

Abstract
Protease-activated receptor-1 (PAR1), a G protein–coupled receptor (GPCR) for thrombin, is irreversibly activated by proteolysis. Consequently, PAR1 trafficking is critical for the fidelity of thrombin signaling. PAR1 displays constitutive and agonist-induced internalization, which are clathrin and dynamin dependent but are independent of arrestins. The clathrin adaptor AP2 (adaptor protein complex-2) is critical for constitutive but not for activated PAR1 internalization. In this study, we show that ubiquitination negatively regulates PAR1 constitutive internalization and specifies a distinct clathrin adaptor requirement for activated receptor internalization. PAR1 is basally ubiquitinated and deubiquitinated after activation. A PAR1 lysineless mutant signaled normally but was not ubiquitinated. Constitutive internalization of ubiquitin (Ub)-deficient PAR1 was markedly increased and inhibited by the fusion of Ub to the cytoplasmic tail. Ub-deficient PAR1 constitutive internalization was AP2 dependent like the wild-type receptor. However, unlike wild-type PAR1, AP2 was required for the internalization of activated Ub-deficient receptor, suggesting that the internalization of ubiquitinated PAR1 requires different endocytic machinery. These studies reveal a novel function for ubiquitination in the regulation of GPCR internalization.
Introduction
Protease-activated receptor-1 (PAR1), a G protein–coupled receptor (GPCR) for thrombin, is important for hemostasis and thrombosis, inflammation, embryonic development, and cancer progression (Coughlin, 2005; Arora et al., 2007). Unlike most GPCRs, PAR1 is irreversibly activated by proteolysis. The PAR1 N terminus is cleaved by thrombin, which unmasks a new N terminus that acts as a tethered ligand and binds intramolecularly to the receptor to trigger transmembrane signaling (Vu et al., 1991). Synthetic peptides that mimic the newly formed N terminus can activate PAR1 independently of proteolysis. Because of the irreversible proteolytic nature of PAR1 activation, rapid desensitization and receptor trafficking tightly regulate PAR1 signaling (Trejo et al., 1998; Paing et al., 2002; Chen et al., 2004).
PAR1 displays two modes of trafficking that are important for the regulation of receptor signaling. Unactivated PAR1 constitutively cycles between the cell surface and an intracellular compartment, generating an intracellular pool of uncleaved receptor that replenishes the cell surface after thrombin exposure and leads to rapid resensitization to thrombin signaling independent of de novo receptor synthesis (Hein et al., 1994; Paing et al., 2006). Unlike most GPCRs, which internalize and recycle, activated PAR1 is internalized, sorted directly to lysosomes, and degraded (Hoxie et al., 1993; Trejo and Coughlin, 1999). Sorting of activated PAR1 to lysosomes is critical for signal termination (Trejo et al., 1998). Constitutive and agonist-induced PAR1 internalization are clathrin and dynamin dependent (Trejo et al., 2000). However, in contrast to most GPCRs, neither constitutive nor activated PAR1 internalization requires arrestins (Paing et al., 2002). Arrestins interact with clathrin and adaptor protein complex-2 (AP2) to facilitate the internalization of activated GPCRs through clathrin-coated pits (Goodman et al., 1996; Laporte et al., 1999). We recently showed that AP2 and not arrestins is critical for PAR1 constitutive internalization and is essential for the cellular recovery of thrombin signaling (Paing et al., 2006). Interestingly, activated PAR1 internalization through clathrin-coated pits is independent of AP2, suggesting that constitutive and activated receptor internalization require different endocytic machinery. The mechanisms that regulate activated PAR1 internalization through clathrin-coated pits is not known.
Ubiquitin (Ub) modification of integral membrane proteins can function as an internalization and endosomal sorting signal (Hicke and Dunn, 2003). Ub, a 76–amino acid protein, is recognized by Ub-binding domains (UBDs), which are found in proteins of the endocytic sorting machinery. Ubiquitination regulates internalization of the yeast Ste2 and Ste3 GPCRs. Studies using yeast strains that lack specific Ub-conjugating enzymes and Ub-defective Ste2 mutants or chimeras indicate that monoubiquitination is both necessary and sufficient for constitutive and agonist-induced receptor internalization (Hicke and Riezman, 1996; Terrell et al., 1998). In contrast, recent studies suggest that mammalian GPCR ubiquitination is essential for lysosomal sorting but not for receptor internalization (Marchese and Benovic, 2001; Shenoy et al., 2001). Direct β2-adrenergic receptor (β2AR) ubiquitination is not required for internalization but regulates activated receptor lysosomal sorting and degradation (Shenoy et al., 2001). Similar to β2AR, ubiquitination of chemokine receptor 4 (CXCR4) is essential for agonist-promoted receptor lysosomal degradation but not for internalization (Marchese and Benovic, 2001). Although ubiquitination does not have a direct role in mammalian GPCR internalization, it has been shown to function indirectly. Indeed, activation-dependent ubiquitination of arrestins is required for β2AR internalization (Shenoy et al., 2001). However, the function of ubiquitination in the regulation of mammalian GPCRs that do not require arrestins for endocytosis is not known.
We have shown that constitutive and agonist-induced PAR1 internalization is clathrin and dynamin dependent and independent of arrestins (Paing et al., 2002). We recently found that the clathrin adaptor AP2 is critical for constitutive but not for agonist-induced PAR1 internalization (Paing et al., 2006). Given these observations and the previous findings that Ub regulates yeast GPCR internalization (Hicke and Riezman, 1996; Terrel et al., 1998), we examined the function of ubiquitination in PAR1 trafficking. Our findings here reveal a novel role for ubiquitination in the negative regulation of PAR1 constitutive internalization and in specifying a distinct clathrin adaptor requirement for activated receptor internalization.
Results
Basal ubiquitination and agonist-induced deubiquitination of PAR1
To define the importance of the posttranslational modification of lysine residues with Ub in the regulation of PAR1 trafficking, we constructed a PAR1 lysineless mutant by replacing the 10 intracytosolic lysine (K) residues with arginines, which is designated PAR1 0K (Fig. 1 A). To ensure that the PAR1 0K mutant was not globally disrupted in receptor function, the capacity of the activated receptor to promote Gαq-stimulated phosphoinositide hydrolysis was examined. HeLa cells expressing comparable amounts of cell surface PAR1 wild type and 0K mutant were incubated with various concentrations of thrombin, and the amounts of [3H]inositol phosphates (IPs) formed were then measured (Fig. 1 B). Both PAR1 wild type and 0K mutant were equally effective at stimulating [3H]IP accumulation as well as at inducing a maximal effect at saturating concentrations of thrombin. The ability of the PAR1 0K mutant to couple to G protein activation like the wild-type receptor indicates that receptor function is intact.
Figure 1.

PAR1 wild-type and lysineless 0K mutant signaling and ubiquitination. (A) The PAR1 intracytosolic lysine (K) residues that serve as potential sites for ubiquitination are highlighted and were mutated to arginine to generate a PAR1 0K mutant. (B) HeLa cells stably expressing FLAG-tagged PAR1 wild type (Wt) and 0K mutant labeled with myo-[3H]inositol were incubated with various concentrations of α-thrombin for 10 min at 37°C in media with lithium chloride. The amounts of accumulated [3H]IPs were then measured. The data shown (mean ± SD [error bars]; n = 3) are expressed as the fold increase in [3H]IP accumulation over basal counts per minute from one experiment and are representative of three separate experiments. The values for PAR1 wild-type and 0K mutant cell surface expression were 0.385 ± 0.023 and 0.376 ± 0.044, respectively. (C) HEK293 cells transiently transfected with either FLAG-tagged PAR1 wild type or 0K mutant together with HA-tagged Ub and dynamin K44A mutant were incubated with or without 100 μM TRAP for 10 min at 37°C. Cells were lysed and immunoprecipitated with M2 anti-FLAG antibody, and ubiquitinated PAR1 was detected by immunoblotting with an anti-HA antibody conjugated to HRP. Membranes were stripped and probed with an anti-FLAG polyclonal antibody to detect PAR1. Cell lysates were immunoblotted for dynamin (Dyn) expression to control for differences in transfection efficiency and for actin to control for sample loading. The asterisk indicates detection of the immunoprecipitating antibody heavy and light chains. Similar findings were observed in three independent experiments.
We next determined whether the receptor was modified with Ub by directly comparing the ubiquitination status of PAR1 wild type to the lysineless 0K mutant. HEK293 cells transiently coexpressing similar amounts of PAR1 wild type or 0K mutant together with HA-tagged Ub and dynamin K44A were incubated in the absence or presence of agonist peptide for 10 min at 37°C. Cells were lysed, PAR1 was immunoprecipitated, and the extent of receptor ubiquitination was determined. Strikingly, in untreated cells, wild-type PAR1 displayed a substantial amount of basal ubiquitination that was detected as prominent high molecular weight species migrating above ∼107 kD (Fig. 1 C, lane 1), which is consistent with the addition of multiple HA-tagged Ubs. Remarkably, incubation with agonist decreased wild-type PAR1 ubiquitination (Fig. 1 C, lane 2). In contrast to wild-type receptor, ubiquitinated species of PAR1 0K were undetectable in both untreated or agonist-treated cells (Fig. 1 C, lanes 3 and 4). These findings suggest that PAR1 is basally ubiquitinated and that activation promotes deubiquitination.
Ub-deficient PAR1 displays enhanced constitutive internalization
To investigate the function of ubiquitination in PAR1 trafficking, we examined the constitutive and agonist-induced loss of cell surface wild-type and Ub-deficient PAR1 by ELISA. HeLa cells and Rat1 fibroblasts stably expressing similar amounts of surface PAR1 wild type or 0K mutant were incubated with M1 anti-FLAG antibody for 1 h at 4°C to label cell surface receptors and were treated with or without agonist for various times at 37°C, and the amount of receptor remaining on the cell surface was then quantified by ELISA. In wild-type PAR1–expressing cells, agonist induced rapid receptor internalization within 10 min, and the receptor continued to slowly internalize, leading to an ∼70% loss of surface PAR1 after 30 min (Fig. 2, A and C). PAR1 0K mutant internalization was comparable with wild-type receptor after agonist exposure in both cell types (Fig. 2, A and C). We next examined the constitutive internalization of wild-type PAR1 and observed a slow rate of internalization resulting in a 10–20% loss of cell surface receptor after 30 min (Fig. 2, B and D), which is consistent with that previously reported for these cell types (Shapiro et al., 1996; Paing et al., 2004). In contrast, Ub-deficient PAR1 0K mutant displayed an increased rate of constitutive internalization in which 50–60% of receptor was lost from the cell surface after 30 min of incubation in both cell types (Fig. 2, B and D). Immunofluorescence microscopy studies also revealed a substantial amount of internalized Ub-deficient PAR1 0K mutant in early endosomes compared with wild-type receptor even without agonist exposure or antibody prebinding (Fig. 2 E), suggesting that the absence of ubiquitination enhances PAR1 constitutive internalization. In contrast, both PAR1 wild type and 0K mutant showed robust internalization after agonist exposure (Fig. 2 E).
Figure 2.

Internalization of wild-type and Ub-deficient PAR1. (A–D) HeLa cells and Rat1 fibroblasts stably expressing similar amounts of cell surface PAR1 wild type (Wt) or 0K mutant were incubated with M1 anti-FLAG antibody for 1 h at 4°C such that only cell surface receptors bound antibody. Cells were then incubated in media with (A and C) or without (B and D) 100 μM TRAP for various times at 37°C. Cells were fixed, and the amount of receptor remaining at the cell surface was quantified by ELISA. The data (mean ± SD [error bars]; n = 3) are expressed as the fraction of initial cell surface antibody bound at 0 min and are representative of three separate experiments. The difference between PAR1 wild-type and 0K constitutive internalization at various times was significant (**, P < 0.01; ***, P < 0.005). (E) HeLa cells expressing PAR1 wild type, 0K mutant, or pBJ vector control were incubated in the absence or presence of 100 μM TRAP for 15 min at 37°C, fixed, permeabilized, and immunostained for PAR1 (green) using anti-FLAG polyclonal antibody and for the early endosome marker EEA1 (red) using monoclonal anti-EEA1 antibody. The cells were then imaged by confocal microscopy. Colocalization of PAR1 with EEA1 is shown as yellow in the merged image. The insets are magnifications of the boxed areas. Bars, 10 μm.
To confirm the enhanced constitutive internalization of Ub-deficient PAR1, we used a receptor-bound antibody capture assay as a measure of receptor internalization. HeLa cells expressing similar amounts of surface PAR1 wild type or 0K mutant were labeled with antibody at 4°C and incubated in the absence or presence of agonist for the indicated times at 37°C. After incubation, antibody bound to cell surface receptor was stripped, cells were lysed, and the amount of internalized receptor-bound antibody was quantified by ELISA. In wild-type PAR1–expressing cells, ∼10% of antibody that initially bound to cell surface receptor was internalized at the steady state (Fig. 3 A), whereas virtually no internalized antibody was detected in untransfected cells (not depicted). Remarkably, PAR1 0K mutant displayed an increased rate of constitutive internalization, with ∼50% of receptor-bound antibody internalized after 15 min (Fig. 3 A). Agonist caused a robust increase in PAR1 wild-type internalization, whereas activated PAR1 0K mutant internalization was comparable with constitutive internalization (Fig. 3 B). A minimal amount of constitutively internalized wild-type PAR1 was detected in endosomes by immunofluorescence microscopy, whereas PAR1 0K mutant showed substantial redistribution to endosomes in the absence of agonist (Fig. 3 C). The addition of agonist triggered comparable wild-type and Ub-deficient PAR1 internalization (Fig. 3 C). Together, these findings provide further evidence that the absence of ubiquitination increases PAR1 constitutive internalization.
Figure 3.

PAR1 C-tail lysine residue mutations enhance constitutive internalization. (A and B) HeLa cells stably expressing similar amounts of PAR1 wild type (Wt), 0K, or 4K/R mutant were labeled with M1 anti-FLAG antibody for 1 h at 4°C, washed, and incubated in media for various times at 37°C (A) or with or without 100 μM TRAP for 15 min at 37°C (B). After incubations, cells were stripped of antibody remaining bound to the cell surface, and internalized antibody bound to receptor was quantified by ELISA. Data (mean ± SD [error bars]; n = 3) are expressed as the fraction of initial cell surface receptor-bound antibody determined at 0 min and are representative of three separate experiments. The difference between PAR1 wild-type and 4K/R mutant constitutive internalization at various times was significant (***, P < 0.005). (C) HeLa cells expressing PAR1 wild type and 0K mutant labeled with polyclonal anti-FLAG antibody were either left untreated (0 min) or incubated in the absence (constitutive) or presence of 100 μM TRAP for 10 min at 37°C, fixed, and processed. Cells were immunostained for PAR1 and imaged by confocal microscopy. These images are representative of many cells examined in three independent experiments. Bars, 10 μm.
To define the lysine residues that are important for PAR1 internalization, we examined mutants in which lysines in the first or third intracytosolic loops or cytoplasmic tail (C tail) were mutated to arginines. A mutant PAR1 in which the four C-tail lysine residues were replaced with arginines (4K/R) displayed an enhanced rate of constitutive internalization similar to Ub-deficient PAR1 (Fig. 3), whereas PAR1 intracytosolic loop lysine mutants internalized like wild-type receptor (not depicted). However, the extent of constitutive internalization displayed by PAR1 4K/R was less than that observed with PAR1 0K mutant, indicating that in addition to PAR1 C-tail lysines, other lysine residues or regulatory domains may contribute to constitutive internalization. To test the role of individual C-tail lysines in PAR1 internalization, we constructed receptor mutants in which K407, K421, and K422 (3K/R) or K421 and K422 (2K/R) were converted to arginines (Fig. 4 A). The rate of PAR1 3K/R and 2K/R constitutive internalization was increased substantially compared with wild-type receptor (Fig. 4 B), suggesting that the distal K421 and K422 are important for constitutive internalization. PAR1 3K/R and 2K/R also showed marked redistribution to endosomes compared with wild-type receptor in immunofluorescence microscopy studies (Fig. 4 D), whereas agonist caused a similar increase in wild-type and mutant receptor internalization (Fig. 4, C and D). These data suggest that K421 and K422 residues are critical for the regulation of PAR1 constitutive internalization.
Figure 4.

PAR1 3K/R and 2K/R display enhanced constitutive internalization. (A–C) HeLa cells expressing PAR1 wild type (Wt) or C-tail lysine mutants as depicted in A were labeled with M1 anti-FLAG antibody and incubated in the media only (B) for various times at 37°C or with or without 100 μM TRAP (C) for 15 min at 37°C. Cells were processed, and PAR1 internalization was quantified as described in Fig. 3. The data (mean ± SD [error bars]; n = 3) shown are representative of three independent experiments. The difference between PAR1 wild-type and K/R mutant constitutive internalization at various times was significant (**, P < 0.01; ***, P < 0.005). (D) PAR1 wild type–, 3K/R–, and 2K/R–expressing HeLa cells labeled with anti-FLAG polyclonal antibody were either left untreated (0 min) or were incubated without (constitutive) or with 100 μM TRAP for 10 min at 37°C, processed, and imaged by confocal microscopy. Bars, 10 μm.
We next examined whether PAR1 C-tail lysines were important for receptor ubiquitination. HEK293 cells transiently coexpressing HA-tagged PAR1 wild type, 4K/R, and 2K/R together with FLAG-Ub and dynamin K44A were treated with or without agonist for 10 min at 37°C, and the extent of receptor ubiquitination was examined. In untreated cells, the major ubiquitinated PAR1 wild-type species migrated above ∼107 kD, whereas minor species were detected below ∼94 kD (Fig. 5, lane 1), which is consistent with multiple ubiquitinated PAR1 species. Strikingly, the addition of agonist caused a marked decrease in wild-type PAR1 ubiquitination (Fig. 5, lane 1). In contrast to wild-type receptor, basal ubiquitination of both PAR1 4K/R and 2K/R C-tail mutants was substantially reduced irrespective of agonist addition (Fig. 5, lanes 3–6). Together, these data suggest that the PAR1 C-tail K421 and K422 residues are the major sites for receptor ubiquitination.
Figure 5.

PAR1 C-tail lysine residues are major sites of ubiquitination. HEK293 cells were transiently transfected with either HA-tagged PAR1 wild type (Wt), 4K/R, 2K/R, or pcDNA together with FLAG-tagged Ub and dynamin K44A and were incubated in the absence or presence of 100 μM TRAP for 10 min at 37°C. Cells were lysed and immunoprecipitated with anti-HA polyclonal antibody, and ubiquitinated PAR1 was detected with M2 anti-FLAG conjugated to HRP. Membranes were stripped and probed with an anti-HA monoclonal antibody conjugated to HRP to detect PAR1. Cell lysates were immunoblotted for dynamin and actin as controls. These data are representative of at least three independent experiments.
Ub fused to the C tail of PAR1 0K mutant inhibits constitutive internalization
Our results suggest an important regulatory role for the ubiquitination of C-tail lysines in PAR1 internalization. Therefore, we tested whether constitutive internalization was affected when Ub was fused in frame to the C tail of the PAR1 0K Ub-deficient mutant. We used a modified Ub in which K48 was mutated to arginine (K48R), and the two terminal glycine residues (ΔGG) were deleted to avoid extensive modification of Ub and to increase the efficiency of PAR1 0K–Ub chimera expression at the cell surface. We first examined PAR1 wild-type and mutant expression in transfected HeLa cells by immunoblotting using anti-PAR1 antibodies. As expected, PAR1 wild type and 0K mutant appeared as one broad major transfection–dependent band migrating between ∼64 and 98 kD (Fig. 6 A), which is indicative of posttranslational glycosylation of the receptor protein as previously reported (Vouret-Craviari et al., 1995). The PAR1 0K–Ub chimera migrated as two high molecular weight species, which is consistent with the appearance of multiple ubiquitinated receptor species (Fig. 6 A). PAR1 0K mutant displayed an increased rate of constitutive internalization (Fig. 6 B). In contrast, the attachment of Ub to the C tail of the PAR1 0K mutant reduced the rate of constitutive internalization comparable with that observed with wild-type PAR1 (Fig. 6 B). The addition of agonist induced similar increases in PAR1 wild type, 0K mutant, and 0K–Ub chimera internalization, indicating that activated PAR1 0K–Ub internalization remained intact (Fig. 6 C). Immunofluorescence microscopy studies were consistent with a marked inhibition of PAR1 0K–Ub chimera constitution internalization compared with Ub-deficient PAR1, whereas agonist induced a comparable increase in wild-type and mutant receptor internalization (Fig. 6 D). Together, these data suggest that modification of PAR1 0K with Ub negatively regulates constitutive internalization.
Figure 6.
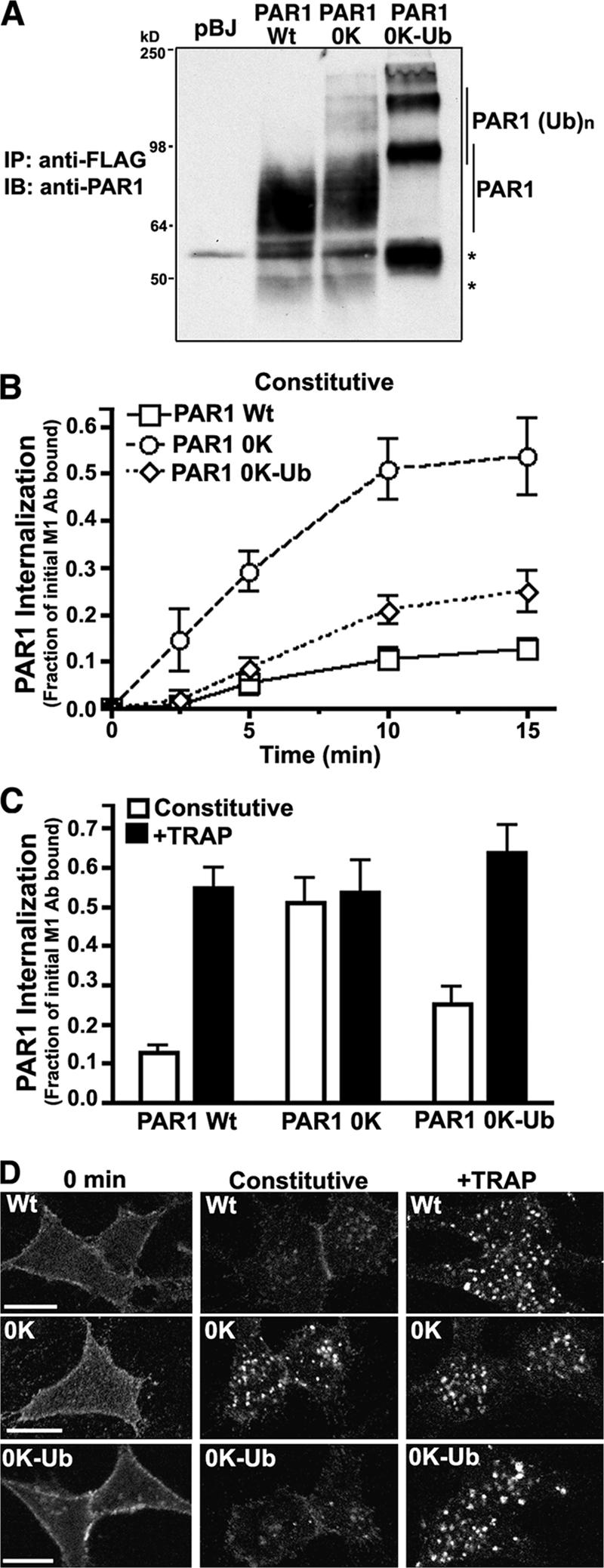
Ub fused to the C tail of PAR1 0K mutant markedly inhibits constitutive internalization. (A) HeLa cells expressing similar amounts of PAR1 wild type (Wt), 0K mutant, or 0K–Ub K48R ΔGG chimera were lysed, immunoprecipitated with M2 anti-FLAG antibody, and immunoblotted for PAR1 using a polyclonal anti-PAR1 antibody. Asterisks indicate unprocessed PAR1. These data are representative of three different experiments. (B and C) HeLa cells expressing PAR1 wild type, 0K mutant, or 0K–Ub chimera labeled with M1 anti-FLAG antibody were incubated in media only (B) for various times at 37°C or with or without 100 μM TRAP (C) for 15 min at 37°C. Cells were processed, and receptor internalization was quantified as described in Fig. 3. The data (mean ± SD [error bars]; n = 3) shown are representative of three independent experiments. (D) HeLa cells expressing PAR1 wild type, 0K mutant, or 0K–Ub chimera labeled with anti-FLAG polyclonal antibody were either left untreated (0 min) or treated in the absence (constitutive) or presence of 100 μM TRAP for 10 min at 37°C, fixed, processed, and imaged by confocal microscopy. Bars, 10 μm.
Ubiquitination of PAR1 negatively regulates constitutive internalization but not receptor recycling
The endosomal accumulation of Ub-deficient PAR1 may be caused by an increased rate of constitutive internalization as well as to an inability of the PAR1 0K mutant to recycle back to the cell surface. To address this possibility, we examined the internalization and recycling of wild-type and Ub-deficient PAR1. HeLa cells expressing PAR1 wild type or 0K mutant were labeled with antibody at 4°C and incubated for 30 min at 37°C to facilitate constitutive internalization. After incubation, surface-bound antibody was stripped, and the recovery of previously internalized receptor-bound antibody was followed for various times. An initial 30-min incubation at 37°C caused an ∼25% decrease in the amount of PAR1 wild type, whereas a substantially greater amount (∼50%) of PAR1 0K mutant was initially lost from the cell surface (Fig. 7, t1), which is consistent with an increased rate of constitutive internalization. In both PAR1 wild-type– and 0K mutant–expressing cells, a comparable amount of previously internalized receptor recycled back to the cell surface (Fig. 7). These results are consistent with the previously described extent of constitutive internalization and recycling of PAR1 (Trejo and Coughlin, 1999) and indicate that PAR1 recycling is not attenuated with Ub-deficient PAR1.
Figure 7.

Ub-deficient PAR1 mutant displays normal recycling. HeLa cells expressing PAR1 wild type (Wt) or 0K mutant labeled with M1 anti-FLAG antibody were incubated in media without agonist for 30 min at 37°C to facilitate constitutive internalization. Cells were stripped of surface-bound antibody, and the amount of receptor-bound antibody reappearing at the cell surface was measured after various recovery times. Surface receptor-bound antibody was quantitated after the initial 4°C labeling (t0), after 30-min incubation at 37°C in the absence of agonist (t1), and after the indicated times of recovery. The data (mean ± SD [error bars]; n = 3) are expressed as follows: (amount of receptor-bound antibody at the indicated time points)/(amount of receptor-bound antibody at t0). Data are representative of three separate experiments.
Internalization of Ub-deficient PAR1 is clathrin and dynamin dependent
We next examined whether Ub-deficient PAR1 internalized through a clathrin- and dynamin-dependent pathway like wild-type receptor (Trejo et al., 2000). HeLa cells expressing PAR1 wild type or 0K mutant were transiently transfected with GFP-tagged dynamin wild type, K44A, or vector, and the amounts of cell surface receptor at steady state were measured. In PAR1 wild-type–expressing cells, neither dynamin wild-type nor K44A mutant expression affected the amounts of cell surface receptor expression when compared with vector (Fig. 8 A). The amount of surface PAR1 0K mutant was also comparable in dynamin wild-type and vector-transfected cells. In contrast, the coexpression of dynamin K44A increased PAR1 0K expression detected on the cell surface compared with wild-type receptor or vector (Fig. 8 A). In immunofluorescence microscopy studies, the constitutive internalization of PAR1 wild type and 0K mutant were virtually abolished in K44A-expressing cells, whereas in adjacent untransfected cells, PAR1-containing endosomes were clearly evident (Fig. 8 C, arrowheads). Internalization of activated PAR1 wild type and 0K mutant were similarly inhibited by dynamin K44A (Fig. 8 C, arrowheads), suggesting that wild-type and Ub-deficient PAR1 internalization are dynamin dependent.
Figure 8.

Internalization of Ub-deficient PAR1 is clathrin and dynamin dependent. (A and B) PAR1 wild type (Wt)– and 0K mutant–expressing HeLa cells were transiently transfected with GFP-tagged dynamin wild type, K44A mutant, or pcDNA (A) or with 50 nM siRNA targeted to either clathrin heavy chain (CHC) or nonspecific (ns) mRNA sequences (B). In B, the inset confirms the loss of clathrin protein in CHC siRNA–transfected cells as detected by immunoblotting. After transfection, the amount of PAR1 residing on the cell surface at steady state was measured by ELISA. The data (mean ± SD [error bars]; n = 3) are expressed as the fold increase over surface PAR1 expression detected in vector or ns-siRNA–transfected cells and are representative of three independent experiments. A significant difference in surface expression between PAR1 wild type and 0K mutant was detected in K44A mutant–transfected cells and CHC siRNA–transfected cells (**, P < 0.01). (C and D) PAR1 wild type– and 0K mutant–expressing HeLa cells transiently transfected with GFP-tagged K44A mutant (C) or 50 nM ns- or CHC siRNA (D) were incubated in the absence (constitutive) or presence of 100 μM TRAP for 10 min at 37°C. Cells were fixed, immunostained for PAR1 or clathrin, and imaged by confocal microscopy. These images are representative of many cells examined in three separate experiments. In cells not expressing dynamin K44A, PAR1 wild type and 0K mutant localize to the cell surface and endocytic vesicles in the absence of agonist (arrowheads), whereas in agonist-treated cells, PAR1 wild type and 0K are retained predominantly on the cell surface in dynamin K44A–expressing cells (arrowheads). Bars, 10 μm.
To determine the role of clathrin in Ub-deficient PAR1 internalization, we used siRNA targeting the clathrin heavy chain (CHC) to deplete HeLa cells of endogenous clathrin. The expression of CHC was considerably decreased in cells transiently transfected with siRNA specifically targeting CHC compared with nonspecific control (Fig. 8 B, inset). In control siRNA-transfected cells, PAR1 wild-type and Ub-deficient 0K mutant surface levels were comparable. In contrast, the amount of PAR1 0K mutant surface expression was considerably increased in CHC siRNA–transfected cells compared with wild-type receptor, which is consistent with the inhibition of Ub-deficient PAR1 constitutive internalization (Fig. 8 B). Constitutive and agonist-induced internalization of wild-type and Ub-deficient PAR1 was virtually ablated in CHC siRNA–transfected cells compared with control cells as assessed by immunofluorescence microscopy (Fig. 8 D). Together, these findings are consistent with a clathrin- and dynamin-dependent regulation of wild-type and Ub-deficient PAR1 internalization.
Ubiquitination specifies a distinct clathrin adaptor requirement for activated PAR1 internalization
We recently reported that the clathrin adaptor AP2 function is critical for constitutive but not agonist-induced internalization of wild-type PAR1 (Paing et al., 2006). To assess AP2 function in Ub-deficient PAR1 internalization, we used siRNA targeting the μ2 subunit to deplete cells of the endogenous AP2 complex (Fig. 9, A [inset] and C). In PAR1 wild-type–expressing cells, constitutive internalization was completely inhibited in μ2-siRNA–transfected cells compared with nonspecific (ns) siRNA control cells (Fig. 9 A). The PAR1 0K mutant displayed enhanced constitutive internalization in ns-siRNA control cells, which was virtually abolished in μ2-siRNA–transfected cells (Fig. 9 B), strongly suggesting a critical role for AP2 in both wild-type and Ub-deficient PAR1 constitutive internalization. In ns-siRNA control cells, agonist caused the substantial internalization of wild-type PAR1 that was partially diminished in μ2-siRNA–transfected cells (Fig. 9 A), indicating that even in the absence of AP2, activated wild-type PAR1 is capable of internalization. Remarkably, activated PAR1 0K mutant internalization was virtually abolished in μ2-siRNA–transfected cells, suggesting that AP2 function is critical for agonist-induced Ub-deficient PAR1 internalization (Fig. 9 B). Immunofluorescence microscopy experiments of PAR1 0K mutant–expressing cells were consistent with a critical role for AP2 in activated Ub-deficient receptor internalization. In the absence of agonist exposure, PAR1 wild type and 0K mutant failed to redistribute to endosomes in AP2-depleted cells (Fig. 9 C). In contrast, agonist peptide caused a marked increase in wild-type PAR1 internalization in μ2-siRNA–transfected cells comparable with siRNA control cells (Fig. 9 C), which is consistent with an AP2-independent pathway for activated wild-type PAR1 internalization. In contrast, activated PAR1 0K mutant internalization was markedly inhibited in μ2-siRNA–transfected cells (Fig. 9 C), suggesting a critical role for AP2 in agonist-promoted Ub-deficient PAR1 internalization. Together, these data suggest a novel function for ubiquitination in specifying a distinct clathrin adaptor requirement for activated PAR1 internalization.
Figure 9.

AP2 function is critical for constitutive and agonist-induced internalization of Ub-deficient PAR1. (A and B) HeLa cells expressing PAR1 wild type (Wt; A) or 0K mutant (B) were transiently transfected with 50 nM siRNA targeted to either μ2 or nonspecific (ns) mRNA sequences. Cells prelabeled with M1 anti-FLAG antibody were then incubated in the absence (constitutive) or presence of 100 μM TRAP for 10 min at 37°C, and the amount of receptor remaining on the cell surface was quantified by ELISA. The data (mean ± SD [error bars]; n = 3) shown are representative of three independent experiments. In A, the inset confirms the loss of AP2 in μ2-siRNA–transfected cells, whereas actin expression was unaffected. The difference between constitutive and agonist-induced PAR1 internalization in ns-siRNA– and μ2-siRNA–transfected cells was significant (**, P < 0.01). (C) HeLa cells expressing PAR1 wild type or 0K mutant transiently transfected with 50 nM of ns- or μ2-siRNA were incubated in the absence (constitutive) or presence of 100 μM TRAP for 10 min at 37°C. Cells were fixed, processed, and immunostained for PAR1 or β2-adaptin and imaged by confocal microscopy. The images shown are representative of many cells observed in at least three different experiments. Bars, 10 μm.
Ubiquitination of PAR1 is not required for agonist-induced lysosomal degradation
We next determined whether ubiquitination functions in agonist-induced PAR1 lysosomal degradation by comparing the extent of wild-type versus Ub-deficient PAR1 degradation after prolonged agonist exposure. HeLa cells stably expressing similar amounts of surface PAR1 wild type or 0K mutant were incubated with or without agonist for 90 min at 37°C, and the amount of receptor remaining was then measured. Remarkably, the extent of activated Ub-deficient PAR1 degradation was comparable with that observed with wild-type receptor (Fig. 10 A), suggesting that PAR1 ubiquitination is not required for lysosomal degradation. To confirm Ub-independent degradation of PAR1, we examined receptor degradation in Rat1 fibroblasts. PAR1 wild-type and 0K mutant exhibited a comparable, ∼60% loss of receptor protein after prolonged agonist exposure (Fig. 10 B), which is consistent with the extent of PAR1 degradation previously reported in these cell types (Trejo and Coughlin, 1999; Trejo et al., 2000). These findings suggest a Ub-independent pathway for agonist-induced PAR1 lysosomal sorting and degradation.
Figure 10.

Agonist-induced degradation of Ub-deficient PAR1. (A and B) PAR1 wild type (Wt)– and 0K mutant–expressing HeLa cells (A) and Rat1 fibroblasts (B) were incubated in the absence or presence of 100 μM TRAP for either 90 or 60 min at 37°C, respectively. Cells were lysed and immunoprecipitated with M2 anti-FLAG antibody, and the amount of PAR1 remaining was detected by immunoblotting. Untransfected (UT) HeLa cells or Rat1 fibroblast controls are shown in adjacent lanes. Asterisks indicate unprocessed PAR1. The data shown (mean ± SEM [error bars]) in the bar graphs are expressed as the fraction of PAR1 remaining compared with untreated cells and represent the mean of three separate experiments.
Discussion
In this study, we have defined a novel function for ubiquitination in the negative regulation of PAR1 constitutive internalization and in specifying a distinct clathrin adaptor requirement for activated receptor internalization. We demonstrate that PAR1 is ubiquitinated under basal conditions and deubiquitinated after activation. The ubiquitination of PAR1 appears to negatively regulate constitutive internalization. This is supported by our findings that the constitutive internalization of PAR1 is markedly increased in the absence of ubiquitination and is inhibited by the fusion of Ub to the C tail of Ub-deficient PAR1. Several GPCRs, including the yeast Ste2 and Ste3 receptors, have been reported to be basally ubiquitinated at the plasma membrane, similar to PAR1. However, in contrast to PAR1, the ubiquitination of Ste2 and Ste3 receptors promotes constitutive and agonist-induced internalization (Hicke and Riezman, 1996; Terrell et al., 1998). The mammalian platelet-activating receptor GPCR has also been shown to be basally ubiquitinated, but the role of ubiquitination in receptor internalization was not examined (Dupre et al., 2003). Our studies also suggest that the ubiquitination of PAR1 specifies a distinct clathrin adaptor requirement for activated receptor internalization that is not critically dependent on AP2. We found that AP2 function was required for the constitutive internalization of both wild-type and Ub-deficient PAR1. Strikingly, AP2 function was also critical for the internalization of activated Ub-deficient PAR1, whereas the internalization of activated wild-type receptor occurred in the absence of AP2. We also demonstrate that ubiquitination is not essential for agonist-promoted PAR1 lysosomal degradation. Thus, our findings with PAR1 suggest a novel function for ubiquitination in the regulation of GPCR trafficking in mammalian cells.
The ubiquitination of PAR1 is likely a highly dynamic and reversible process, and, under basal conditions, the receptor probably exists as a ubiquitinated and deubiquitinated species. Our findings raise the intriguing possibility that the ubiquitination of PAR1 might affect the ability of AP2 to regulate constitutive internalization. We previously reported that a PAR1 tyrosine-based motif (Y420KKL423) localized at the extreme C terminus directly binds to the μ2 subunit of AP2 using surface plasmon resonance (Paing et al., 2006). Moreover, both the μ2 subunit and the tyrosine-based motif are essential for promoting PAR1 constitutive internalization in multiple cell types. In this study, we show that the highly conserved K421 and K422 residues located within the PAR1 C-tail tyrosine-based motif are the major sites of ubiquitination and negatively regulate constitutive internalization, suggesting that receptor ubiquitination at these sites might affect AP2 binding. In addition, the fusion of Ub to the C tail of Ub-deficient PAR1 0K mutant places the Ub moiety within five residues of the tyrosine-based motif, which could also affect AP2 binding and, thereby, diminish constitutive internalization. However, the low micromolar affinity binding of the PAR1 C tail with the μ2 subunit is typical of μ2 subunit weak interactions with proteins bearing tyrosine-based motifs and has precluded our ability to directly test the role of ubiquitination in PAR1 and AP2 interaction in cells using coimmunoprecipitations or pull downs. Thus, we cannot exclude the possibility that other ubiquitination sites or regulatory domains could also contribute to the regulation of PAR1 constitutive internalization. It is also possible that the ubiquitinated PAR1 conformation is simply not compatible with AP2 interaction or that ubiquitinated PAR1 is bound to another protein important for localization at the plasma membrane. Regardless, our findings suggest that PAR1 ubiquitination provides a mechanism to retain the majority of the receptor at the cell surface so that it is readily available for proteolytic activation by extracellular proteases.
PAR1 ubiquitination also appears to have a critical role in specifying a distinct clathrin adaptor requirement for activated receptor internalization. Several clathrin adaptors, including epsins and eps15, contain UBDs that recognize ubiquitinated cargo and facilitate clathrin-dependent internalization. Interestingly, the yeast homologues of the mammalian epsins Ent1 and Ent2 contain UBDs and facilitate the endocytosis of ubiquitinated Ste2 receptor (Chen et al., 1998; Shih et al., 2002). In mammalian cells, epsin is ubiquitinated under basal conditions, which may prevent its interaction with AP2, clathrin, and membrane lipids, and the deubiquitination of epsin appears to enhance its endocytic activity (Chen and De Camilli, 2005). Ub may also negatively regulate epsin function by binding to its UBDs intramolecularly, similar to other endocytic adaptor proteins (Hoeller et al., 2006). In Drosophila melanogaster, the deubiquitinating enzyme Fat facets/USP9X regulates Delta/Notch receptor internalization by deubiquitinating Liquid facets, a homologue of epsin, which is consistent with a function for epsin deubiquitination in the regulation of receptor endocytosis (Cadavid et al., 2000; Chen et al., 2002). In contrast, the ubiquitination of arrestins is critical for the internalization of certain GPCRs (Shenoy et al., 2001). However, arrestins are not essential for PAR1 internalization (Paing et al., 2002), suggesting that activated PAR1 internalization may require epsins similar to the Ste2 receptor. However, whether epsin and/or Ub regulation of epsin is important for PAR1 internalization remains to be determined.
A role for ubiquitination in mammalian GPCR lysosomal sorting and degradation has been demonstrated. Agonist-induced ubiquitination of β2AR and CXCR4 is critical for lysosomal degradation but is not required for internalization (Marchese and Benovic, 2001; Shenoy et al., 2001). The Ub moiety on CXCR4 is thought to interact with UBDs in some endocytic adaptor proteins, such as hepatocyte growth factor–regulated kinase substrate (Hrs), to be efficiently degraded in the lysosome (Marchese et al., 2003). Hrs interacts with Tsg101 (tumor suppressor gene product 101) and promotes the assembly of a multiprotein ESCRT (endosomal sorting complex required for transport) complex that binds and sorts ubiquitinated cargo into the involuting membrane of multivesicular endosomes in a highly coordinated manner (Raiborg et al., 2003). In contrast to β2AR and CXCR4, the ubiquitination of PAR1 is not required for agonist-induced lysosomal degradation because Ub-deficient PAR1 is degraded comparably with wild-type receptor in HeLa cells and Rat1 fibroblasts. A δ opioid receptor mutant lacking all intracytosolic lysines was also shown to undergo efficient agonist-induced degradation, indicating that lysosomal degradation of certain GPCRs occurs independently of ubiquitination (Tanowitz and Von Zastrow, 2002). We show that after activation, PAR1 is deubiquitinated, suggesting that deubiquitinated rather than ubiquitinated receptor transits through the endocytic sorting pathway to lysosomes for degradation (Fig. 10). Moreover, we recently reported that agonist-induced PAR1 lysosomal degradation is independent of Hrs and Tsg101 but requires sorting nexin 1 (Gullapalli et al., 2006), which is consistent with a Ub-independent PAR1 lysosomal sorting pathway. These data, in conjunction with the ability of the Ub-deficient PAR1 mutant to be efficiently degraded, strongly suggest that the conventional Ub-dependent ESCRT-mediated pathway is not required for agonist-induced PAR1 lysosomal sorting and degradation. However, we cannot exclude the possibility that the ubiquitination of PAR1 has a role in basal turnover of the receptor. The efficient trafficking of proteolytically activated PAR1 to lysosomes is essential for the termination of receptor signaling (Trejo et al., 1998); thus, further delineation of the lysosomal sorting pathway of activated PAR1 is important.
Our studies reveal a novel function for ubiquitination in the negative regulation of PAR1 constitutive internalization and in specifying a distinct clathrin adaptor requirement for activated receptor internalization. PAR1 is uniquely activated by proteolytic cleavage that results in irreversible activation, unlike normal ligand-activated GPCRs. Thus, rapid desensitization and receptor trafficking tightly regulate PAR1 signaling. We have shown that PAR1 trafficking does not require arrestins and is essential for the disposal of irreversibly activated receptor and for replenishing the cell surface with uncleaved receptor after protease exposure. The novel regulation of PAR1 internalization by ubiquitination has a critical role in these distinct endocytic pathways. Interestingly, the regulation of PAR1 internalization by ubiquitination is not observed with all PARs because the ubiquitination of PAR2, a second protease-activated GPCR, functions in lysosomal degradation but not in receptor internalization (Jacob et al., 2005). Unlike PAR1, arrestins are required for PAR2 internalization (Stalheim et al., 2005). However, whether other GPCRs that do not require arrestins for endocytic sorting are similarly regulated by ubiquitination remains to be determined. Our studies provide new insight into novel mechanisms by which ubiquitination functions in the endocytic sorting of GPCRs in mammalian cells. The challenge is to now identify the physiologically relevant Ub ligases and deubiquitinating enzymes that function in PAR1 trafficking.
Materials and methods
Reagents and antibodies
Human α-thrombin was obtained from Enzyme Research Laboratories. Thrombin receptor–activating peptides (TRAPs), SFLLRN, and TFLLRNPNDK were synthesized as the carboxyl amide and purified by reverse-phase high pressure liquid chromatography (University of North Carolina Peptide Facility, Chapel Hill, NC).
Monoclonal M1 and M2 anti-FLAG and polyclonal anti-FLAG antibodies and the anti–β-actin antibody were purchased from Sigma-Aldrich. Monoclonal anti-HA antibody conjugated to HRP was obtained from Roche, and polyclonal anti-HA was obtained from Covance. Anti-PAR1 rabbit polyclonal antibody was previously described (Paing et al., 2006). The anti-AP50 (μ2), anti–β2 adaptin, anti–early endosomal antigen-1 (EEA1), and anti-CHC monoclonal antibodies were purchased from BD Biosciences. Antidynamin monoclonal antibody was obtained from Santa Cruz Biotechnology, Inc. HRP-conjugated goat anti–mouse and goat anti–rabbit antibodies were purchased from Bio-Rad Laboratories. AlexaFluor488 and -594-conjugated anti–mouse and anti–rabbit antibodies were obtained from Invitrogen.
cDNAs and cell lines
A human PAR1 cDNA containing an N-terminal FLAG or HA epitope was used to generate mutants. Mutations were introduced by site-directed mutagenesis using the QuikChange Mutagenesis kit (Stratagene) and confirmed by dideoxy sequencing. PAR1 0K and K/R mutants were generated by replacing intracytosolic lysine (K) residues with arginine (R). A PAR1 0K mutant with Ub fused in frame to the C tail was generated as follows. A Pm1I site was introduced at the 3′ end of the PAR1 0K C tail and positioned such that the PmlI sequence, CACGTG, coincided with the native stop codon. A SacII site was introduced in the 3′ untranslated region 21 bp from the PmlI site. PCR amplification was then used to generate the Ub cDNA fragment with the 5′ Pm1I site and 3′ SacII sites. A 222-bp PmlI–SacII fragment encoding Ub containing a K48 to R48 mutation and glycine deletions was then ligated in frame using PAR1 0K 3′ end compatible sites. PAR1 and Ub coding regions were separated by a spacer sequence, HVV. Insertion of Ub in frame after the PAR1 0K C tail was confirmed by dideoxy sequencing.
HeLa cells and Rat1 fibroblasts stably expressing PAR1 wild type and mutants were generated and maintained as previously described (Trejo et al., 1998, 2000). HeLa cells were transiently transfected with a total plasmid amount of 0.4 μg per 24 wells, 0.8 μg per 12 wells, and 2 μg per six wells using LipofectAMINE reagent (Invitrogen) according to the manufacturer's instructions and were assayed 48 h after transfection. HEK293 cells plated at ∼1 × 106 cells per 10-cm2 dish were transiently transfected with a total of 7.4 μg of plasmids (consisting of 5 μg of PAR1, 0.4 μg of tagged Ub, and 2 μg of dynamin K44A) using FuGene-6 reagent or LipofectAMINE as described previously (Marchese and Benovic, 2001) and according to the manufacturer's instructions. Cells were then split into 6-cm plates and assayed 48 h after transfection.
siRNAs
HeLa cells were transiently transfected with 50 nM of ns or μ2-specific siRNAs using LipofectAMINE 2000 according to the manufacturer's instructions. The μ2-siRNA targeting the mRNA sequence 5′-GTGGATGCCTTTCGGGTCA-3′ and the ns-siRNA 5′-CTACGTCCAGGAGCGCACC-3′ were previously described (Paing et al., 2006). The CHC siRNA targeting the mRNA sequence 5′-GCAATGAGCTGTTTGAAGA-3′ was used at 50 nM and was previously described (Huang et al., 2004). All siRNAs were synthesized by Dharmacon.
Phosphoinositide hydrolysis
HeLa cells stably expressing PAR1 wild type or 0K mutant were labeled with 1.0 μCi/ml myo-[3H]inositol (American Radiolabeled Chemicals), and accumulated [3H]IPs were measured as previously described (Paing et al., 2002).
Immunofluorescence confocal microscopy
HeLa cells stably expressing PAR1 wild type or mutants were processed, fixed, permeabilized, and immunostained with species-specific secondary antibodies conjugated to AlexaFluor488 or -594 and were mounted in FluorSave reagent (Calbiochem) and imaged by confocal microscopy as we previously described (Paing et al., 2002). Images were acquired using a laser-scanning confocal imaging system (FluoView 300; Olympus) configured with a fluorescence microscope (IX70; Olympus) fitted with a planApo 60× NA 1.4 oil objective (Olympus). Confocal images (x-y section at 0.28 μm) were collected sequentially at 800 × 600 resolution with 2× optical zoom using FluoView software at room temperature. The final composite image was created using Photoshop 7.0 (Adobe).
Internalization and recycling assays
Constitutive and agonist-induced PAR1 internalization were assessed using our previously described assays for loss of surface receptor and receptor-bound antibody uptake (Paing et al., 2002, 2004). PAR1 recycling was measured as we previously described (Trejo and Coughlin, 1999).
PAR1 ubiquitination and degradation
HEK293 cells transiently transfected with FLAG-tagged PAR1 wild type or mutants, HA- or FLAG-tagged Ub, and dynamin K44A were incubated with or without agonists and lysed in buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, pH 8.0, 0.5% (wt/vol) sodium deoxycholate, 0.1% (vol/vol) NP-40, 0.1% (wt/vol) SDS, 100 μM sodium orthovanadate, 20 mM N-ethylmaleimide, and protease inhibitor tablet (Roche). Equivalent amounts of protein lysates were then immunoprecipitated with M2 anti-FLAG or anti-HA antibody. Immunoprecipitates were resolved by SDS-PAGE, transferred to nitrocellulose membranes, and probed with an anti-HA or M2 anti-FLAG antibody conjugated to HRP. Blots were then stripped and probed with a polyclonal anti-FLAG or -HA antibody to detect PAR1. Immunoblots were developed with ECL (GE Healthcare) and imaged by autoradiography. PAR1 degradation was determined in HeLa cells and Rat1 fibroblast cells stably expressing the PAR1 wild type or mutants as previously described (Trejo and Coughlin, 1999; Trejo et al., 2000).
Data analysis
Data were analyzed using Prism 4.0 software (GraphPad), and statistical significance was determined using InStat 3.0 (GraphPad). Group comparisons were made using an unpaired t test.
Acknowledgments
We thank members of the J. Trejo, T.K. Harden, and R. Nicholas laboratories for comments and helpful discussions.
This work was supported by National Institutes of Health grant HL073328 (to J. Trejo), an American Heart Association Established Investigator Award (to J. Trejo), and American Heart Association Scientist Development grant 0530185N (to A. Marchese). B.L. Wolfe is supported by an American Heart Association Predoctoral Fellowship Award.
Abbreviations used in this paper: AP2, adaptor protein complex-2; β2AR, β2-adrenergic receptor; CHC, clathrin heavy chain; C tail, cytoplasmic tail; CXCR4, chemokine receptor 4; EEA1, early endosomal antigen-1; GPCR, G protein–coupled receptor; Hrs, hepatocyte growth factor–regulated kinase substrate; IP, inositol phosphate; PAR, protease-activated receptor; TRAP, thrombin receptor–activating peptide; Ub, ubiquitin; UBD, Ub-binding domain.
References
- Arora, P., T.K. Ricks, and J. Trejo. 2007. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J. Cell Sci. 120:921–928. [DOI] [PubMed] [Google Scholar]
- Cadavid, A.L., A. Ginzel, and J.A. Fischer. 2000. The function of the Drosophila fat facets deubiquitinating enzyme in limiting photoreceptor cell number is intimately associated with endocytosis. Development. 127:1727–1736. [DOI] [PubMed] [Google Scholar]
- Chen, C.H., M.M. Paing, and J. Trejo. 2004. Termination of protease-activated receptor-1 signaling by beta-arrestins is independent of receptor phosphorylation. J. Biol. Chem. 279:10020–10031. [DOI] [PubMed] [Google Scholar]
- Chen, H., and P. De Camilli. 2005. The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proc. Natl. Acad. Sci. USA. 102:2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H., S. Fre, V.I. Slepnev, M.R. Capua, K. Takei, M.H. Butler, P.P. Di Fiore, and P. De Camilli. 1998. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 394:793–797. [DOI] [PubMed] [Google Scholar]
- Chen, X., B. Zhang, and J.A. Fischer. 2002. A specific protein substrate for a deubiquitinating enzyme: liquid facets is the substrate of Fat facets. Genes Dev. 16:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin, S.R. 2005. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3:1800–1814. [DOI] [PubMed] [Google Scholar]
- Dupre, D.J., Z. Chen, C. Le Gouill, C. Theriault, J.L. Parent, M. Rola-Pleszczynski, and J. Stankova. 2003. Trafficking, ubiquitination, and down-regulation of the human platelet-activating factor receptor. J. Biol. Chem. 278:48228–48235. [DOI] [PubMed] [Google Scholar]
- Goodman, O.B., Jr., J.G. Krupnick, F. Santini, V.V. Gurevich, R.B. Penn, A.W. Gagnon, J.H. Keen, and J.L. Benovic. 1996. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 383:447–450. [DOI] [PubMed] [Google Scholar]
- Gullapalli, A., B.L. Wolfe, C.T. Griffin, T. Magnuson, and J. Trejo. 2006. An essential role for SNX1 in lysosomal sorting of protease-activated receptor-1: evidence for retromer-, Hrs-, and Tsg101-independent functions of sorting nexins. Mol. Biol. Cell. 17:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein, L., K. Ishii, S.R. Coughlin, and B.K. Kobilka. 1994. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. J. Biol. Chem. 269:27719–27726. [PubMed] [Google Scholar]
- Hicke, L., and R. Dunn. 2003. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 19:141–172. [DOI] [PubMed] [Google Scholar]
- Hicke, L., and H. Riezman. 1996. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell. 84:277–287. [DOI] [PubMed] [Google Scholar]
- Hoeller, D., N. Crosetto, B. Blagoev, C. Raiborg, R. Tikkanen, S. Wagner, K. Kowanetz, R. Breitling, M. Mann, H. Stenmark, and I. Dikic. 2006. Regulation of ubiquitin-binding proteins by monoubiquitination. Nat. Cell Biol. 8:163–169. [DOI] [PubMed] [Google Scholar]
- Hoxie, J.A., M. Ahuja, E. Belmonte, S. Pizarro, R. Parton, and L.F. Brass. 1993. Internalization and recycling of activated thrombin receptors. J. Biol. Chem. 268:13756–13763. [PubMed] [Google Scholar]
- Huang, F., A. Khvorova, W. Marshall, and A. Sorkin. 2004. Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J. Biol. Chem. 279:16657–16661. [DOI] [PubMed] [Google Scholar]
- Jacob, C., G.S. Cottrell, D. Gehringer, F. Schmidlin, E.F. Grady, and N.W. Bunnett. 2005. c-Cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 280:16076–16087. [DOI] [PubMed] [Google Scholar]
- Laporte, S.A., R.H. Oakley, J. Zhang, J.A. Holt, S.S. Ferguson, M.G. Caron, and L.S. Barak. 1999. The beta2-adrenergic receptor/betaarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. USA. 96:3712–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese, A., and J.L. Benovic. 2001. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J. Biol. Chem. 276:45509–45512. [DOI] [PubMed] [Google Scholar]
- Marchese, A., C. Raiborg, F. Santini, J.H. Keen, H. Stenmark, and J.L. Benovic. 2003. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev. Cell. 5:709–722. [DOI] [PubMed] [Google Scholar]
- Paing, M.M., A.B. Stutts, T.A. Kohout, R.J. Lefkowitz, and J. Trejo. 2002. beta-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 277:1292–1300. [DOI] [PubMed] [Google Scholar]
- Paing, M.M., B.R. Temple, and J. Trejo. 2004. A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. J. Biol. Chem. 279:21938–21947. [DOI] [PubMed] [Google Scholar]
- Paing, M.M., C.A. Johnston, D.P. Siderovski, and J. Trejo. 2006. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell. Biol. 26:3231–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg, C., T.E. Rusten, and H. Stenmark. 2003. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 15:446–455. [DOI] [PubMed] [Google Scholar]
- Shapiro, M.J., J. Trejo, D. Zeng, and S.R. Coughlin. 1996. Role of the thrombin receptor's cytoplasmic tail in intracellular trafficking. Distinct determinants for agonist-triggered versus tonic internalization and intracellular localization. J. Biol. Chem. 271:32874–32880. [DOI] [PubMed] [Google Scholar]
- Shenoy, S.K., P.H. McDonald, T.A. Kohout, and R.J. Lefkowitz. 2001. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 294:1307–1313. [DOI] [PubMed] [Google Scholar]
- Shih, S.C., D.J. Katzmann, J.D. Schnell, M. Sutanto, S.D. Emr, and L. Hicke. 2002. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat. Cell Biol. 4:389–393. [DOI] [PubMed] [Google Scholar]
- Stalheim, L., Y. Ding, A. Gullapalli, M.M. Paing, B.L. Wolfe, D.R. Morris, and J. Trejo. 2005. Multiple independent functions of arrestins in regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 67:78–87. [DOI] [PubMed] [Google Scholar]
- Tanowitz, M., and M. Von Zastrow. 2002. Ubiquitination-independent trafficking of G protein-coupled receptors to lysosomes. J. Biol. Chem. 277:50219–50222. [DOI] [PubMed] [Google Scholar]
- Terrell, J., S. Shih, R. Dunn, and L. Hicke. 1998. A function for monoubiquitination in the internalization of a G protein-coupled receptor. Mol. Cell. 1:193–202. [DOI] [PubMed] [Google Scholar]
- Trejo, J., and S.R. Coughlin. 1999. The cytoplasmic tails of protease-activated receptor-1 and substance P receptor specify sorting to lysosomes versus recycling. J. Biol. Chem. 274:2216–2224. [DOI] [PubMed] [Google Scholar]
- Trejo, J., S.R. Hammes, and S.R. Coughlin. 1998. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc. Natl. Acad. Sci. USA. 95:13698–13702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo, J., Y. Altschuler, H.W. Fu, K.E. Mostov, and S.R. Coughlin. 2000. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J. Biol. Chem. 275:31255–31265. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari, V., D. Grall, J.C. Chambard, U.B. Rasmussen, J. Pouyssegur, and E. Van Obberghen-Schilling. 1995. Post-translational and activation-dependent modifications of the G protein-coupled thrombin receptor. J. Biol. Chem. 270:8367–8372. [DOI] [PubMed] [Google Scholar]
- Vu, T.K., D.T. Hung, V.I. Wheaton, and S.R. Coughlin. 1991. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 64:1057–1068. [DOI] [PubMed] [Google Scholar]