent HUVECs (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1).
Figure 6.
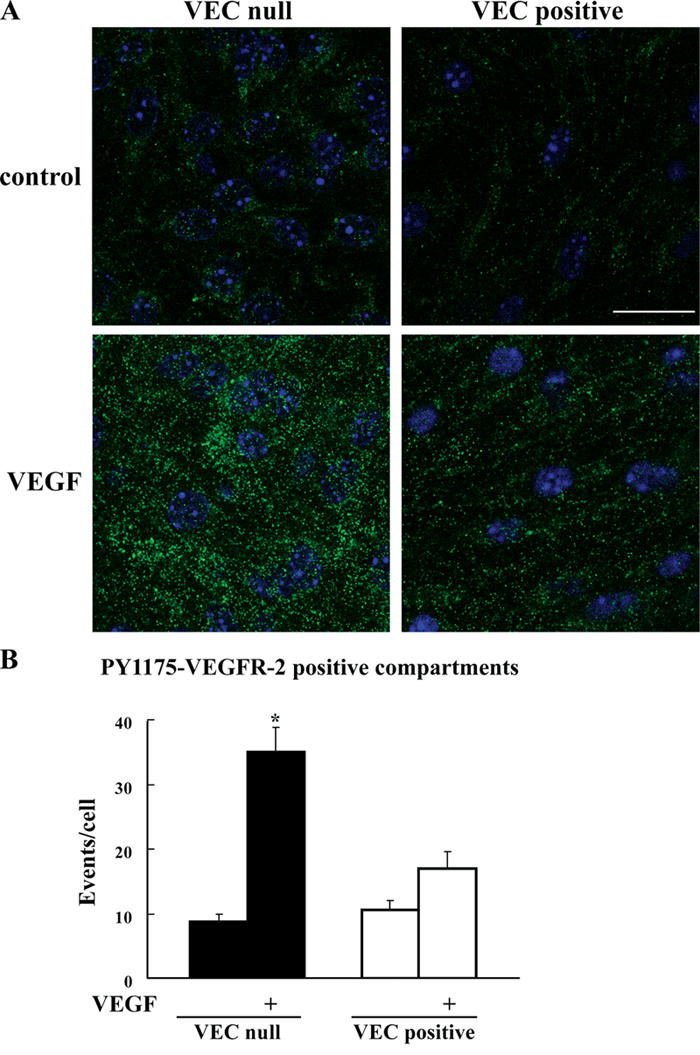
Internalized VEGFR-2 is phosphorylated at tyrosine 1175. (A) VEC-null and -positive cells were stimulated with VEGF as in Fig. 1, fixed, and processed for immunofluorescence microscopy. The vesicular labeling pattern observed with PY1175–VEGFR-2 antibody after stimulation with VEGF was significantly more abundant in VEC-null than in VEC-positive cells. Nuclei appear blue after DAPI staining. Bar, 20 μm. (B) Images were analyzed by ImageJ to quantify PY115–VEGFR-2–positive compartments (see Materials and methods). The graph presents the mean ± SD (error bars) calculated from 18 random fields that were analyzed through ImageJ in five independent experiments and is normalized to the number of cells per field. In each experiment, at least three random fields were analyzed. *, P ≤ 0.01 comparing VEC-null with -positive cells after VEGF by analysis of variance and the Duncan test.
To further prove that PLC-γ could be activated by the internalized receptor, we stained the cells with antibodies directed to the active PY783–PLC-γ. As shown in Fig. 7, active PLC-γ codistributes with internalized VEGFR-2 more effectively in the absence than in the presence of VEC.
Figure 7.

Colocalization of internalized VEGFR-2 and activated PLC-γ. (A) VEC-null and -positive cells were labeled in vivo with antibodies to VEGFR-2, stimulated with VEGF as in Fig. 6, acid washed, fixed, and processed for immunofluorescence microscopy. Cells were double labeled with an antibody that recognizes PLC-γ only when phosphorylated at tyrosine 783. VEGFR-2 was revealed with an AlexaFluor488-conjugated secondary antibody and is shown in green. PY783–PLC-γ, which was revealed with an AlexaFluor647-conjugated secondary antibody, is shown in red. For each cell type, the bottom panel on the right (threefold magnification of the boxed areas) shows the colocalization of VEGFR-2 and PY783–PLC-γ (pink) set upon the VEGFR-2 background (gray). This was obtained through the ImageJ colocalization plugin (see Materials and methods for details). Arrows point to the colocalization of PY783–PLC-γ and VEGFR-2. Bars, 10 μm. (B) The confocal images were analyzed through ImageJ to calculate the number of colocalization events. These values, which were normalized over the number of VEGFR-2–positive compartments per cell, are presented in the graph. After VEGF treatment, colocalization was about fivefold higher in VEC-null than in VEC-positive cells. Values in the graph are the mean from three independent experiments ± SD (error bars). In each experiment, each point was calculated from at least five random fields. *, P ≤ 0.01 comparing VEC-null with -positive cells after VEGF by analysis of variance and the Duncan test.
Inhibition of VEGFR-2 internalization affects its proliferative signaling
Overall, the aforementioned data indicate that VEC limits receptor activation and internalization. In the absence of this protein, the receptor is internalized more efficiently and retains its active state for a longer time, leading to continuous proliferative signaling.
To further test this hypothesis, we prevented receptor internalization by silencing clathrin with two specific siRNAs (Figs. 4, 8, and S3) that target independent sequences of clathrin heavy chain messenger. As expected, VEGF-induced phosphorylation of VEGFR-2 and activation of p44/42 MAPK were higher in the absence than in the presence of VEC (Fig. 8). When siRNA clathrin was applied to VEC-null cells, both receptor and MAPK phosphorylation dropped to values comparable with those of VEC-positive cells. In contrast, in the presence of VEC, the effect of siRNA clathrin was either weak or undetectable. Consistently, the inhibition of MAPK activation was also observed by treating the cells with hypertonic medium, whereas treatment with filipin did not modify MAPK activation in response to VEGF either in VEC-positive or -null cells (unpublished data). Overall, these results strongly suggest that internalization protects the receptor from dephosphorylation and, therefore, increases and prolongs its proliferative signaling.
VEC association with VEGFR-2 is required for receptor retention at the plasma membrane
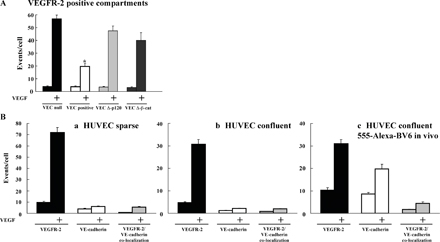
We then investigated the mechanism through which VEC inhibits VEGFR-2 internalization. In a previous study, we found that VEC forms a complex with VEGFR-2, and we analyzed the domains of VEC involved in this process (Lampugnani et al., 2003). VEC mutants lacking either the β-catenin– or p120-binding domains were unable or less efficient, respectively, to coimmunoprecipitate VEGFR-2. We found that these mutants were also unable to significantly prevent VEGFR-2 internalization (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1), suggesting that receptor internalization is reduced as a consequence of binding to VEC.
Similar to E-cadherin (for review see Bryant and Stow, 2004), VEC can be internalized through clathrin-coated pits (Xiao et al., 2005). We tested whether VEC codistributes with VEGFR-2 in intracellular compartments. As reported in Fig. 9 upon VEGF activation, no significant codistribution of VEC with VEGFR-2–positive vesicles is detected in VEC-positive cells and HUVECs (Fig. S4). Only junctional colocalization can be observed (Fig. 9). The lack of codistribution in internal compartments was confirmed by immuno-EM (unpublished data). Collectively, these data suggest that the receptor internalizes upon dissociation from VEC.
Figure 9.
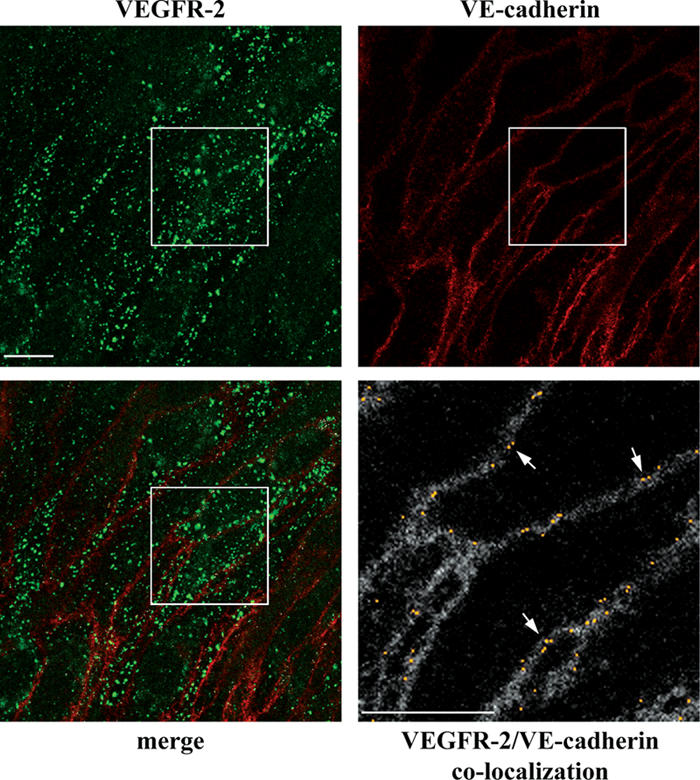
VEC does not codistribute with VEGFR-2 in internal compartments. VEC-positive cells were treated with VEGF for 10 min, double stained for VEGFR-2 and VEC, and analyzed by confocal microscopy. Besides junctional staining, VEC did not show any obvious vesicular pattern. VEGFR-2 and VEC appeared to codistribute only at cell–cell contacts and not in intracellular compartments. The bottom panel on the right (2.6-fold magnification of the boxed areas) shows the colocalization of VEGFR-2 and VEC (yellow) set upon the VEC background (gray). This was obtained through the colocalization plugin of ImageJ (see Materials and methods for details). Arrows point to the junctional colocalization of VEGFR-2 and VEC. Bars, 10 μm.
VEGFR-2 phosphorylation is required for internalization and signaling
In previous studies, we showed that the phosphatase DEP-1/CD148 can reduce VEGFR-2 signaling. DEP-1/CD148 can associate with β-catenin and p120 (Holsinger et al., 2002; Palka et al., 2003) and with the VEC–VEGFR-2 complex, reducing VEGFR-2 phosphorylation (Lampugnani et al., 2003). Therefore, we tested whether DEP-1/CD148 could also reduce receptor internalization.
As reported in Fig. 10 A, in endothelial cells transfected with DEP-1/CD148 siRNA, VEGFR-2 internalization is significantly higher. This effect is accompanied by an increase in VEGFR-2 phosphorylation and MAPK activation (Lampugnani et al., 2003). These data suggest that retention of VEGFR-2 at the membrane by VEC allows its dephosphorylation by DEP-1/CD148 and limits its internalization and signaling.
Figure 10.

DEP-1/CD148 silencing in VEC-positive cells increases internalization, tyrosine phosphorylation, and activity of VEGFR-2. VEC-positive cells were transfected with either Stealth siRNA-targeting mouse DEP-1/CD148 or a negative Stealth siRNA control duplex. After 72 h, cells were treated with VEGF for 10 min and either fixed and processed for immunofluorescence microscopy or extracted and processed for Western blotting. DEP-1/CD148 siRNA reduced DEP-1/CD148 protein by ∼45% (B). (A) VEGFR-2–positive vesicular compartments were found to be significantly higher (>80%) in VEGF-treated cells after transfection with DEP-1/CD148 siRNA. Means of three independent experiments ± SD (error bars) are shown. In each experiment, at least five random fields were analyzed. *, P ≤ 0.01 comparing negative with DEP-1 siRNA interference by analysis of variance and the Duncan test. (B) Phosphorylation of VEGFR-2 at tyrosine 1214 and of p44/42 MAPK was increased by ∼80 and 60%, respectively, after DEP-1/CD148 siRNA transfection in VEGF-treated cells.
Discussion
In this study, we report a novel aspect of the mechanism through which VEC expression and clustering inhibits VEGFR-2 proliferative signaling. We found that in the absence of VEC or in conditions in which VEC is not clustered at adherens junctions as in sparse cells, VEGFR-2 is endocytosed to a higher extent in intracellular compartments, from where it maintains its signaling activity. VEC could therefore reduce receptor activity by inhibiting VEGFR-2 internalization and promoting its inactivation at the cell surface.
In our experimental conditions, VEGFR-2 is internalized in early endosomes mostly through a clathrin-dependent pathway. We were unable to detect caveolin-1–positive vesicles containing VEGFR-2, and we could not inhibit receptor internalization using a caveolae-perturbing drug such as filipin (Schnitzer et al., 1994; Andriopoulou et al., 1999). Other studies found the codistribution of VEGFR-2 with caveolin-1 (Labrecque et al., 2003; Bhattacharya et al., 2005; Ikeda et al., 2005), and we cannot exclude that under different experimental conditions, VEGFR-2 may be internalized through caveolae. However, the observed association with caveolin-1 may also represent a mechanism of receptor compartmentalization at the plasma membrane. It was found that caveolin-1 would form a molecular complex with VEGFR-2 that inhibits receptor activation in resting cells. Upon activation of the cells with VEGF, caveolin-1 is phosphorylated, and the complex rapidly dissociates (Labrecque et al., 2003). Thus, it is tempting to speculate that similar to TGF-β receptor (Di Guglielmo et al., 2003), once VEGFR-2 is released from the caveolin-1 complex, it becomes available for internalization in clathrin-coated pits. In agreement with this model, the overexpression of caveolin-1 in transgenic mice reduces permeability and angiogenic response to VEGF (Bauer et al., 2005).
It has been reported that VEGFR-2 internalization and degradation are regulated by ubiquitination through a Cbl-dependent mechanism (Duval et al., 2003) or C-tail serine phosphorylation by activated PKC (Singh et al., 2005). These mechanisms may coexist and may be responsible for the amount of receptor degradation reported here.
Our observations support the hypothesis that internalized VEGFR-2 maintains its activity. These data are in agreement with recent publications indicating that signaling through growth factor receptors does not occur only at the cell membrane but may continue even more effectively from intracellular compartments (Di Fiore and De Camilli, 2001; Sorkin and Von Zastrow, 2002; Miaczynska et al., 2004; Le Roy and Wrana, 2005). Clathrin-dependent internalization of TGF-β in early endosomes, where the Smad2 anchor SARA is enriched, promotes TGF-β signaling (Di Guglielmo et al., 2003). EGF receptor is internalized shortly after ligand addition in intracellular compartments together with its downstream signaling factors shc, Grb2, and mSOS (Di Guglielmo et al., 1994) and maintains its signaling activity (Pennock and Wang, 2003). Similarly, the specific activation of endosome-associated PDGF receptor leads to the activation of its major signaling pathways (Wang et al., 2004). Thus, as for VEGFR-2, endocytic transport is important not only for receptor turnover but also for regulating signal transduction and for mediating the formation of specialized signaling complexes.
A novel aspect of our work is that VEC inhibits VEGFR-2 internalization, thereby reducing its cell growth signaling activity. How can VEC inhibit receptor endocytosis? A likely hypothesis is that VEC retains VEGFR-2 at the membrane by binding to it. In addition to our studies, others have reported (Shay-Salit et al., 2002; Lampugnani et al., 2003; Weis et al., 2004; Lambeng et al., 2005) that VEGFR-2 couples with VEC. This process requires the binding of VEC to β-catenin and, to a lesser extent, to p120. In the present study, we found that mutants of VEC lacking the cytoplasmic domain responsible for binding either β-catenin or p120 and unable to associate with VEGFR-2 (Lampugnani et al., 2003) do not prevent VEGFR-2 internalization. This supports the idea that VEC–VEGFR-2 coupling is required to inhibit receptor endocytosis.
Cadherins themselves are endocytosed via several different routes, including clathrin-dependent (Le et al., 1999; Palacios et al., 2002; Ivanov et al., 2004; Izumi et al., 2004) and -independent pathways (Akhtar and Hotchin, 2001; Paterson et al., 2003). Therefore, cotrafficking of receptor and cadherin complexes is possible (for reviews see Bryant and Stow, 2004, 2005; Cavallaro and Christofori, 2004). However, under our experimental conditions, we could detect VEC in intracellular compartments (Xiao et al., 2005), but we could not see codistribution with the receptor. Thus, it is likely that the receptor dissociates from VEC before internalization (Fig. 9).
In a previous study, the phosphatase DEP-1/CD148 was found to play a role in the inhibitory effect of VEC on VEGR-2 signaling (Lampugnani et al., 2003). This phosphatase associates with VEC through its binding to β-catenin and p120 and, in this way, reduces VEGFR-2 phosphorylation (Lampugnani et al., 2003). We report that DEP-1/CD148 could prevent VEGFR-2 internalization along with the reduction of receptor phosphorylation and signaling. Therefore, it is possible that VEC, by retaining VEGFR-2 at the cell surface, allows its dephosphorylation by DEP-1/CD148, which, in turn, inhibits its internalization and signaling.
Besides VEC, VEGFR-2 was found to bind to integrins (Soldi et al., 1999). Another study reported that when cells are plated on collagen I, the phosphatase SHP2 can associate with VEGFR-2 and stimulate its internalization. SHP2 activates Src, which in turn activates dynamin II–dependent receptor internalization (Mitola et al., 2006). Interestingly, this phenomenon does not occur when cells are plated on vitronectin, and SHP2 does not bind to VEGFR-2 (Mitola et al., 2006). These observations suggest that the capacity of different adhesive proteins to complex with growth factor receptors and modulate their internalization and signaling may be a general paradigm. In this way, cells may modulate their growth and survival as a function of density and interaction with specific matrix proteins.
In conclusion, the results reported in this study are consistent with the idea that VEGFR-2 proliferative signaling is increased by endocytosis. The inhibitory role of VEC is likely that of binding and retaining the receptor at the cell surface, preventing its endocytosis, and favoring inactivation by DEP-1/CD148. This suggests that the modulation of VEC–VEGFR-2 complex formation may be a novel strategy to regulate VEGF proliferative signaling and, therefore, to inhibit or stimulate angiogenesis.
Materials and methods
Primary antibodies
For the detection of VEGFR-2, anti–human VEGFR-2 (single chain recombinant; clone scFvA7 with E tag; RDI and Fitzgerald) and anti–mouse VEGFR-2 (rat clone Avas12α1; RDI and Fitzgerald) were used for immunofluorescence; rabbit polyclonal C-1158 (sc504; Santa Cruz Biotechnology, Inc.) was used for Western blotting. Antibodies to tyrosine-phosphorylated VEGFR-2 were rabbit polyclonal PY1214 and PY1054/59 (Biosource International) and rabbit polyclonal PY1175, which was provided by M. Shibuya (University of Tokyo, Tokyo, Japan). Antibodies to clathrin heavy chain were mouse monoclonal cloneX22 (Affinity BioReagents, Inc.) for immunofluorescence and mouse monoclonal clone 23 (R&D Systems) for Western blotting. Antibody to EEA-1 was goat polyclonal N-19 (sc-6415; Santa Cruz Biotechnology, Inc.); antibody to caveolin-1 was rabbit polyclonal N-20 (sc-894; Santa Cruz Biotechnology, Inc.); and antibody to VEC was goat polyclonal C-19 (sc-6458; Santa Cruz Biotechnology, Inc.) and mouse monoclonal BV6 and BV9 (produced in our laboratory; Corada et al., 2001). Antibody to PY783-PLCγ, total p42/44 MAPK, and phospho-p42/44 MAPK was rabbit polyclonal (Cell Signaling). Antibody to DEP-1/CD148/CD148 was goat polyclonal (R&D Systems), and antibody to PV-1 was rat monoclonal (provided by R. Stan, Dartmouth Medical School, Lebanon, NH).
Cell types and culture conditions
Endothelial cells with a homozygous null mutation of the VEC gene (VEC null) and the cell lines derived from them through retroviral gene transfer and expressing wild-type (VEC positive) or various VEC mutant constructs were generated and characterized as described previously in detail (Lampugnani et al., 2003). For all of the experiments, 50,000 cells/cm2 (to reach confluence within 24 h) were seeded in complete culture medium and cultured without medium change for 72 h. Cells were then washed once with MCDB 131 (Life Technologies) and starved in 1% BSA in MCDB 131 (starving medium) for 18–20 h. 2 h before activation, cells were washed once with MCDB 131 and further incubated in fresh starving medium. Cells were treated with 80 ng/ml VEGF (human recombinant VEGF 165; PeproTech) in fresh starving medium (fresh starving medium alone was used for controls) for the indicated intervals at 37°C. If not otherwise indicated, VEGF treatment was for 10 min.
HUVECs were cultured in MCDB 131 with endothelial cell supplements as described previously (Lampugnani et al., 2003). For the experiments 1,800 and 42,000 cells/cm2 were seeded to obtain sparse and confluent cultures, respectively. HUVECs were then treated as described in this section for mouse endothelial cells except that starving was reduced to 6 h before stimulation with VEGF.
Filipin and hypertonic treatments
Caveolae organization was altered by treatment with 1 μg/ml filipin (filipin III from Streptomyces filipinensis; Sigma-Aldrich) for 1 h (Schnitzer et al., 1994; Andriopoulou et al., 1999). Clathrin pit–mediated endocytosis was inhibited using hypertonic medium (0.45 M sucrose in MCDB 131 with 1% BSA) for 30 min to affect as described previously (Heuser and Anderson, 1989; Ehrlich et al., 2001). Cells were then stimulated and assayed as indicated in the specific sections.
RNA interference
Cells were seeded as described in Cell types and culture conditions. 20 h later, cells were washed once with OptiMEM (Life Technologies) and transfected with 40 nM of Stealth oligonucleotides (Invitrogen) using 2 μl/ml LipofectAMINE 2000 (Invitrogen) in OptiMEM according to the manufacturer's instructions. After 5 h, the transfection medium was removed, and complete culture medium was added. Cells were cultured for a further 72 h before stimulation and processing for immunofluorescence and extraction.
Stealth oligonucleotides were used as follows: a, 5′-GCAGUUGUUCAUACCCAUCUUCUUA-3′ (start nucleotide was 2,118 bases downstream of the start codon); b, 5′-GAAGAACUCUUUGCCCGGAAAUUUA-3′ (start nucleotide was 1,284 bases downstream of the start codon) to target mouse clathrin heavy chain; and 5′-UCGAGCCAGUGAGCAUGUUUGGAAA-3′ (start nucleotide was 4,023 bases downstream of the start codon) to target mouse DEP-1/CD148. As a control, a Stealth siRNA-negative control duplex oligonucleotide (Invitrogen) with a C/G content equivalent to the positive oligonucleotide was used.
Internalization assays
Microscopy.
Cells were treated in vivo with anti–VEGFR-2 antibody and acid washed before fixation (Ehrlich et al., 2001; Di Guglielmo et al., 2003) as follows. We used monoclonal antibodies to the extracellular domain of human and mouse VEGFR-2 (see Primary antibodies) that are described by the manufacturers to be devoid of biological activity. As this aspect was crucial to the assay, we tested the effect of these antibodies on basal and VEGF-stimulated phosphorylation and internalization of VEGFR-2. We found that these antibodies do not stimulate the phosphorylation of VEGFR-2 in either condition. These results are reported in Fig. S5 (available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1). Anti–mouse VEGFR-2 was dialyzed for 3 h against PBS (with one change) to remove azide. Cells were precooled for 30 min on ice and treated with 10 μg/ml of antibody for 1 h on ice with gentle agitation. Before stimulation, cells were washed with ice-cold 1% BSA in MCDB 131 to remove unbound antibody, and fresh medium was added. Cells were stimulated with 80 ng/ml VEGF and transferred to 37°C. After the indicated time intervals, cells were placed on ice and acid washed (three washes with ice-cold 50 mM glycine in Ca2+/Mg2+ HBSS, pH 2.5, and two washes with Ca2+/Mg2+ HBSS, pH 7.5) to remove the antibody from the cell surface. Cells were then fixed as described in Immunofluorescence microscopy. To reveal the distribution of the primary antibody, AlexaFluor488-conjugated donkey anti–rat (Invitrogen) was used for the rat anti–mouse VEGFR-2. For the recombinant E-tagged anti–human VEGFR-2, rabbit anti–E-tag (Abcam) followed by AlexaFluor488-conjugated donkey anti–rabbit (Invitrogen) were used.
Cell surface biotinylation and immunoprecipitation.
Internalization, recycling, and degradation were measured as described previously by Fabbri et al. (1999) with the following modifications. Cells were put on ice and washed three times with ice-cold PBS containing Ca2+ and Mg2+ (Ca2+/Mg2+ PBS). For surface biotinylation, cells in Ca2+/Mg2+ PBS were treated with 0.5 mg/ml of thiol-cleavable Sulfo-NHS-S-S-Biotin (Pierce Chemical Co.) for 1 h on ice. They were then washed on ice twice with Ca2+/Mg2+ PBS, once with MCDB 131, and once with 1% BSA MCDB 131. 80 ng/ml VEGF in fresh 1% BSA MCDB 131 was added, and cells were incubated at 37°C for the time indicated to allow internalization. The cultures were then put back on ice and washed three times with ice-cold Ca2+/Mg2+ PBS. Samples were incubated twice for 20 min with 45 mM of the membrane-nonpermeable reducing agent GSH in 75 mM NaCl, with 75 mM NaOH and 1% BSA added just before use (stripping buffer). Cells were further washed twice on ice with Ca2+/Mg2+ PBS and incubated for 15 min with iodoacetamide (in Ca2+/Mg2+ PBS with 1% BSA; quenching buffer) to quench free sulfo-reactive groups. To evaluate total labeling, a sample for each cell type was not reduced with GSH. To control background, a sample was labeled and reduced without incubation at 37°C.
For immunoprecipitation, cells were washed with Ca2+/Mg2+ PBS and extracted on ice in 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Triton X-100, 1% NP-40, and a cocktail of protease inhibitors (Set III; Calbiochem). Extracts were precleared for 90 min with protein A–agarose beads and incubated overnight with 5 μg anti–VEGFR-2 (rabbit sc-504), and the immunocomplexes were collected on protein A–agarose beads for 90 min. After five washes in extraction buffer (the last one containing 0.1% Triton X-100), proteins were eluted by boiling for 10 min in nonreducing laemmli sample buffer. Samples were analyzed by SDS-PAGE followed by Western blotting on nitrocellulose membrane and revealed by ECL chemiluminescence. Band intensity was quantified by ImageJ analysis (National Institutes of Health; freely available at http://rsb.info.nih.gov/ij/).
To quantify VEGFR-2 recycling and degradation, cells were labeled as described in the first paragraph of this section, and endocytosis was allowed for 10 min in the presence of 80 ng/ml VEGF (peak time for VEGF-induced VEGFR-2 internalization both in VEC-null and -positive cells as determined in internalization experiments). Samples were then reduced as described in the first paragraph of this section to remove the label from the residual cell surface receptor. The internalized fraction was chased by reincubation at 37°C for 10 and 20 min in duplicate samples. One sample was reduced to evaluate the amount of VEGFR-2 that recycled back to the plasma membrane, and the other sample was left unreduced to measure degradation. The samples were then processed as described in the first paragraph of this section. VEGFR-2 degradation was calculated by subtracting the value of residual biotinylated receptor after incubation at 37°C without reduction (i.e., internalized + recycled − degraded) from the total pool of internalized receptor. VEGFR-2 recycling was calculated by subtracting both the degradation value and the value of residual biotinylated receptor after incubation at 37°C and reduction (i.e., internalized − recycled − degraded) from the total pool of internalized receptor.
Immunofluorescence microscopy
Cells were cultured in 35-mm diameter petri dishes as described in Cell types and culture conditions. After the treatments indicated in the specific sections, culture medium was removed, and cells were fixed in 1% PFA in 2.5 mM triethanolamine, pH 7.5, containing 0.1% Triton X-100 and 0.1% NP-40 for 25 min at RT (Lallemand et al., 2003). Before staining, 0.5% Triton X-100 in PBS was added for 10 min at RT. In some experiments, immunofluorescence microscopy for VEGFR-2 was performed using the Avas12α1 antibody after cell fixation (in vitro staining). The fixation/permeabilization method applied (Lallemand et al., 2003) allows an optimal observation of VEGFR-2 in internal compartments with in vitro staining (primary antibody after cell fixation). Data obtained with in vivo (see Internalization assays) and in vitro staining were superimposable for both VEC's effect on VEGFR-2 vesicular labeling and the codistribution of VEGFR-2 with markers of specific compartments such as EEA-1 and caveolin-1. A comparison of VEGFR-2 vesicular distribution after in vivo staining (with and without acid wash before fixation) is shown in Fig. S5.
In some experiments (in particular for clathrin detection), fixation was performed in 1% PFA, and permeabilization was performed with 0.02% saponin that was maintained for all of the staining procedure. Fluorophore-labeled secondary antibodies produced in donkey had minimal cross-reactivity to other species except for the targeted species (Invitrogen). Anti–VEGFR-2 was revealed with AlexaFluor488-conjugated secondary antibodies (anti–rat for anti–mouse VEGFR-2 and anti–rabbit for anti–human VEGFR-2). In double labeling experiments, AlexaFluo488- and -647 fluorochromes were used to stain each of the two antigens, respectively.
Samples were observed under a fluorescence microscope (DMR; Leica) using 63× and 100× lenses. Images were captured using a charge-coupled camera (model 3; Hamamatsu) before processing through Adobe Photoshop for MacIntosh. Quantification of vesicular labeling was performed using the ImageJ program (version 10.2). For comparison purposes, different sample images of the same antigen were acquired under constant acquisition settings. The best-fit lower threshold to eliminate most of the signal background was determined using the threshold tool and confirmed by visual inspection and count of one-pixel dimension particles. In some cases, the same sample was analyzed at different lower thresholds to determine the best fit. Upper threshold was always set at 255. Particles with a minimum size of five pixels were counted. For colocalization analysis, images were acquired using a confocal microscope (TCS SP2 AOBS; Leica) with a 63× objective and a 3× zoom. Colocalization was quantified using the colocalization plugin of ImageJ. The channel ratio was always set at 90%. For both channels, the best-fit lower threshold value to remove most background signal was determined using the threshold tool as described above.
EM
For immunogold labeling, 4% PFA/0.4% glutaraldehyde in PBS was added in a 1:1 ratio to culture medium. After 2 h at RT, the fixative was discarded, and cells were scraped in 1% PFA in PBS, collected in Eppendorf tubes, and processed for ultrathin cryosectioning as described previously (Confalonieri et al., 2000). Double immunogold labeling was performed as described previously (Slot et al., 1991).
Cell fractionation, extraction, and Western blotting
Subcellular fractionation on an iodixanol gradient was performed as described previously (Yeaman et al., 2001). Cells were cultured in 150 cm2 flasks as described in Cell types and culture conditions. Three flasks per gradient were used. After the indicated treatments, cultures were put on ice and washed twice with ice-cold PBS. Cells were scraped in 1.2 ml of ice-cold isotonic buffer/flask (20 mM Hepes-KOH, pH 7.5, 0.25 M sucrose, 90 mM KO-acetate, 2 mM Mg-acetate, 0.5 mM Na-vanadate, 1 mM NaF, 10 mM pyrophosphate, 3 mM β-glycerophasphate, 1 mM pefabloc, 40 U/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin) and homogenized with a Dounce homogenizer. Nuclei and residual intact cells were pelleted by centrifugation at 1,400 g for 5 min at 4°C. The supernatants were separated in three equal aliquots and mixed with iodixanol (OptiPrep; Axis-Shield) and homogenization buffer to generate 30, 20, and 10% iodixanol solutions. They were then loaded into 11.2-ml OptiSeal tubes (Beckman Coulter) and ultracentrifuged at 353,000 g for 3 h in a rotor (VT 65.1; Beckman Coulter). 600-μl fractions were collected from the top of the gradient, and protein concentration (bicinchoninic acid reagent; Pierce Chemical Co.) and density (OD at 244 nm as indicated by the OptiPrep manufacturer) were determined. The fractions were then boiled in the presence of reducing laemmli sample buffer. Samples of each fraction containing the same amount of protein for the different cell types to be compared and representative of the total protein content of each fraction were analyzed by SDS-PAGE followed by Western blotting.
For total cell extracts, cells were washed twice in PBS, extracted in 2× boiling laemmli sample buffer (200 μl for a 35-mm petri dish) containing 100 mM DTT, scraped, and boiled for a further 10 min. Parallel samples were extracted without DTT, and protein concentration was determined by bicinchoninic acid analysis. A total of 15 μg of protein was loaded in each lane and separated by SDS-PAGE, transferred onto nitrocellulose, and immunoblotted with the indicated antibodies.
For detection, HRP-conjugated horse anti–mouse, goat anti–rabbit (Cell Signaling), and rabbit anti–goat (DakoCytomation) secondary antibodies and ECL chemiluminescence reagent (GE Healthcare) were used. Films were scanned and bands were quantified using ImageJ set on the uncalibrated OD function. Adobe Photoshop 7.0, Excel X for MacIntosh, and Adobe Illustrator 11 for the PC were used to produce the figures presented.
Online supplemental material
Fig. S1 shows that PV-1 colocalizes with caveolin in VEC-null and -positive cells. Fig. S2 shows that cell confluence modulates VEGF-induced PY1175–VEGFR-2–positive compartments in HUVECs. Fig. S3 shows that clathrin siRNA inhibits the formation of clathrin-positive vesicular compartments. Fig. S4 shows that the cytoplasmic domain of VEC is required to modulate VEGFR-2 internalization from the plasma membrane. Fig. S5 shows that the antibody Avas12α1 does not modify either the basal or VEGF-stimulated tyrosine phosphorylation of VEGFR-2 and does not induce VEGFR-2 internalization. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200602080/DC1.
Supplementary Material
Acknowledgments
We are most grateful to Kendra Swirsding for editing the manuscript.
This work was supported by the Associazione Italiana per la Ricerca sul Cancro, the European Community (grants QLRT-2001-02059, Integrated Project Contract LSHG-CT-2004-503573, NoE MAIN 502935, and NoE EVGN 503254), Italian Ministry of Health, Ministero dell'Università e della Ricerca/Fondo degli Investimenti della Ricerca di Base (grants RBNE01MWA_009 and RBNE01F8LT_007), and the Cariplo Foundation. C. Tacchetti received a grant from the Telethon Foundation (GTF03001).
Abbreviations used in this paper: EEA-1, early endosomal antigen-1; DEP-1, density-enhanced phosphatase-1; GSH, glutathione; HUVEC, human umbilical vein endothelial cell; PY, phosphotyrosine; VEC, vascular endothelial cadherin; VEGFR, VEGF receptor.
References
- Akhtar, N., and N.A. Hotchin. 2001. RAC1 regulates adherens junctions through endocytosis of E-cadherin. Mol. Biol. Cell. 12:847–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo, K., T. Tammela, and T.V. Petrova. 2005. Lymphangiogenesis in development and human disease. Nature. 438:946–953. [DOI] [PubMed] [Google Scholar]
- Andriopoulou, P., P. Navarro, A. Zanetti, M.G. Lampugnani, and E. Dejana. 1999. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler. Thromb. Vasc. Biol. 19:2286– 2297. [DOI] [PubMed] [Google Scholar]
- Bauer, P.M., J. Yu, Y. Chen, R. Hickey, P.N. Bernatchez, R. Looft-Wilson, Y. Huang, F. Giordano, R.V. Stan, and W.C. Sessa. 2005. Endothelial-specific expression of caveolin-1 impairs microvascular permeability and angiogenesis. Proc. Natl. Acad. Sci. USA. 102:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister, U., R. Funke, K. Ebnet, H. Vorschmitt, S. Koch, and D. Vestweber. 2005. Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J. 24:1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya, R., N. Kang-Decker, D.A. Hughes, P. Mukherjee, V. Shah, M.A. McNiven, and D. Mukhopadhyay. 2005. Regulatory role of dynamin-2 in VEGFR-2/KDR-mediated endothelial signaling. FASEB J. 19:1692–1694. [DOI] [PubMed] [Google Scholar]
- Bryant, D.M., and J.L. Stow. 2004. The ins and outs of E-cadherin trafficking. Trends Cell Biol. 14:427–434. [DOI] [PubMed] [Google Scholar]
- Bryant, D.M., and J.L. Stow. 2005. Nuclear translocation of cell-surface receptors: lessons from fibroblast growth factor. Traffic. 6:947–954. [DOI] [PubMed] [Google Scholar]
- Bryant, D.M., F.G. Wylie, and J.L. Stow. 2005. Regulation of endocytosis, nuclear translocation, and signaling of fibroblast growth factor receptor 1 by E-cadherin. Mol. Biol. Cell. 16:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet, P. 2005. Angiogenesis in life, disease and medicine. Nature. 438:932–936. [DOI] [PubMed] [Google Scholar]
- Carmeliet, P., M.G. Lampugnani, L. Moons, F. Breviario, V. Compernolle, F. Bono, G. Balconi, R. Spagnuolo, B. Oostuyse, M. Dewerchin, et al. 1999. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 98:147–157. [DOI] [PubMed] [Google Scholar]
- Cavallaro, U., and G. Christofori. 2004. Cell adhesion and signaling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 4:118–132. [DOI] [PubMed] [Google Scholar]
- Confalonieri, S., A.E. Salcini, C. Puri, C. Tacchetti, and P.P. Di Fiore. 2000. Tyrosine phosphorylation of Eps15 is required for ligand-regulated, but not constitutive, endocytosis. J. Cell Biol. 150:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada, M., F. Liao, M. Lindgren, M.G. Lampugnani, F. Breviario, R. Frank, W.A. Muller, D.J. Hicklin, P. Bohlen, and E. Dejana. 2001. Monoclonal antibodies directed to different regions of vascular endothelial cadherin extracellular domain affect adhesion and clustering of the protein and modulate endothelial permeability. Blood. 97:1679–1684. [DOI] [PubMed] [Google Scholar]
- Dejana, E. 2004. Endothelial cell-cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5:261–270. [DOI] [PubMed] [Google Scholar]
- Di Fiore, P.P., and P. De Camilli. 2001. Endocytosis and signaling. an inseparable partnership. Cell. 106:1–4. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo, G.M., P.C. Baass, W.J. Ou, B.I. Posner, and J.J. Bergeron. 1994. Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 13:4269–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guglielmo, G.M., C. Le Roy, A.F. Goodfellow, and J.L. Wrana. 2003. Distinct endocytic pathways regulate TGF-beta receptor signaling and turnover. Nat. Cell Biol. 5:410–421. [DOI] [PubMed] [Google Scholar]
- Duval, M., S. Bedard-Goulet, C. Delisle, and J.P. Gratton. 2003. Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J. Biol. Chem. 278:20091–20097. [DOI] [PubMed] [Google Scholar]
- Ehrlich, M., A. Shmuely, and Y.I. Henis. 2001. A single internalization signal from the di-leucine family is critical for constitutive endocytosis of the type II TGF-beta receptor. J. Cell Sci. 114:1777–1786. [DOI] [PubMed] [Google Scholar]
- Fabbri, M., L. Fumagalli, G. Bossi, E. Bianchi, J.R. Bender, and R. Pardi. 1999. A tyrosine-based sorting signal in the beta2 integrin cytoplasmic domain mediates its recycling to the plasma membrane and is required for ligand-supported migration. EMBO J. 18:4915–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara, N., and R.S. Kerbel. 2005. Angiogenesis as a therapeutic target. Nature. 438:967–974. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., E. Wong, and B.M. Gumbiner. 2001. E-cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J. Cell Biol. 153:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B.M. 2005. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6:622–634. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E., and R.G. Anderson. 1989. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger, L.J., K. Ward, B. Duffield, J. Zachwieja, and B. Jallal. 2002. The transmembrane receptor protein tyrosine phosphatase DEP1 interacts with p120(ctn). Oncogene. 21:7067–7076. [DOI] [PubMed] [Google Scholar]
- Ikeda, S., M. Ushio-Fukai, L. Zuo, T. Tojo, S. Dikalov, N.A. Patrushev, and R.W. Alexander. 2005. Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 96:467–475. [DOI] [PubMed] [Google Scholar]
- Ivanov, A.I., A. Nusrat, and C.A. Parkos. 2004. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol. Biol. Cell. 15:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi, G., T. Sakisaka, T. Baba, S. Tanaka, K. Morimoto, and Y. Takai. 2004. Endocytosis of E-cadherin regulated by Rac and Cdc42 small G proteins through IQGAP1 and actin filaments. J. Cell Biol. 166:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque, L., I. Royal, D.S. Surprenant, C. Patterson, D. Gingras, and R. Beliveau. 2003. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol. Biol. Cell. 14:334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand, D., M. Curto, I. Saotome, M. Giovannini, and A.I. McClatchey. 2003. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 17:1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeng, N., Y. Wallez, C. Rampon, F. Cand, G. Christe, D. Gulino-Debrac, I. Vilgrain, and P. Huber. 2005. Vascular endothelial-cadherin tyrosine phosphorylation in angiogenic and quiescent adult tissues. Circ. Res. 96:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani, M.G., A. Zanetti, F. Breviario, G. Balconi, F. Orsenigo, M. Corada, R. Spagnuolo, M. Betson, V. Braga, and E. Dejana. 2002. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol. Biol. Cell. 13:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampugnani, G.M., A. Zanetti, M. Corada, T. Takahashi, G. Balconi, F. Breviario, F. Orsenigo, A. Cattelino, R. Kemler, T.O. Daniel, and E. Dejana. 2003. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, β-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, T.L., A.S. Yap, and J.L. Stow. 1999. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J. Cell Biol. 146:219–232. [PMC free article] [PubMed] [Google Scholar]
- Le Roy, C., and J.L. Wrana. 2005. Clathrin- and non-clathrin-mediated endocytic regulation of cell signaling. Nat. Rev. Mol. Cell Biol. 6:112–126. [DOI] [PubMed] [Google Scholar]
- Matsumoto, T., S. Bohman, J. Dixelius, T. Berge, A. Dimberg, P. Magnusson, L. Wang, C. Wikner, J.H. Qi, C. Wernstedt, et al. 2005. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 24:2342–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska, M., L. Pelkmans, and M. Zerial. 2004. Not just a sink: endosomes in control of signal transduction. Curr. Opin. Cell Biol. 16:400–406. [DOI] [PubMed] [Google Scholar]
- Mitola, S., B. Brenchio, M. Piccinini, L. Tertoolen, L. Zammataro, G. Breier, M.T. Rinaudo, J. den Hertog, M. Arese, and F. Bussolino. 2006. Type I collagen limits VEGFR-2 signaling by a SHP2 protein-tyrosine phosphatase-dependent mechanism 1. Circ. Res. 98:45–54. [DOI] [PubMed] [Google Scholar]
- Mueller, S., E. Cadenas, and A.H. Schonthal. 2000. p21WAF1 regulates anchorage- independent growth of HCT116 colon carcinoma cells via E-cadherin expression. Cancer Res. 60:156–163. [PubMed] [Google Scholar]
- Palacios, F., J.K. Schweitzer, R.L. Boshans, and C. D'Souza-Schorey. 2002. ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat. Cell Biol. 4:929–936. [DOI] [PubMed] [Google Scholar]
- Palka, H.L., M. Park, and N.K. Tonks. 2003. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J. Biol. Chem. 278:5728–5735. [DOI] [PubMed] [Google Scholar]
- Paterson, A.D., R.G. Parton, C. Ferguson, J.L. Stow, and A.S. Yap. 2003. Characterization of E-cadherin endocytosis in isolated MCF-7 and Chinese hamster ovary cells: the initial fate of unbound E-cadherin. J. Biol. Chem. 278:21050–21057. [DOI] [PubMed] [Google Scholar]
- Pennock, S., and Z. Wang. 2003. Stimulation of cell proliferation by endosomal epidermal growth factor receptor as revealed through two distinct phases of signaling. Mol. Cell. Biol. 23:5803–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzer, J.E., P. Oh, E. Pinney, and J. Allard. 1994. Filipin-sensitive caveolae- mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J. Cell Biol. 127:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay-Salit, A., M. Shushy, E. Wolfovitz, H. Yahav, F. Breviario, E. Dejana, and N. Resnick. 2002. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. USA. 99:9462–9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund, S., T. Woelk, C. Puri, E. Maspero, C. Tacchetti, P. Transidico, P.P. Di Fiore, and S. Polo. 2005. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA. 102:2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A.J., R.D. Meyer, H. Band, and N. Rahimi. 2005. The carboxyl terminus of VEGFR-2 is required for PKC-mediated down-regulation. Mol. Biol. Cell. 16:2106–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot, J.W., H.J. Geuze, S. Gigengack, G.E. Lienhard, and D.E. James. 1991. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldi, R., S. Mitola, M. Strasly, P. Defilippi, G. Tarone, and F. Bussolino. 1999. Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 18:882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin, A., and M. Von Zastrow. 2002. Signal transduction and endocytosis: close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 3:600–614. [DOI] [PubMed] [Google Scholar]
- St Croix, B., C. Sheehan, J.W. Rak, V.A. Florenes, J.M. Slingerland, and R.S. Kerbel. 1998. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell Biol. 142:557–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan, R.V. 2005. Structure of caveolae. Biochim. Biophys. Acta. 1746:334–348. [DOI] [PubMed] [Google Scholar]
- Stan, R.V., E. Tkachenko, and I.R. Niesman. 2004. PV1 is a key structural component for the formation of the stomatal and fenestral diaphragms. Mol. Biol. Cell. 15:3615–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger, A., A. Eger, J. Wolf, H. Beug, and R. Foisner. 2001. E-cadherin regulates cell growth by modulating proliferation-dependent β-catenin transcriptional activity. J. Cell Biol. 154:1185–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama, K., I. Shapiro, M. Guttman, and R.B. Hazan. 2002. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2:301–314. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., and M. Shibuya. 1997. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblsts. Oncogene. 14:2079–2089. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., H. Ueno, and M. Shibuya. 1999. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 18:2221–2230. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., S. Yamaguchi, K. Chida, and M. Shibuya. 2001. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20:2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., S.D. Pennock, X. Chen, A. Kazlauskas, and Z. Wang. 2004. Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J. Biol. Chem. 279:8038–8046. [DOI] [PubMed] [Google Scholar]
- Weis, S., S. Shintani, A. Weber, R. Kirchmair, M. Wood, A. Cravens, H. McSharry, A. Iwakura, Y.S. Yoon, N. Himes, et al. 2004. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest. 113:885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, K., J. Garner, K.M. Buckley, P.A. Vincent, C.M. Chiasson, E. Dejana, V. Faundez, and A.P. Kowalczyk. 2005. p120-Catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell. 16:5141–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman, C., K.K. Grindstaff, J.R. Wright, and W.J. Nelson. 2001. Sec6/8 complexes on trans-Golgi network and plasma membrane regulate late stages of exocytosis in mammalian cells. J. Cell Biol. 155:593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}