

Abstract
Ras activates Raf, leading to the extracellular-regulated kinase (ERK)–mitogen-activated protein kinase pathway, which is involved in a variety of cellular, physiological, and pathological responses. Thus, regulators of this Ras–Raf interaction play crucial roles in these responses. In this study, we report a novel regulator of the Ras–Raf interaction named DA-Raf1. DA-Raf1 is a splicing isoform of A-Raf with a wider tissue distribution than A-Raf. It contains the Ras-binding domain but lacks the kinase domain, which is responsible for activation of the ERK pathway. As inferred from its structure, DA-Raf1 bound to activated Ras as well as M-Ras and interfered with the ERK pathway. The Ras–ERK pathway is essential for the negative regulation of myogenic differentiation induced by growth factors. DA-Raf1 served as a positive regulator of myogenic differentiation by inducing cell cycle arrest, the expression of myogenin and other muscle-specific proteins, and myotube formation. These results imply that DA-Raf1 is the first identified competent, intrinsic, dominant-negative antagonist of the Ras–ERK pathway.
Introduction
The small GTPase Ras (H-, K-, and N-Ras) activated by extracellular signals exerts its cellular or physiological functions through binding to a variety of target proteins (Campbell et al., 1998; Mitin et al., 2005). Raf is one of the best characterized target proteins of Ras. The Raf family consists of three members: Raf-1 (C-Raf), A-Raf, and B-Raf. They share the three conserved regions CR1, 2, and 3. CR1 contains the Ras-binding and cysteine-rich domains (CRDs), both of which participate in binding to active Ras and membrane recruitment, whereas CR3 encompasses the Ser/Thr protein kinase domain (Hagemann and Rapp, 1999; Mercer and Pritchard, 2003; Wellbrock et al., 2004). The binding of active Ras to Raf triggers activation of the extracellular-regulated kinase (ERK)–MAPK pathway (Raf–MAPK and ERK kinase [MEK]–ERK), which participates in a variety of cellular, physiological, and pathological responses (Schaeffer and Weber, 1999; Chang and Karin, 2001; Pearson et al., 2001). For instance, the growth factor–induced activation of Ras leads to ERK pathway–mediated cell proliferation and morphogenesis during animal development. Oncogenic Ras causes constitutive activation of the ERK pathway, resulting in cellular transformation and tumorigenesis.
The Ras–ERK pathway also regulates differentiation in a variety of cell types (Schramek, 2002). This pathway is essential for NGF-induced neuronal differentiation in PC12 cells (Marshall, 1995). In contrast, this pathway, which is activated by serum mitogens or growth factors as well as by oncogenic Ras, negatively regulates skeletal muscle cell differentiation (Olson et al., 1987; Bennett and Tonks, 1997). Skeletal muscle cell differentiation necessitates irreversible cell cycle arrest in G0 phase, the expression of a battery of muscle-specific genes, and cell fusion to form multinucleated myotubes (Endo and Nadal-Ginard, 1987; Lassar et al., 1994). The tumor suppressor protein Rb is indispensable for cell cycle arrest (Endo and Goto, 1992; Schneider et al., 1994; Novitch et al., 1996), whereas MyoD family and MEF2 family transcription factors bring about the myogenic program by inducing muscle-specific gene expression (Black and Olson, 1998; Tapscott, 2005). The negative regulation of skeletal muscle cell differentiation by the Ras–ERK pathway is ascribable to interference with the expression or functions of MyoD and MEF2 families (Lassar et al., 1989; Winter and Arnold, 2000; Perry et al., 2001; Tortorella et al., 2001) and with the activation of Rb (Mittnacht et al., 1997; Peeper et al., 1997; Lee et al., 1999). Thus, it is reasonable that the deprivation of serum mitogens or growth factors leads to inactivation of the Ras–ERK pathway and, consequently, results in skeletal muscle cell differentiation. However, detailed mechanisms of this inactivation of the Ras–ERK pathway have not been fully elucidated.
The activity of the Ras–ERK pathway is tightly controlled through the coordinated action of both positive and negative regulators that function at various levels of the pathway and at different time points of the responses. Recently, several suppressor proteins of the Ras–ERK pathway have been identified. These include Sprouty (Spry)/Spred proteins (Wakioka et al., 2001; Kim and Bar-Sagi, 2004; for review see Mason et al., 2006), RKIP (Raf kinase inhibitor protein; Yeung et al., 1999; Odabaei et al., 2004), and IMP (Matheny et al., 2004; for review see Ory and Morrison, 2004). Both Spry and RKIP bind to Raf to inhibit the ERK pathway by preventing the binding of MEK. Spry also intercepts growth factor receptor–Ras–ERK signaling by interacting with distinct proteins, depending on the signaling. In contrast, IMP uncouples signal transduction from Raf to MEK by inactivating the scaffold protein KSR (kinase suppressor of Ras). However, it has been poorly understood whether regulatory proteins that directly bind to Ras to prevent the ERK pathway are present.
M-Ras, a member of the Ras family small GTPases, induces the reorganization of actin cytoskeleton, cellular transformation, inhibition of myogenic differentiation, and neuronal differentiation in PC12 cells (Matsumoto et al., 1997; Quilliam et al., 1999; Kimmelman et al., 2002; Sun et al., 2006). To elucidate the molecular mechanisms of these cellular functions of M-Ras, we have identified proteins that bind to a constitutively activated mutant of M-Ras. One of them was a novel splicing isoform of A-Raf, designated as DA-Raf1, which contains the Ras-binding domain (RBD) but lacks the kinase domain. DA-Raf1 bound to both active Ras and M-Ras and interfered with the ERK pathway. Its expression level was prominently elevated, and it positively regulated myogenic differentiation by inducing cell cycle arrest, muscle-specific protein expression, and myotube formation. Thus, DA-Raf1 is the first identified intrinsic dominant-negative antagonist of the Ras–ERK pathway.
Results
DA-Raf1 binds to active Ras and M-Ras
To elucidate the molecular mechanisms of the cellular functions of M-Ras, we have screened a mouse brain cDNA library by a yeast two-hybrid system and cloned cDNAs of proteins that bind to a constitutively activated mutant of M-Ras(G22V) (Gly22 is substituted by Val). These proteins include B-Raf, RalGDS, Rgl2, AF-6, and a C terminus–truncated isoform of A-Raf consisting of N-terminal 186 amino acids (Fig. 1 A). We designate this truncated protein DA-Raf1 (deleted A-Raf).
Figure 1.

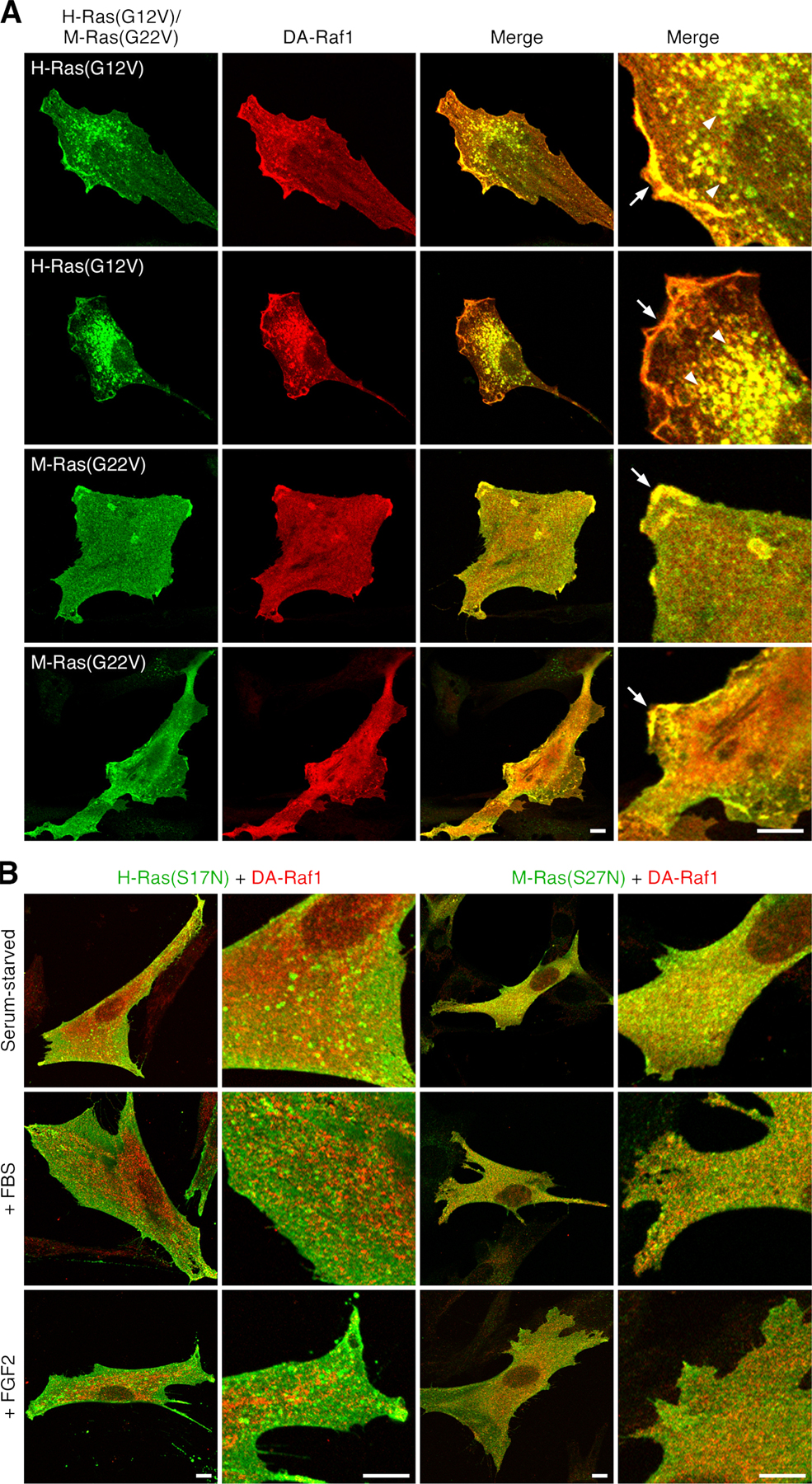
Binding of DA-Raf1 to M-Ras and Ras proteins. (A) Yeast two-hybrid binding assay detecting the binding of DA-Raf1 and other proteins to M-Ras and H-Ras. The binding was detected by β-galactosidase colony-lift filter assay. wt, wild-type. (B) Pull-down assay with GST–DA-Raf1. Binding of myc-tagged H-Ras, M-Ras, and v–K-Ras in COS-1 cell lysates to GST–DA-Raf1 was detected by immunoblotting with the anti-myc mAb myc1-9E10. (C) Coimmunoprecipitation assay. Myc-tagged H-Ras and M-Ras in COS-1 cell lysates were immunoprecipitated with the anti-myc mAb, and coprecipitated HA–DA-Raf1 was detected by immunoblotting with the anti-HA tag pAb. (D) Colocalization of DA-Raf1 with activated H-Ras and M-Ras to the membrane ruffles and vesicles. C2C12 myoblasts cotransfected with myc–Hras or myc–Mras and HA–DAraf1 were serum starved and stimulated with 10% FBS or 50 ng/ml FGF2 for 30 min. The localization of myc–H-Ras or myc–M-Ras and HA–DA-Raf1 was detected by staining with the anti-myc mAb (green) and the anti-HA tag pAb (red), respectively. Their merged images are shown. Arrows point to the membrane ruffles where DA-Raf1 is colocalized with activated H-Ras or M-Ras. Arrowheads indicate the vesicles where DA-Raf1 is colocalized with activated H-Ras. Bars, 10 μm.
DA-Raf1 bound not only to M-Ras(G22V) but also to H-Ras (both wild type and a constitutively active G12V mutant), whereas it did not bind to their dominant-negative mutants M-Ras(S27N) and H-Ras(S17N), as determined by a two-hybrid binding assay (Fig. 1 A). A pull-down assay also showed that GST-tagged DA-Raf1 bound to the constitutively active mutants of M-Ras and H-Ras as well as v–K-Ras but not to their dominant-negative mutants (Fig. 1 B). Moreover, immunoprecipitation of myc-tagged H-Ras(G12V) or M-Ras(G22V) exogenously expressed in COS-1 cells with the anti-myc mAb myc1-9E10 resulted in the coprecipitation of HA-tagged DA-Raf1 (Fig. 1 C), indicating their interaction in vivo. Immunoprecipitation of H-Ras in differentiating C2C12 skeletal muscle cell lysates and in mouse brain extracts brought about the coprecipitation of endogenous DA-Raf1 (see Fig. 5 G), also implying the binding of endogenous Ras and DA-Raf1.
Figure 5.

Prominent induction of DA-Raf1 and reduction in B-Raf–MEK–ERK activities during myogenic differentiation of C2C12 cells. (A) Induction of DAraf1/2 and Myog mRNAs during C2C12 cell differentiation detected by RT-PCR. (B) Relative amounts of the mRNAs. The amounts of DAraf1 and Myog mRNAs are normalized with that of Gapdh mRNA. The amounts of each mRNA are indicated as multiples of the amount at 24 h. The values are means ± SD (error bars) of three independent experiments. *, P < 0.05; **, P < 0.01 by t test compared with the corresponding mRNA at 24 h. (C) Induction of DA-Raf1 and muscle- specific proteins (myogenin, MyHC, and TnT) during C2C12 cell differentiation detected by immunoblotting. β-Tubulin is shown as a standard. (D) Relative amounts of the proteins. The amounts of each protein are indicated as multiples of the amount at 24 h. The values are means ± SD of three independent experiments. (E) Decrease in MEK and ERK phosphorylation and increase in Akt phosphorylation during differentiation. MEK1/2, phospho-MEK1/2, ERK1/2, phospho-ERK1/2, Akt, and phospho-Akt were detected by immunoblotting. (F) Reduction in MEK phosphorylation by B-Raf. B-Raf and Raf1 were immunoprecipitated with anti–B-Raf and anti-Raf1, respectively. Their in vitro kinase assay was conducted by using MEK1 as a substrate. (G) Binding of DA-Raf1 to H-Ras in differentiated C2C12 cells. H-Ras was immunoprecipitated with anti–H-Ras from C2C12 cells cultured in the differentiation medium (DM) and from mouse brain. Precipitated H-Ras and coprecipitated DA-Raf1 and B-Raf as well as DA-Raf1 and B-Raf in brain extract were detected by immunoblotting.
Immunofluorescent staining in combination with myc epitope tagging located both H-Ras(G12V) and M-Ras(G22V) to membrane ruffles and H-Ras(G12V) to vesicles such as macropinosomes as well (Bar-Sagi and Feramisco, 1986) as in C2C12 myoblasts (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200703195/DC1). HA–DA-Raf1 was colocalized with the constitutively active H-Ras and M-Ras to these membrane-associated structures (Fig. S1 A). On the other hand, in serum-starved C2C12 cells expressing wild-type myc–H-Ras or M-Ras, HA–DA-Raf1 was diffusely distributed throughout the cytoplasm and coincided with neither wild-type H-Ras nor M-Ras (Fig. 1 D). When the cells were stimulated with FBS or FGF2, however, HA–DA-Raf1 was colocalized with the activated H-Ras and M-Ras to the plasma membrane or vesicle membrane–associated structures (Fig. 1 D). In contrast, if myc–H-Ras(S17N) or M-Ras(S27N) was expressed, the membrane ruffles or vesicles were not formed even by stimulation with FBS or FGF2 (Fig. S1 B). HA–DA-Raf1 was diffusely distributed throughout the cytoplasm and coexisted with neither H-Ras(S17N) nor M-Ras(S27N) in these stimulated cells (Fig. S1 B). These results support the interaction of DA-Raf1 with active Ras and M-Ras in vivo.
DA-Raf1 and 2 are generated by alternative splicing of Araf pre-mRNA
All known Raf family proteins (Raf-1, A-Raf, and B-Raf) have three conserved regions: CR1, 2, and 3 (Fig. 2 A; Hagemann and Rapp, 1999; Mercer and Pritchard, 2003; Wellbrock et al., 2004). CR1 contains the RBD and CRD, both of which participate in binding to active Ras and membrane recruitment, whereas CR3 represents the kinase domain. The amino acid sequence of DA-Raf1 deduced from its nucleotide sequence indicated that DA-Raf1 exactly corresponds to the N-terminal 186 amino acids of A-Raf, which contains CR1 but lacks CR2 and 3 (Fig. 2 A). Comparison of the nucleotide sequence of mouse DAraf1 cDNA with that of the genomic sequence of the mouse Araf gene revealed that DAraf1 mRNA is generated by the alternative splicing of Araf pre-mRNA (Fig. 2 B). DAraf1 cDNA retains intron 6–7 linked to exon 6 and, consequently, gives rise to the termination codon TAG starting from the second nucleotide of intron 6–7 (Fig. 2, B and C). Poly(A) tail is added 12 bases downstream of the putative poly(A) addition signal AATAAA.
Figure 2.

Domain structure of DA-Raf1/2 and splicing patterns of Araf/DAraf pre-mRNA. (A) Functional domains of Raf proteins and DA-Raf1/2. The numbers indicate amino acid residues of mouse proteins. (B) Alternative splicing of Araf/DAraf pre-mRNA generating Araf and DAraf1/2 mRNAs. Pink boxes, exons; purple boxes, introns; yellow boxes, introns retained in DAraf1/2 mRNAs but not in Araf mRNA; asterisks, termination codons for DA-Raf2 (in intron 5–6) and DA-Raf1 (in intron 6–7). (C) Nucleotide sequence of exons (red) and introns (blue) retained in mouse DAraf1/2 mRNAs and the corresponding amino acid sequence. Asterisks in intron 5–6 and intron 6–7 represent termination codons of DAraf2 and DAraf1 mRNAs, respectively. Red and green underlines indicate putative poly(A) addition signals of DAraf1 and DAraf2 mRNAs, respectively.
To confirm that DAraf1 is actually expressed in vivo, we conducted RT-PCR and Northern blotting. Quantitative RT-PCR using the sense primer (nt 62–79), which is common to both DAraf1 and Araf, and the antisense primer (nt 620–639), which is specific for DAraf1 (Fig. 3 A), amplified major 578-bp and minor 695-bp DNA bands from mRNAs of various mouse tissues (Fig. 3 B). Nucleotide sequences of these DNAs indicated that the former band represented expected DAraf1, whereas the latter band corresponded to the DNA containing intron 5–6 (117 nt) between exons 5 and 6. The retention of intron 5–6 generates the termination codon TGA starting from the second nucleotide of intron 5–6 (Fig. 2, B and C). We refer to this splicing isoform as DA-Raf2. DAraf2 encodes a protein consisting of the N-terminal 153 amino acids of A-Raf, which also includes RBD and CRD (Fig. 2 A). Examination of the genomic sequences of rat and human Araf genes revealed that both of the genes have exon–intron junctional arrangements and sequences very similar to those of the mouse gene. Consequently, mRNAs encoding DA-Raf1 and 2 seem to be generated by the alternative splicing of rat and human Araf pre-mRNAs as well (Fig. S2, A and B; available at http://www.jcb.org/cgi/content/full/jcb.200703195/DC1). Indeed, we have cloned rat and human DAraf1 cDNAs by RT-PCR from rat PC12 cells and human HeLa cells, respectively.
Figure 3.

Expression of DAraf1/2 mRNAs and the proteins in mouse tissues. (A) DAraf1/2 mRNAs together with primers and probes used for RT-PCR and Northern blotting, respectively. Pink boxes, coding regions including termination codons; purple boxes, untranslated regions; Ex, exon; Int, intron. Asterisks represent termination codons. The numbers indicate nucleotide numbers starting from the 5′ ends of the cDNAs. (B) Quantitative RT-PCR detecting DAraf1/2, Araf, Braf, and Raf1 mRNAs. Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) mRNA is shown as a standard. (C) Northern blotting detecting DAraf1/2 and Araf mRNAs with probe 1 and DAraf1 mRNA with probe 2. Ethidium bromide (EtBr) staining of a gel shows 28S and 18S rRNAs as a standard. (D) Immunoblotting detecting DA-Raf1/2 and A-Raf proteins with the anti–DA-Raf pAb. The A-Raf bands were verified by immunoblotting with the anti–A-Raf pAb. Each lane contains 50 μg of proteins. The asterisks indicate unidentified extra protein bands reacted with the anti–DA-Raf pAb.
DA-Raf1 is widely expressed
Quantitative RT-PCR showed that both DAraf1 and DAraf2 were ubiquitously expressed in a variety of tissues examined and that they were prominently expressed in the brain and heart, among others (Fig. 3 B). In contrast, Araf had more limited tissue distribution. It was most highly expressed in the liver among the tissues examined, as has been previously reported (Huleihel et al., 1986), whereas it was scarcely expressed in skeletal muscle (Fig. 3 B). Braf was also highly expressed in the brain, whereas Raf1 was widely expressed except in the brain. Next, we conducted Northern blotting with probe 1, which can hybridize to DAraf1, DAraf2, and Araf mRNAs in common, and conducted blotting with probe 2, which specifically recognizes DAraf1 mRNA (Fig. 3, A and C). DAraf1 mRNA (1.65 kb) and DAraf2 mRNA (1.35 kb) were abundantly present in the brain and heart and moderately in other tissues, which is consistent with the results of RT-PCR analysis. On the other hand, Araf mRNA (2.4 kb) was expressed in the liver at a moderate level and had a more restricted tissue distribution (Fig. 3 C). Immunoblotting analysis with an anti–DA-Raf pAb showed that DA-Raf1 protein was abundantly present in the brain and moderately in the heart, skeletal muscle, uterus, and spleen (Fig. 3 D). DA-Raf2 was also detected at least in the brain. In contrast, A-Raf was predominantly present in the liver and spleen, as corroborated by immunoblotting with an anti–A-Raf pAb (Fig. 3 D). Collectively, DAraf1 mRNA and protein are actually present in mouse tissues with a wider tissue distribution than A-Raf.
DA-Raf1 antagonizes the Ras–ERK pathway
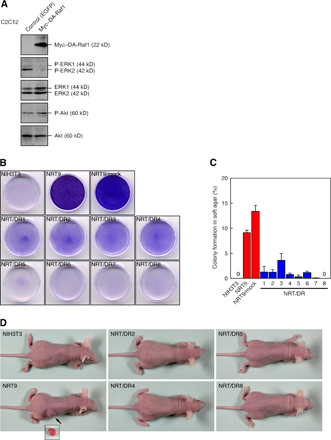
The structure of the DA-Raf1 protein, which contains both RBD and CRD but lacks the kinase domain, suggests that it serves as an intrinsic dominant-negative antagonist of the Ras–ERK pathway and possibly as an antagonist of other Ras-activated signaling pathways such as Ras–phosphatidylinositol 3-kinase (PI3K)–Akt signaling. To assess this possibility, we transfected NRT9 cells (the mouse NIH3T3 fibroblasts stably transformed with the oncogenic v-Kras gene) with myc-tagged DAraf1 cDNA and selected eight stable transfectants (NRT/DR1–8). The expression of exogenous myc–DA-Raf1 in these cell lines was detected by immunoblotting with the anti-myc mAb (Fig. 4 A). The levels of ERK activation and Akt activation were estimated by immunoblotting with an antiphospho-ERK1/2 mAb and an antiphospho-Akt pAb, respectively. NRT9 and mock-transfected NRT9 cells had much higher activating phosphorylation levels of ERK1/2 than did normal NIH3T3 cells, whereas all of the NRT/DR cell lines showed ERK1/2 phosphorylation levels comparable with that in normal NIH3T3 cells (Fig. 4 A). In contrast, activating phosphorylation levels of Akt in NRT9 and mock-transfected NRT9 cells were almost equivalent to that in normal NIH3T3 cells. This is consistent with the results that K-Ras efficiently activates Raf but not PI3K (Yan et al., 1998; Voice et al., 1999). The Akt phosphorylation levels were substantially unchanged in any of the NRT/DR cell lines (Fig. 4 A). Transiently expressed myc–DA-Raf1 in C2C12 myoblasts also reduced the ERK1/2 phosphorylation level but not the Akt phosphorylation level (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200703195/DC1). Consequently, the exogenously expressed DA-Raf1 is likely to antagonize the Ras–ERK pathway but not Ras–PI3K–Akt signaling.
Figure 4.

Suppression of v–K-Ras–induced ERK activation, morphological alteration, and facilitated proliferation rate by DA-Raf1. (A) Suppression of ERK phosphorylation without affecting Akt phosphorylation in NRT/DR1–8 cells. Proteins were detected by immunoblotting. NRT9/mock, mock-transfected NRT9 cells; P-ERK1/2, ERK1/2 with activating phosphorylation on Thr202 and Tyr204, P-Akt, Akt with activating phosphorylation on Ser473. (B) Recovery of actin stress fibers and extended flattened cell shape in NRT/DR cells. Stress fibers were detected by staining with rhodamine-phalloidin. (C) Reduction of the proliferation rate in NRT/DR cells. Cells were cultured in DME containing 0.5% FBS for the indicated times. The values are means ± SD (error bars) of three independent experiments.
Activation of the Raf-mediated ERK pathway by oncogenic Ras induces transformation in mouse fibroblasts (Kolch et al., 1991; Cowley et al., 1994; Mansour et al., 1994). Because the activation of ERK was abrogated by DA-Raf1 expression, we examined whether the v–K-Ras–induced transformed phenotype in NRT9 cells was suppressed by DA-Raf1. Normal NIH3T3 cells contained well-developed actin stress fibers and showed an extended flattened cell shape. Although NRT9 cells exhibited transformed morphology (i.e., the loss of stress fibers and consequent round cell shape), all of the NRT/DR cell lines restored the normal flattened cell shape with stress fibers (Fig. 4 B). Next, we analyzed the cell proliferation rate under a serum starvation condition in 0.5% FBS by counting the cell number. NRT9 and mock-transfected NRT9 cells showed growth rates several fold higher than that of normal NIH3T3 cells, whereas all of the NRT/DR cell lines exhibited growth rates similar to or lower than that of NIH3T3 cells (Fig. 4 C).
When NRT9 and mock-transfected NRT9 cells were cultured for 7 d after reaching confluence in the growth medium, they formed innumerable foci, which merged with each other because of the loss of contact inhibition of transformed cells (Fig. S3 B). In contrast, all of the NRT/DR cell lines as well as normal NIH3T3 cells scarcely formed foci. We further analyzed the anchorage-independent growth potential of these cells in soft agar. Although ∼10–15% of NRT9 and mock-transfected NRT9 cells formed colonies, all of the NRT/DR cell lines formed colonies at very low efficiencies (Fig. S3 C). Next, we injected 1 × 106 cells subcutaneously into dorsal flanks of athymic nude mice. All mice (three out of three) injected with NRT9 or mock-transfected NRT9 cells formed tumors 6–7 mm in diameter within 4 wk after the injection (Fig. S3 D). However, no mice injected with normal NIH3T3 cells or any of the NRT/DR cell lines formed tumors even 8 wk after the injection (Fig. S3 D). These results indicate that the exogenously expressed DA-Raf1 can suppress all of the transformed phenotypes, including tumorigenicity induced by oncogenic Ras.
DA-Raf1 prominently induced during skeletal myocyte differentiation is responsible for reduction in MEK–ERK activity
The Ras–ERK pathway negatively regulates skeletal muscle cell differentiation by suppressing the expression or functions of the muscle-specific transcription factors MyoD and MEF2 (Lassar et al., 1989; Winter and Arnold, 2000; Perry et al., 2001; Tortorella et al., 2001). This suppression results in the aborted expression of myogenin, which induces muscle differentiation. Thus, to assess the involvement of DA-Raf1 in the regulation of skeletal muscle cell differentiation, we first analyzed the expression of DAraf1 during the differentiation of C2C12 skeletal muscle cells by quantitative RT-PCR and immunoblotting. Undifferentiated myoblasts terminally differentiate to form multinucleated myotubes by cell fusion by shift from the mitogen-rich growth medium containing 10% FBS to the mitogen-poor differentiation medium containing 5% horse serum. Well-differentiated myotubes are developed by 96 h under this culture condition. DAraf1 mRNA was present at a low level in myoblasts but was markedly induced about 10-fold during differentiation (Fig. 5, A and B). Myog mRNA was almost absent in myoblasts but highly induced during differentiation in accordance with the DAraf1 expression (Fig. 5, A and B). Immunoblotting showed that DA-Raf1 was almost absent in myoblasts but prominently induced during differentiation (Fig. 5, C and D). Similarly, myogenin and late muscle-specific proteins, myosin heavy chain (MyHC) and troponin T (TnT), were also induced (Fig. 5, C and D). Although the band derived from DAraf2 mRNA was detected by RT-PCR, DA-Raf2 protein was hardly detected by immunoblotting even in differentiated C2C12 cells, which is different from in the brain. The increased rate of DA-Raf1 protein during differentiation was much higher than that of DAraf1 mRNA. In contrast, the amounts of Raf-1, B-Raf, and A-Raf were almost constant during differentiation (Fig. 5, C and D). These results suggest the possibility that DA-Raf1 positively regulates skeletal muscle cell differentiation.
In proliferating myoblasts, the activating phosphorylation of MEK and ERK was easily detected by immunoblotting with an antiphospho-MEK1/2 pAb and the antiphospho-ERK1/2 mAb, respectively (Fig. 5 E). In contrast, phospho-Akt was marginally present in myoblasts. The phosphorylation levels of both MEK and ERK were reduced, whereas that of Akt was elevated during differentiation (Fig. 5 E). These results are consistent with previous studies (Rommel et al., 1999; Tortorella et al., 2001). Immunoprecipitated B-Raf from myoblasts efficiently phosphorylated MEK, but the kinase activity was markedly diminished during differentiation (Fig. 5 F). In contrast, immunoprecipitated Raf1 and A-Raf scarcely phosphorylated MEK regardless of the differentiation stage (Fig. 5 F and not depicted). These results are compatible with the notion that B-Raf is the main activator of MEK in most cell types (Mercer and Pritchard, 2003; Wellbrock et al., 2004). DA-Raf1 was not coimmunoprecipitated with H-Ras in myoblasts (Fig. 5 G). As differentiation proceeded, however, DA-Raf1 was coimmunoprecipitated with H-Ras at the level similar to that in the brain (Fig. 5 G). In contrast, B-Raf was coimmunoprecipitated with H-Ras in myoblasts, and the amounts of precipitated B-Raf decreased as differentiation proceeded (Fig. 5 G). Collectively, these results imply that the decrease in MEK–ERK activity during C2C12 cell differentiation is ascribed to the retardation of B-Raf activity and that the association of DA-Raf1 with Ras is likely to be responsible for the decline in Ras–B-Raf interaction and the consequent reduction in B-Raf–MEK–ERK activity during the differentiation.
Highly expressed DA-Raf1 induces skeletal myocyte differentiation
Because DA-Raf1 seems to negatively regulate the B-Raf– MEK–ERK pathway during C2C12 cell differentiation, it may be involved in the induction of differentiation. Skeletal muscle cell differentiation entails cell cycle arrest in G0 phase together with muscle-specific gene expression and myotube formation. To examine whether DA-Raf1 is implicated in the induction of cell cycle arrest, C2C12 cells were transfected with HA–DAraf1, and proliferating cells in S phase were detected by the incorporation of BrdU in the nuclei. DAraf1 expression markedly reduced the ratio of BrdU-incorporating cells (Fig. 6, A and B).
Figure 6.

Induction of the myogenic differentiation phenotype in C2C12 cells by highly expressed DA-Raf1. (A) Induction of the cell cycle arrest in C2C12 cells exogenously expressing DAraf1. C2C12 cells were transfected with HA–DAraf1 and incubated with BrdU for 2 h. The anti-HA staining detecting the expression of HA–DA-Raf1 (green) is merged with anti-BrdU staining (red). (B) Ratio of the BrdU-incorporating C2C12 cells in the analysis of A. (C) Facilitation of myogenin expression in C2C12 cells exogenously expressing DAraf1. The cells were cultured in the differentiation medium. Control EGFP fluorescence (green) or anti-HA staining detecting the expression of HA–DA-Raf1 (green) is merged with antimyogenin staining (red). (D) Ratio of the myogenin-expressing cells in the analysis of C. (E) Promotion of MyHC expression in C2C12 cells exogenously expressing DAraf1. Control EGFP fluorescence (green) or anti-HA staining detecting the expression of HA–DA-Raf1 (green) is merged with anti-MyHC staining (red). (F) Ratio of the MyHC-expressing cells in the analysis of E. (G) Facilitation of TnT expression in C2C12 cells exogenously expressing DAraf1. Control EGFP fluorescence (green) or anti-HA staining detecting the expression of HA–DA-Raf1 (green) is merged with anti-TnT staining (red). (H) Ratio of the TnT-expressing cells in the analysis of G. The values in the graphs are means ± SD (error bars) of three independent experiments. *, P < 0.05; **, P < 0.01 by t test compared with the control at each time point. Bars, 20 μm.
Next, we addressed whether DA-Raf1 participated in the induction of myogenin and late muscle-specific protein expression. The ratio of myogenin-expressing cells was facilitated 1.5–2-fold by the transfection of DAraf1 during 48–72 h in the differentiation medium (Fig. 6, C and D). The transfection of DAraf1 also elevated the ratio of MyHC-expressing cells (Fig. 6, E and F) and TnT-expressing cells (Fig. 6, G and H) two- to several fold during differentiation. The effects of DAraf1 transfection to induce myogenin and the late muscle-specific proteins were more obvious around 48–72 h after the transfection than at later time points probably because of the up-regulation of endogenous DA-Raf1 protein in control cells and breakdown of the transfected plasmids expressing DAraf1 in the process of time. Therefore, highly expressed DA-Raf1 can bring on the myogenic differentiation program by inducing the cell cycle arrest and expression of myogenin and late muscle-specific proteins.
Intrinsic DA-Raf1 induces skeletal myocyte differentiation by suppressing the ERK pathway
We further examined the role of endogenous DA-Raf1 by RNAi through expression of the two siRNAs, M1 and M2, which target the sequences in the 3′ untranslated regions of mouse DAraf1/2 mRNAs. Expression of these siRNAs markedly reduced the amount of endogenous DA-Raf1 protein during the differentiation of C2C12 cells (Fig. 7 A). In contrast, expression of the other two siRNAs, R1 and R2, targeting rat DAraf1/2 mRNA sequences, which are distinct from the mouse counterparts, did not affect the amount of DA-Raf1 protein in C2C12 cells (Fig. 7 A).
Figure 7.

Suppression of the myogenic differentiation phenotype in C2C12 cells by knockdown of endogenous DA-Raf. (A) Knockdown of endogenous DA-Raf by mouse-specific siRNAs but not by rat-specific siRNAs in C2C12 cells. The mouse-specific DAraf siRNAs (M1 and M2) and the rat-specific siRNAs (R1 and R2) were expressed in C2C12 cells by the retroviral vector. The amounts of endogenous DA-Raf1 were analyzed by immunoblotting with the anti–DA-Raf pAb. β-Tubulin is shown as a standard. (B) Prevention of the cell cycle arrest by RNAi of DAraf1/2 in C2C12 cells under the differentiation condition. C2C12 cells were transfected with the empty pSilencer vector (control) or the siRNA-expressing pSilencer vectors together with pEGFP vector, cultured in the differentiation medium for 48 h, and incubated with BrdU for 8 h. EGFP fluorescence (green) monitoring the siRNA expression is merged with anti-BrdU staining (red). The cells expressing M1 or M2 incorporated BrdU in the nuclei, but those expressing R1 or R2 did not. (C) Ratio of the BrdU-incorporating C2C12 cells in the analysis of B. (D) Inhibition of myogenin expression and myotube formation by RNAi of DAraf1/2 in C2C12 cells. EGFP fluorescence (green) monitoring the siRNA expression is merged with antimyogenin staining (red). The cells expressing M1 or M2 were prevented from differentiation, but those expressing the rat-specific siRNAs (R1 and R2) were not. (E) Ratio of the myogenin-expressing cells in the analysis of D. (F) The number of nuclei in a C2C12 cell at 72 and 96 h in the differentiation medium in the analysis of D. The values in the graphs are means ± SD (error bars) of three independent experiments. *, P < 0.05; **, P < 0.01 by t test compared with the control at each time point. Bars, 20 μm.
C2C12 cells cultured in the differentiation medium for 48 h became quiescent, and only ∼2% of the mock-transfected control cells incorporated BrdU for a further 8 h. However, expression of the mouse-specific siRNAs M1 and M2 resulted in BrdU incorporation in ∼8–12% of the cells for 8 h after the 48-h culture in the differentiation medium (Fig. 7, B and C). On the other hand, expression of the rat-specific R1 and R2 did not facilitate the BrdU incorporation (Fig. 7, B and C). These results indicate that intrinsic DA-Raf1 is required for the cell cycle arrest that is indispensable for skeletal myocyte differentiation.
When the mouse-specific siRNA M1 was expressed, the ratio of myogenin-expressing cells strikingly decreased during 24–96 h in the differentiation medium (Fig. 7, D and E). The expression of another siRNA, M2, was less effective than that of M1 but significantly reduced the ratio of myogenin expression. In contrast, the expression of siRNAs R1 and R2 did not reduce the ratio. Both the expression of M1 and that of M2 almost completely suppressed myotube formation by cell fusion, and, consequently, myocytes with the knockdown of DA-Raf were mononucleated (Fig. 7, D and F). The mean number of nuclei in individual C2C12 cells expressing R1 or R2 was not so high (2.5–3 at 96 h). This is probably because myotubes generated by cell fusion of a small number of myoblasts are more easily detected than those generated by cell fusion of a large number of myoblasts as a result of limited transfection efficiency. Altogether, these results imply that not only exogenously expressed DA-Raf1 but also intrinsic DA-Raf1 play crucial roles in the induction of myocyte differentiation represented by cell cycle arrest, by the expression of myogenin and a set of late muscle-specific proteins, and by cell fusion–mediated myotube formation.
Transfection of DAraf1 in C2C12 cells considerably reduced the ratio of the cells containing active phospho-ERK as detected by immunofluorescent staining (Fig. 8, A and B). In contrast, expression of the siRNA M1 or M2 facilitated the ratio of the cells containing phospho-ERK at 72 and 96 h in the differentiation medium (Fig. 8, A and B). These results corroborate the notion that the function of DA-Raf1 to induce myocyte differentiation is mediated by suppression of the ERK pathway.
Figure 8.
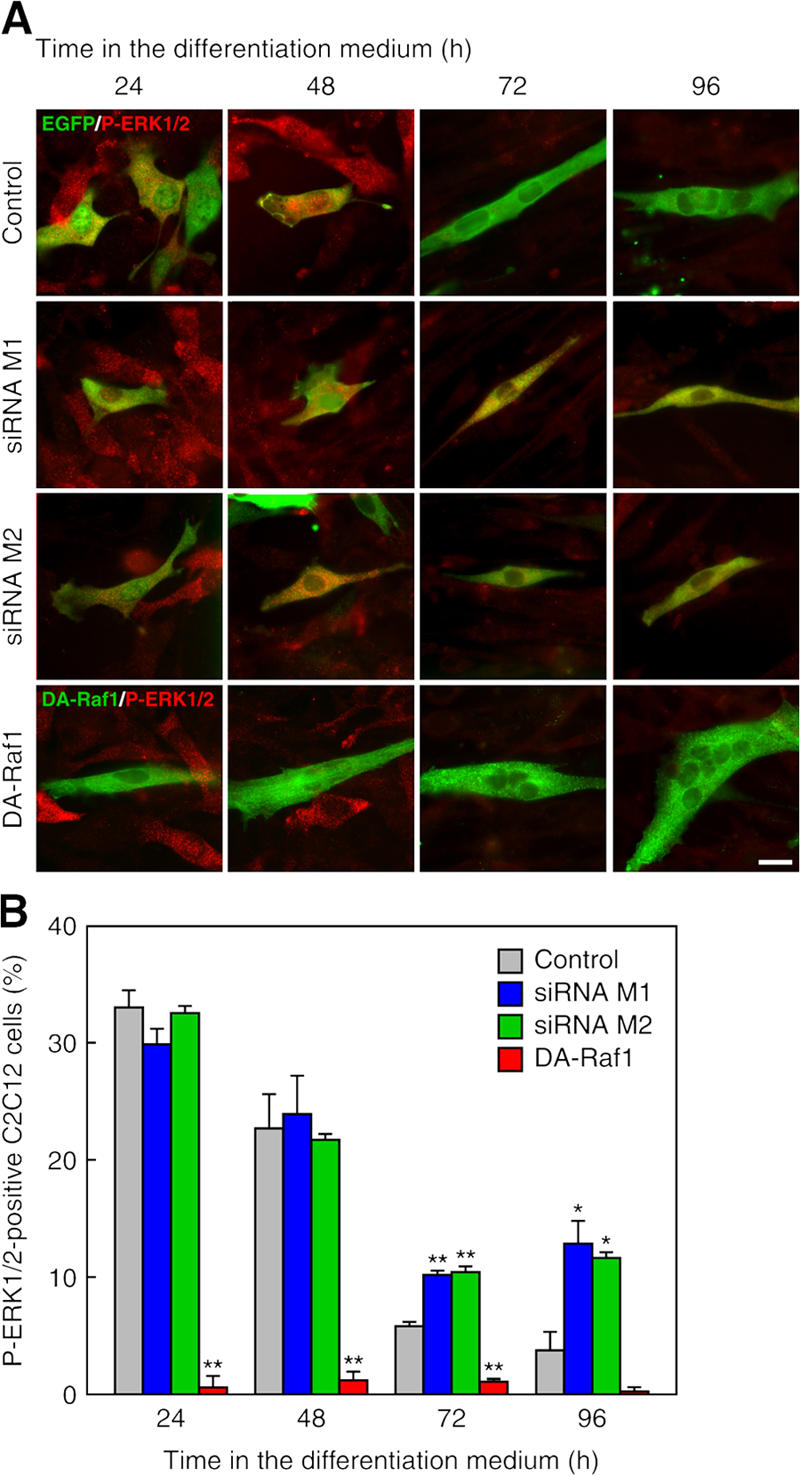
Suppression of ERK phosphorylation by DA-Raf1 and facilitation of ERK phosphorylation by the knockdown of endogenous DA-Raf during C2C12 cell differentiation. (A) C2C12 cells were transfected with the pSilencer vectors expressing control scrambled siRNAs (control) or the siRNA M1 or M2 together with pEGFP vector. Other C2C12 cells were transfected with myc–DAraf1 and were cultured in the differentiation medium. EGFP fluorescence (green) monitoring the siRNA expression or anti-myc staining detecting myc–DA-Raf1 expression (green) is merged with antiphospho-ERK1/2 staining (red). (B) Ratio of the cells with phospho-ERK1/2 in the analysis of A. The values in the graphs are means ± SD (error bars) of three independent experiments. *, P < 0.05; **, P < 0.01 by t test compared with the control at each time point. Bar, 20 μm.
Discussion
There are crucial differences among the canonical members of the Raf family (Raf-1, A-Raf, and B-Raf) in the regulatory mechanisms, target proteins, activities, and biological functions (Hagemann and Rapp, 1999; Mercer and Pritchard, 2003; Wellbrock et al., 2004). Furthermore, alternative splicing generates multiple splicing variants of B-Raf with different activities (Barnier et al., 1995; Papin et al., 1998). Nevertheless, all of the known Raf family proteins, including these splicing variants, share the three conserved regions CR1, 2, and 3. In marked contrast, DA-Raf1/2 that we have reported here almost exclusively comprises CR1, which encompasses Ras-binding and membrane-targeting RBD and CRD, and lacks CR2 and 3 representing the kinase domain. As expected from its structure, DA-Raf1 bound to active Ras and M-Ras and interfered with the ERK pathway. The binding to active Ras and M-Ras in vivo was confirmed by the findings that DA-Raf1 was translocated to the cellular membranes to colocalize with Ras and M-Ras activated by mitogenic signals.
Induction of endogenous DA-Raf1 during C2C12 cell differentiation was accompanied by reduction in the activating phosphorylation levels of MEK and ERK but not that of Akt. In addition, the exogenous expression of DAraf1 in v-Kras–transformed fibroblasts and in C2C12 myoblasts interfered with the Ras–ERK pathway but not with Ras–PI3K–Akt signaling. This is probably because the domains in Ras involved in the binding of Raf and PI3K p110 are partially overlapping but distinct (Rodriguez-Viciana et al., 1997; Vetter and Wittinghofer, 2001). Thus, DA-Raf1 is likely to compete with the binding of Raf proteins but not with that of PI3K p110. Consequently, DA-Raf1 may exclusively antagonize the Ras–ERK pathway but not the signaling pathways of other target proteins. Several suppressor proteins of the Ras–ERK pathway have been identified, including Spry/Spred, RKIP, and IMP (Kim and Bar-Sagi, 2004; Odabaei et al., 2004; for reviews see Ory and Morrison, 2004; Mason et al., 2006). These proteins antagonize the Ras–ERK pathway by interacting with a variety of components of the growth factor receptor–Ras–ERK pathway, but none of them directly bind to Ras. Thus, the mechanism of DA-Raf1 to interfere with the Ras–ERK pathway is unique and distinct from those of the other suppressor proteins.
Exogenously expressed DA-Raf1 suppressed all of the transformed phenotypes, including the tumorigenic potential of the v-Kras–transformed NIH3T3 fibroblasts in nude mice. Accordingly, we can expect that DA-Raf1 might serve as a tumor suppressor protein if the gene has loss of function mutations or if the expression levels are noticeably reduced in tumors of certain tissues. Even if DA-Raf is not a bona fide tumor suppressor protein, it might be applicable for the treatment of Ras-induced tumors, which comprise 15–30% of human cancers (Mercer and Pritchard, 2003; Wellbrock et al., 2004), by promoting its expression or alternative splicing efficiency to generate DA-Raf1.
The binding of DA-Raf1 to Ras/M-Ras and suppression of the ERK pathway in the cells highly expressing DA-Raf1 imply that DA-Raf can serve as a dominant-negative antagonist of the Ras–ERK pathway. DA-Raf1 seems to bind to Ras/M-Ras faster and more easily than the typical Raf family members because of its small size and its structure that is comprised almost exclusively of the RBD without regulatory sequences. The binding of DA-Raf1 to Ras/M-Ras may not be regulated by a variety of regulatory mechanisms for the typical Raf proteins, including phosphorylation and the binding of 14-3-3 proteins, scaffolding proteins such as KSR, and the suppressor proteins Spry and RKIP (Hagemann and Rapp, 1999; Chong et al., 2003; Mercer and Pritchard, 2003; Wellbrock et al., 2004). Thus, DA-Raf1 might accomplish the dominant-negative functions in vivo even if its amount is lower than those of the typical Raf proteins. A-Raf is specifically located to mitochondria (Yuryev et al., 2000), whereas DA-Raf1 was diffusely present throughout the cytoplasm and translocated to the plasma membrane and vesicle membranes by the mitogenic signals. This suggests that DA-Raf1 exerts the dominant-negative functions against Raf proteins close to these membranes rather than against mitochondrion-associated A-Raf. Among the Raf family members, B-Raf is the main protein that couples Ras to MEK (Mercer and Pritchard, 2003; Wellbrock et al., 2004), and this was true for C2C12 cells. In contrast, most of the signaling mechanisms and cellular functions of A-Raf remain enigmatic. Considering that DAraf1 is more highly expressed than Araf, particularly in the brain and myotubes, DA-Raf1 is the primary product of the Araf/DAraf gene by alternative splicing, at least in these tissues and cells. Furthermore, the binding of endogenous DA-Raf1 to Ras lead to the decline in Ras–B-Raf interaction and B-Raf activity during C2C12 cell differentiation. Consequently, DA-Raf1 is likely to serve as the intrinsic dominant-negative antagonist of the Ras–B-Raf–MEK–ERK pathway.
Because the amount of DA-Raf1 was prominently elevated during C2C12 cell differentiation, this elevated amount of DA-Raf1 seems to be required to achieve the positive regulation of differentiation. Indeed, DA-Raf1 exogenously expressed in C2C12 cells facilitated myogenic differentiation by inducing myogenin and a set of late muscle-specific protein expression as well as the cell cycle arrest. Moreover, the knockdown of endogenous DA-Raf interfered with myogenic differentiation by suppressing cell cycle arrest, myogenin expression, and myotube formation by cell fusion. Therefore, DA-Raf1 is essential for skeletal myocyte differentiation. This essential role of DA-Raf1 is probably through suppression of the Ras–ERK pathway because endogenous DA-Raf1 bound to Ras, and the B-Raf–MEK–ERK pathway was suppressed during differentiation. Furthermore, overexpressed DA-Raf1 abrogated ERK activation, whereas the knockdown of DA-Raf facilitated ERK activation. The Ras–ERK pathway negatively regulates muscle-specific gene expression by interfering with the expression of the MyoD family and MEF2 family transcription factors (Lassar et al., 1989; Winter and Arnold, 2000; Perry et al., 2001; Tortorella et al., 2001). The Ras–ERK pathway also counteracts the cell fusion and activation of Rb (Mittnacht et al., 1997; Peeper et al., 1997; Lee et al., 1999). Rb is required for myogenic differentiation by inducing not only the irreversible cell cycle arrest (Endo and Goto, 1992; Schneider et al., 1994; Novitch et al., 1996) but also the MEF2-dependent expression of late muscle-specific proteins (Novitch et al., 1999). The Ras–ERK pathway causes the inactivation of Rb by transactivating cyclin D1 (Downward, 1997), which participates in inactivating the phosphorylation of Rb by forming the complex with Cdk4/6. Insulin-like growth factors activate PI3K–Akt signaling that induces skeletal muscle myogenesis and hypertrophy (Glass, 2003; Charge and Rudnicki, 2004). Thus, suppression of the Ras–ERK pathway by DA-Raf1 without affecting Ras–PI3K–Akt signaling is favorable for skeletal muscle cell differentiation.
Culturing skeletal muscle myoblasts under mitogen-poor differentiation conditions leads to inactivation of the Ras–ERK pathway and, consequently, results in myocyte differentiation. However, the mechanisms of this inactivation of the Ras–ERK pathway have not been fully understood. The prominent induction of DA-Raf1 and its binding to Ras under the differentiation condition and suppression of the Ras–ERK pathway by DA-Raf1 explain the mechanisms of this inactivation of the Ras–ERK pathway. The knockdown of DA-Raf by RNAi interfered with ERK activation and with myocyte differentiation under the differentiation condition. This implies that DA-Raf1 is indeed required for adequate inactivation of the Ras–ERK pathway for myocyte differentiation even under the mitogen-poor condition. We have unraveled the essential role of DA-Raf1 in skeletal muscle cell differentiation. Likewise, scrutiny of the cellular and physiological functions of DA-Raf in other cell types or tissues, in which DA-Raf is highly expressed or highly induced under particular conditions, may lead to the elucidation of unexpected roles of this novel isoform of the Raf family.
Materials and methods
cDNA cloning
Mouse brain cDNA library constructed in pACT2 vector (CLONTECH Laboratories, Inc.) was screened by a yeast two-hybrid system with the constitutively active rat Mras(G22V) cDNA (Matsumoto et al., 1997) ligated to the Gal4 DNA-binding domain in pGBD9 vector (CLONTECH Laboratories, Inc.). The yeast strain Y190 was transformed with the pGBT9/Mras(G22V) bait plasmid and then with the pACT2/cDNA library prey plasmids. Double transformants were selected on plates of the minimal synthetic dropout medium. Large colonies were further selected by β-galactosidase colony- lift filter assay. The nucleotide sequence of the obtained 11 positive cDNA clones was determined with a DNA sequencing system (4200G; LI-COR). One of the cDNAs encoded mouse DA-Raf1. Rat and human DAraf1 cDNAs were cloned from PC12 and HeLa cell mRNAs, respectively, by RT-PCR using Omniscript reverse transcriptase and ProofStart DNA polymerase (QIAGEN). Primers used were sense primer 5′-GACAACATGGAACCACG-3′ and antisense primer 5′-TGAAACCTGGAGTGACCAGG-3′ for rat DAraf1 and sense primer 5′-AAGGCTCCATGGAGCCACCA-3′ and antisense primer 5′-GTACCAGATCCTGTTCTAGGC-3′ for human DAraf1. The sequences of mouse, rat, and human DAraf1 cDNAs have been deposited to the GenBank/ EMBL/DDBJ database with the accession nos. AB158252, AB158253, and AB158254, respectively.
RT-PCR and Northern blotting
RNAs were prepared from a variety of mouse tissues and from cultured cells as described previously (Endo and Nadal-Ginard, 1987; Abe et al., 2003). Quantitative RT-PCR and Northern blotting were conducted as described previously (Endo and Nadal-Ginard, 1987; Abe et al., 2003).
Antibody preparation and immunoblotting
Mouse DAraf1 cDNA was inserted into pQE32 vector (QIAGEN) in frame with the 6× His tag. The recombinant His-tagged DA-Raf1 was expressed in the Escherichia coli strain XL1-Blue and affinity purified with Talon Metal Affinity Resin (CLONTECH Laboratories, Inc.). The protein was separated by SDS-PAGE and recovered from the gel. A New Zealand white rabbit was immunized with the purified His–DA-Raf1 emulsified with Freund's complete or incomplete adjuvant (Difco Laboratories). The anti–DA-Raf pAb was affinity purified through formyl-cellulofine (Seikagaku Corp.) coupled with the antigen. Mouse tissues were extracted with radioimmunoprecipitation assay (RIPA) lysis buffer, their protein concentrations were determined by Bradford method, and the extracts were treated with SDS sample buffer. Cultured cells were directly lysed with the SDS sample buffer. These samples were subjected to SDS-PAGE and immunoblotting. Antibodies used for the immunoblotting were the anti–DA-Raf pAb; pAbs to Raf-1, B-Raf, A-Raf (Santa Cruz Biotechnology, Inc.), MEK, phospho-MEK (Ser217/221), p44/42 MAPK, Akt, and phospho-Akt (Ser473; Cell Signaling Technology); and mAbs to myogenin (F5D), MyHC (A4.1025), β-tubulin (E7; Developmental Studies Hybridoma Bank), TnT (NT302; Abe et al., 1986), and phospho-p44/42 MAPK (Thr202/Tyr204; E10; Cell Signaling Technology). The immunological reaction was detected with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer) and analyzed with a luminoimage analyzer (LAS-1000mini; Fuji).
Pull-down assay
DAraf1 cDNA was inserted into pGEX-6P2 vector (GE Healthcare) in frame with the GST tag. The recombinant GST–DA-Raf1 was expressed in E. coli XL1-Blue and affinity purified with glutathione–Sepharose 4B (GE Healthcare). The cDNAs encoding the constitutively active and dominant-negative mutants of M-Ras and H-Ras as well as the v-Kras gene were fused in frame to the N-terminal myc tag in pEF-BOS/myc vector. These recombinant plasmids were transfected to COS-1 cells with FuGENE 6 Transfection Reagent (Roche). The cells were lysed, and the GST pull-down assay was conducted as described previously (Abe et al., 2003). Binding proteins were detected by immunoblotting with the anti-myc mAb myc1-9E10 (American Type Culture Collection).
Coimmunoprecipitation assay
COS-1 cells were cotransfected with the recombinant plasmids pEF-BOS/HA-DAraf1 and pEF-BOS/myc-Hras(G12V) or pEF-BOS/myc-Mras(G22V). 24 h after the transfection, the cells were lysed with 800 μl of lysis buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 1 mM DTT, 0.1 mM PMSF, 2 μg/ml leupeptin, and 1 μg/ml pepstatin) and centrifuged at 16,000 g for 15 min. C2C12 cells cultured in the differentiation medium were lysed with the lysis buffer. Mouse brain was extracted with RIPA lysis buffer. The supernatant solutions were preabsorbed with protein G–Sepharose 4 Fast Flow (GE Healthcare). Protein G–Sepharose was mixed with the mAb myc1-9E10 or mAb to H-Ras (238; Santa Cruz Biotechnology, Inc.) for 1 h and then mixed with the preabsorbed supernatant solution for 1 h. The beads were washed five times with the lysis buffer. Proteins bound to the beads were eluted with 9 M urea and detected by immunoblotting with the pAbs to HA tag (Medical and Biological Laboratories), DA-Raf, and B-Raf or with the anti–pan-Ras mAb RAS10 (Calbiochem).
In vitro kinase assay
C2C12 cells cultured in the differentiation medium were lysed with RIPA lysis buffer. B-Raf, Raf-1, and A-Raf were immunoprecipitated with each specific pAb prebound to protein A–Sepharose 4 Fast Flow (GE Healthcare). The Sepharose beads were washed five times with RIPA lysis buffer and suspended in 20 μl of the assay buffer (20 mM MOPS, pH 7.2, 75 mM MgCl2, 25 mM EGTA, 25 mM glycerol 2-phosphate, 1 mM sodium orthovanadate, and 1 mM DTT) containing 0.5 μg of recombinant GST–MEK1 (Upstate Cell Signaling Solutions). They were mixed with 20 μl of the assay buffer containing 0.74 MBq γ-[32P]ATP (166.5 TBq/mmol; MP Biomedicals) and incubated for 30 min at 30°C. The supernatant was mixed with 40 μl of 2× SDS sample buffer and subjected to SDS-PAGE. Phosphorylation of MEK1 was detected by autoradiography and analyzed with a bioimaging analyzer (BAS-1500 Mac; Fuji).
Epitope-tagging immunofluorescent localization
Mouse C2C12 myoblasts were cotransfected with pEF-BOS/HA-DAraf1 and pEF-BOS/myc-Hras or pEF-BOS/myc-Mras by using LipofectAMINE 2000 (Invitrogen). 24 h after the transfection, the cells were cultured under a serum-free condition for 48 h and stimulated with 10% FBS or 50 ng/ml recombinant human FGF2 (Wako) for 30 min. They were fixed with 4% PFA in PBS and permeabilized with 0.2% Triton X-100 in PBS. The cells were then incubated with the anti-HA tag pAb, the mAb myc1-9E10, and with AlexaFluor546-conjugated goat anti–rabbit IgG and AlexaFluor488–goat anti–mouse IgG (Invitrogen). Fluorescent images were acquired at room temperature using a confocal laser-scanning microscope (LSM 410; Carl Zeiss MicroImaging, Inc.) with a 63× NA 1.40 plan-Apochromat objective lens and were assembled with Photoshop 9 (Adobe). In each plate, photographs were trimmed, and each fluorochrome was adjusted identically for brightness and contrast to represent the observed images.
Cell transformation and transformation assays
The NRT9 cell line was established by stably transfecting the mouse NIH3T3 fibroblasts with the v-Kras gene in pLNCX vector (CLONTECH Laboratories, Inc.) followed by selecting with G418. To establish NRT9 cell clones stably transfected with DAraf1, an XbaI fragment of myc–DAraf1 in pEF-BOS/myc-DAraf1 was subcloned in pMIKHygB vector. The stable transfectants (NRT/DR cells) were selected with 500 μg/ml hygromycin B. The exogenously expressed myc–DA-Raf1 was detected by immunoblotting with the mAb myc1-9E10. The amounts of ERK1/2, Akt, and their activating phosphorylation levels were analyzed by immunoblotting with the p44/42 MAPK pAb, Akt pAb, phospho-p44/42 MAPK (Thr202/Tyr204) mAb E10, and phospho-Akt (Ser473) pAb, respectively. Cell morphology and actin cytoskeleton were analyzed by staining the cells with rhodamine–phalloidin (Invitrogen). Focus formation assay and colony formation assay in soft agar were performed as described previously (Clark et al., 1995). To detect tumorigenicity, 1 × 106 cells were suspended in 100 μl of sterile PBS and subcutaneously injected into dorsal flanks of nude mice. All mouse protocols were approved by the guidelines of Chiba University.
Skeletal myocyte differentiation
To induce terminal differentiation in C2C12 cells, undifferentiated myoblasts maintained in the growth medium (DME containing 10% FBS) were shifted to the differentiation medium (DME containing 5% horse serum) as described previously (Abe et al., 2003). The undifferentiated myoblasts were transfected with pEF-BOS/HA-DAraf1 in the growth medium and were left under the differentiation condition. DA-Raf1 and muscle-specific proteins (myogenin, MyHC, and TnT) were detected by staining with the anti-HA tag pAb and the mAbs to myogenin, MyHC, and TnT followed by AlexaFluor488–goat anti–rabbit IgG and AlexaFluor546–goat anti–mouse IgG. Fluorescent images were acquired at room temperature using a microscope (Axioskop; Carl Zeiss MicroImaging, Inc.) with a 40× NA 1.30 plan-Neofluar objective lens, CCD camera (CoolSNAP; Roper Scientific), and Openlab 3.0.3 software (Improvision). The photographs were processed with Photoshop 9 (Adobe) as described in the Epitope-tagging immunofluorescent localization section.
RNAi
RNAi of DA-Raf was conducted by expressing siRNAs with pSilencer 2.1-U6 neo vector (Ambion) as described previously (Sun et al., 2006). The target sequences of mouse DAraf siRNA M1 and M2 were 5′-AAAGAAGGATTGGAGGACCCT-3′ (nt 595–615 from the initiation codon) and 5′-AAAGCAAGATCTGATACAGAG-3′ (nt 818–838), respectively. The target sequences of rat DAraf siRNA R1 and R2 were 5′-AAGTATTGAATAAGGGCATGG-3′ (nt 670–690; 90.5% homologous to the corresponding mouse sequence) and 5′-AACAAGATCTGATGTAGAGTG-3′ (nt 820–840; 85% homologous to the corresponding mouse sequence), respectively. C2C12 myoblasts were cotransfected with these siRNA-expressing pSilencer vectors and one tenth of the amount of pEGFP-C1 vector (CLONTECH Laboratories, Inc.) to monitor the cells expressing siRNAs. They were shifted to the differentiation medium 24 h after the transfection and were cultured for a further 96 h. To assess the interfering effect of these siRNAs, pSilencer 5.1-U6 Retro vector (Ambion) expressing these siRNAs was constructed, and the retroviruses were produced. C2C12 cells were infected with these retroviruses, and infected cells were selected with puromycin. Lysates of the selected cells cultured in the differentiation medium were subjected to immunoblotting with the anti–DA-Raf pAb.
Cell cycle determination
Proliferating or cell cycle–arrested cells were determined by the incorporation of BrdU. C2C12 myoblasts transfected with pEF-BOS/HA-DAraf1 were cultured for 24 h in the growth medium. Then, they were incubated for 2 h in the same medium supplemented with 10 μM BrdU and 1 μM 5-fluoro 2′-deoxyuridine. C2C12 cells transfected with the pSilencer plasmids encoding DAraf siRNAs together with one tenth of the amount of pEGFP-C1 vector were cultured for 24 h in the growth medium. They were shifted to the differentiation medium and cultured for 48 h and for 8 h in the same medium supplemented with BrdU/5-fluoro 2′-deoxyuridine. The BrdU-incorporating nuclei were detected by staining with an anti-BrdU mAb (GE Healthcare) supplemented with 10 μg/ml DNase I followed by AlexaFluor546–goat anti–mouse IgG.
Online supplemental material
Fig. S1 shows the colocalization of DA-Raf1 with constitutively active H-Ras and M-Ras but not with dominant-negative H-Ras and M-Ras. Fig. S2 indicates the nucleotide sequences and corresponding amino acid sequences of rat and human DA-Raf1/2. Fig. S3 shows the suppression of ERK phosphorylation without affecting Akt phosphorylation in C2C12 myoblasts and suppression of the v–K-Ras–induced transformed phenotype in NRT9 cells by DA-Raf1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703195/DC1.
Supplementary Material
Acknowledgments
This work was partly supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by Research Grants for Nervous and Mental Disorders (14B-4 and 17A-10) from the Ministry of Health, Labor, and Welfare of Japan, and by the Research Grant from Futaba Electronics Memorial Foundation (to T. Endo). P. Sun is supported by the Postdoctoral Fellowship for Foreign Researchers of the Japan Society for the Promotion of Science.
Abbreviations used in this paper: CRD, cysteine-rich domain; ERK, extracellular-regulated kinase; MEK, MAPK and ERK kinase; MyHC, myosin heavy chain; PI3K, phosphatidylinositol 3-kinase; RBD, Ras-binding domain; RIPA, radioimmunoprecipitation assay; Spry, Sprouty; TnT, troponin T.
References
- Abe, H., T. Komiya, and T. Obinata. 1986. Expression of multiple troponin T variants in neonatal chicken breast muscle. Dev. Biol. 118:42–51. [DOI] [PubMed] [Google Scholar]
- Abe, T., M. Kato, H. Miki, T. Takenawa, and T. Endo. 2003. Small GTPase Tc10 and its homologue RhoT induce N-WASP-mediated long process formation and neurite outgrowth. J. Cell Sci. 116:155–168. [DOI] [PubMed] [Google Scholar]
- Barnier, J.V., C. Papin, A. Eychene, O. Lecoq, and G. Calothy. 1995. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J. Biol. Chem. 270:23381–23389. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi, D., and J.R. Feramisco. 1986. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 233:1061–1068. [DOI] [PubMed] [Google Scholar]
- Bennett, A.M., and N.K. Tonks. 1997. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 278:1288–1291. [DOI] [PubMed] [Google Scholar]
- Black, B.L., and E.N. Olson. 1998. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14:167–196. [DOI] [PubMed] [Google Scholar]
- Campbell, S.L., R. Khosravi-Far, K.L. Rossman, G.J. Clark, and C.J. Der. 1998. Increasing complexity of Ras signaling. Oncogene. 17:1395–1413. [DOI] [PubMed] [Google Scholar]
- Chang, L., and M. Karin. 2001. Mammalian MAP kinase signalling cascades. Nature. 410:37–40. [DOI] [PubMed] [Google Scholar]
- Charge, S.B.P., and M.A. Rudnicki. 2004. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84:209–238. [DOI] [PubMed] [Google Scholar]
- Chong, H., H.G. Vikis, and K.-L. Guan. 2003. Mechanisms of regulating the Raf kinase family. Cell. Signal. 15:463–469. [DOI] [PubMed] [Google Scholar]
- Clark, G.J., A.D. Cox, S.M. Graham, and C.J. Der. 1995. Biological assays for Ras transformation. Methods Enzymol. 255:395–412. [DOI] [PubMed] [Google Scholar]
- Cowley, S., H. Paterson, P. Kemp, and C.J. Marshall. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 77:841–852. [DOI] [PubMed] [Google Scholar]
- Downward, J. 1997. Cell cycle: routine role for Ras. Curr. Biol. 7:R258–R260. [DOI] [PubMed] [Google Scholar]
- Endo, T., and S. Goto. 1992. Retinoblastoma gene product Rb accumulates during myogenic differentiation and is deinduced by the expression of SV40 large T antigen. J. Biochem. (Tokyo). 112:427–430. [DOI] [PubMed] [Google Scholar]
- Endo, T., and B. Nadal-Ginard. 1987. Three types of muscle-specific gene expression in fusion-blocked rat skeletal muscle cells: translational control in EGTA-treated cells. Cell. 49:515–526. [DOI] [PubMed] [Google Scholar]
- Glass, D.J. 2003. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat. Cell Biol. 5:87–90. [DOI] [PubMed] [Google Scholar]
- Hagemann, C., and U.R. Rapp. 1999. Isotype-specific functions of Raf kinases. Exp. Cell Res. 253:34–46. [DOI] [PubMed] [Google Scholar]
- Huleihel, M., M. Goldsborough, J. Cleveland, M. Gunnell, T. Bonner, and U.R. Rapp. 1986. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell. Biol. 6:2655–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.J., and D. Bar-Sagi. 2004. Modulation of signalling by Sprouty: a developing story. Nat. Rev. Mol. Cell Biol. 5:441–450. [DOI] [PubMed] [Google Scholar]
- Kimmelman, A.C., N. Nunez Rodriguez, and A.M.-L. Chan. 2002. R-Ras3/ M-Ras induces neuronal differentiation of PC12 cells through cell-type-specific activation of the mitogen-activated protein kinase cascade. Mol. Cell. Biol. 22:5946–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch, W., G. Heidecker, P. Lloyd, and U.R. Rapp. 1991. Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature. 349:426–428. [DOI] [PubMed] [Google Scholar]
- Lassar, A.B., M.J. Thayer, R.W. Overell, and H. Weintraub. 1989. Transformation by activated ras or fos prevents myogenesis by inhibiting expression of MyoD1. Cell. 58:659–667. [DOI] [PubMed] [Google Scholar]
- Lassar, A.B., S.X. Skapek, and B. Novitch. 1994. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 6:788–794. [DOI] [PubMed] [Google Scholar]
- Lee, K.Y., M.H. Ladha, C. McMahon, and M.E. Ewen. 1999. The retinoblastoma protein is linked to the activation of Ras. Mol. Cell. Biol. 19:7724–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, S.J., W.T. Matten, A.S. Hermann, J.M. Candia, S. Rong, K. Fukasawa, G.F. Vande Woude, and N.G. Ahn. 1994. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 265:966–970. [DOI] [PubMed] [Google Scholar]
- Marshall, C.J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 80:179–185. [DOI] [PubMed] [Google Scholar]
- Mason, J.M., D.J. Morrison, M.A. Basson, and J.D. Licht. 2006. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 16:45–54. [DOI] [PubMed] [Google Scholar]
- Matheny, S.A., C. Chen, R.L. Kortum, G.L. Razidlo, R.E. Lewis, and M.A. White. 2004. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature. 427:256–260. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K., T. Asano, and T. Endo. 1997. Novel small GTPase M-Ras participates in reorganization of actin cytoskeleton. Oncogene. 15:2409–2417. [DOI] [PubMed] [Google Scholar]
- Mercer, K.E., and C.A. Pritchard. 2003. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta. 1653:25–40. [DOI] [PubMed] [Google Scholar]
- Mitin, N., K.L. Rossman, and C.J. Der. 2005. Signaling interplay in Ras superfamily function. Curr. Biol. 15:R563–R574. [DOI] [PubMed] [Google Scholar]
- Mittnacht, S., H. Paterson, M.F. Olson, and C.J. Marshall. 1997. Ras signalling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr. Biol. 7:219–221. [DOI] [PubMed] [Google Scholar]
- Novitch, B.G., G.J. Mulligan, T. Jacks, and A.B. Lassar. 1996. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J. Cell Biol. 135:441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch, B.G., D.B. Spicer, P.S. Kim, W.L. Cheung, and A.B. Lassar. 1999. pRb is required for MEF2-dependent gene expression as well as cell-cycle arrest during skeletal muscle differentiation. Curr. Biol. 9:449–459. [DOI] [PubMed] [Google Scholar]
- Odabaei, G., D. Chatterjee, A.R. Jazirehi, L. Goodglick, K. Yeung, and B. Bonavida. 2004. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv. Cancer Res. 91:169–200. [DOI] [PubMed] [Google Scholar]
- Olson, E.N., G. Spizz, and M.A. Tainsky. 1987. The oncogenic forms of N-ras or H-ras prevent skeletal myoblast differentiation. Mol. Cell. Biol. 7:2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory, S., and D.K. Morrison. 2004. Signal transduction: implications for Ras-dependent ERK signaling. Curr. Biol. 14:R277–R278. [DOI] [PubMed] [Google Scholar]
- Papin, C., A. Denouel-Galy, D. Laugier, G. Calothy, and A. Eychene. 1998. Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J. Biol. Chem. 273:24939–24947. [DOI] [PubMed] [Google Scholar]
- Pearson, G., F. Robinson, T.B. Gibson, B. Xu, M. Karandikar, K. Berman, and M.H. Cobb. 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22:153–183. [DOI] [PubMed] [Google Scholar]
- Peeper, D.S., T.M. Upton, M.H. Ladha, E. Neuman, J. Zalvide, R. Bernards, J.A. DeCaprio, and M.E. Ewen. 1997. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature. 386:177–181. [DOI] [PubMed] [Google Scholar]
- Perry, R.L.S., M.H. Parker, and M.A. Rudnicki. 2001. Activated MEK1 binds the nuclear MyoD transcriptional complex to repress transactivation. Mol. Cell. 8:291–301. [DOI] [PubMed] [Google Scholar]
- Quilliam, L.A., A.F. Castro, K.S. Rogers-Graham, C.B. Martin, C.J. Der, and C. Bi. 1999. M-Ras/R-Ras3, a transforming Ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. J. Biol. Chem. 274:23850–23857. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana, P., P.H. Warne, A. Khwaja, B.M. Marte, D. Pappin, P. Das, M.D. Waterfield, A. Ridley, and J. Downward. 1997. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 89:457–467. [DOI] [PubMed] [Google Scholar]
- Rommel, C., B.A. Clarke, S. Zimmermann, L. Nunez, R. Rossman, K. Reid, K. Moelling, G.D. Yancopoulos, and D.J. Glass. 1999. Differentiation stage-specific inhibition of the Raf–MEK–ERK pathway by Akt. Science. 286:1738–1741. [DOI] [PubMed] [Google Scholar]
- Schaeffer, H.J., and M.J. Weber. 1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 19:2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, J.W., W. Gu, L. Zhu, V. Mahdavi, and B. Nadal-Ginard. 1994. Reversal of terminal differentiation mediated by p107 in Rb−/– muscle cells. Science. 264:1467–1471. [DOI] [PubMed] [Google Scholar]
- Schramek, H. 2002. MAP kinases: from intracellular signals to physiology and disease. News Physiol. Sci. 17:62–67. [DOI] [PubMed] [Google Scholar]
- Sun, P., H. Watanabe, K. Takano, T. Yokoyama, J. Fujisawa, and T. Endo. 2006. Sustained activation of M-Ras induced by nerve growth factor is essential for neuronal differentiation of PC12 cells. Genes Cells. 11:1097–1113. [DOI] [PubMed] [Google Scholar]
- Tapscott, S.J. 2005. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 132:2685–2695. [DOI] [PubMed] [Google Scholar]
- Tortorella, L.L., D.J. Milasincic, and P.F. Pilch. 2001. Critical proliferation-independent window for basic fibroblast growth factor repression of myogenesis via the p42/p44 MAPK signaling pathway. J. Biol. Chem. 276:13709–13717. [DOI] [PubMed] [Google Scholar]
- Vetter, I.R., and A. Wittinghofer. 2001. The guanine nucleotide-binding switch in three dimensions. Science. 294:1299–1304. [DOI] [PubMed] [Google Scholar]
- Voice, J.K., R.L. Klemke, A. Le, and J.H. Jackson. 1999. Four human Ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J. Biol. Chem. 274:17164–17170. [DOI] [PubMed] [Google Scholar]
- Wakioka, T., A. Sasaki, R. Kato, T. Shouda, A. Matsumoto, K. Miyoshi, M. Tsuneoka, S. Komiya, R. Baron, and A. Yoshimura. 2001. Spred is a Sprouty-related suppressor of Ras signalling. Nature. 412:647–651. [DOI] [PubMed] [Google Scholar]
- Wellbrock, C., M. Karasarides, and R. Marais. 2004. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5:875–885. [DOI] [PubMed] [Google Scholar]
- Winter, B., and H.-H. Arnold. 2000. Activated Raf kinase inhibits muscle cell differentiation through a MEF2-dependent mechanism. J. Cell Sci. 113:4211–4220. [DOI] [PubMed] [Google Scholar]
- Yan, J., S. Roy, A. Apolloni, A. Lane, and J.F. Hancock. 1998. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J. Biol. Chem. 273:24052–24056. [DOI] [PubMed] [Google Scholar]
- Yeung, K., T. Seitz, S. Li, P. Janosch, B. McFerran, C. Kaiser, F. Fee, K.D. Katsanakis, D.W. Rose, H. Mischak, et al. 1999. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 401:173–177. [DOI] [PubMed] [Google Scholar]
- Yuryev, A., M. Ono, S.A. Goff, F. Macaluso, and L.P. Wennogle. 2000. Isoform-specific localization of A-RAF in mitochondria. Mol. Cell. Biol. 20:4870–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}