

Abstract
Long-term memory requires transcriptional regulation by a combination of positive and negative transcription factors. Aplysia activating factor (ApAF) is known to be a positive transcription factor that forms heterodimers with ApC/EBP and ApCREB2. How these heterodimers are regulated and how they participate in the consolidation of long-term facilitation (LTF) has not, however, been characterized. We found that the functional activation of ApAF required phosphorylation of ApAF by PKA on Ser-266. In addition, ApAF lowered the threshold of LTF by forming a heterodimer with ApCREB2. Moreover, once activated by PKA, the ApAF–ApC/EBP heterodimer transactivates enhancer response element–containing genes and can induce LTF in the absence of CRE- and CREB-mediated gene expression. Collectively, these results suggest that PKA-activated ApAF–ApC/EBP heterodimer is a core downstream effector of ApCREB in the consolidation of LTF.
Introduction
The formation of long-term memory requires both new RNA and protein synthesis, whereas short-term memory requires only covalent modifications of constitutively expressed preexisting proteins (Barondes and Cohen, 1968; Rainbow, 1979; Montarolo et al., 1986; Patterson et al., 1989; Sweatt and Kandel, 1989; Pedreira et al., 1995). On the cellular level, transcriptional regulation is thought to be the starting point for a series of biological amplifications, which are steps necessary for the induction and maintenance of long-term facilitation (LTF). Thus, during consolidation of LTF, a cascade of gene activation is tightly regulated by various transcription factors, including positive and negative regulators (Kandel, 2001).
The core molecular features of the transcriptional regulation involved in long-term memory seem to be evolutionarily conserved in Aplysia, Drosophila melanogaster, and the mouse (Mayford and Kandel, 1999). A growing body of evidence indicates that gene regulation by different combinations of transcriptional factors may be involved in specific forms of long-term memory (Herdegen and Leah, 1998; Hinoi et al., 2002). Moreover, the activation and inactivation of transcription factors by various kinases can regulate the transactivational potency of preexisting transcription factors and the recruitment of other factors. For example, the activation of CREB by PKA, MAPK, or CaMKIV is particularly important for its contribution to transcriptional activation and long-term memory in Aplysia and in the mammalian hippocampus (Kaang et al., 1993; Bartsch et al., 1998; West et al., 2001; Pittenger et al., 2002).
In the marine snail Aplysia, the molecular mechanisms of long-term memory have been extensively studied in the sensory neuron–to–motor neuron synapses of the gill-withdrawal reflex. Five pulses of 5-hydroxytryptamine (5-HT) spaced at 15 min produce LTF that lasts >24 h and depends on transcription and translation. These pulses of 5-HT up-regulate the levels of cAMP within the sensory cell via G protein–coupled receptors and activate protein kinase A (PKA) and MAPK (Brunelli et al., 1976; Kandel et al., 1976; Castellucci et al., 1980; Byrne and Kandel, 1996; Martin et al., 1997; Dyer et al., 2003; Ormond et al., 2004). Both kinases then translocate into the nucleus, where they can activate transcription factors expressed in sensory neurons, such as ApCREB1 and ApCREB2. PKA can activate the transcriptional activator ApCREB1 (Kaang et al., 1993; Bartsch et al., 1998) and relieve the repression of ApCREB1-mediated transcription by the repressor ApCREB2 (Bartsch et al., 1995; Lee et al., 2003). PKA-mediated activation of ApCREB1 can recruit CBP, and this leads to the activation of immediate-early genes, such as ApC/EBP, which are involved in the gene expression required for the consolidation and stabilization of LTF (Alberini et al., 1994; Lee et al., 2001).
Although several transcription factors are important for the consolidation of LTF, phosphorylated ApCREB1 is sufficient (Bartsch et al., 1998). This implies that the CRE-driven downstream genes activated by CREB should also be essential for the consolidation of LTF. Indeed, induction of ApC/EBP by activated ApCREB1 is necessary for LTF (Alberini et al., 1994; Lee et al., 2001). However, even though ApC/EBP is necessary, it does not seem to be sufficient because overexpression of ApC/EBP alone does not induce LTF (Lee et al., 2001). This implies that other transcription factors activated by 5-HT must also be required for the consolidation of LTF. Therefore, Bartsch et al. (2000) suggested that the constitutively expressed transcription factor Aplysia activating factor (ApAF) may be a potential transcriptional cofactor in LTF via its interaction with ApC/EBP or ApCREB2, but not with ApCREB1. However, the transcriptional regulation initiated by these interactions, and the identity of the core transcriptional component that is important for the consolidation of LTF, have not been characterized.
We demonstrate that ApAF relieves the repression mediated by ApCREB2 by interacting with ApCREB2 and induces LTF by forming heterodimers with ApC/EBP. We further show that PKA-activated ApAF–ApC/EBP can induce LTF without CRE-driven gene expression. We further characterize the critical phosphorylation of ApAF by PKA at Ser-266. Thus, our study suggests that PKA-activated ApAF–ApC/EBP heterodimer is both necessary and sufficient in the consolidation of LTF.
Results
The nuclear protein ApAF interacts with ApC/EBP or ApCREB2
In the aforementioned study, Bartsch et al. (2000) found that ApAF could interact with ApC/EBP or ApCREB2 in vitro and might function as a transcriptional cofactor by interacting with other transcription factors. However, it was unclear whether ApAF can actually interact with ApC/EBP or ApCREB2 in Aplysia neurons.
To directly examine interactions between memory-related transcription factors at the cellular level, we used an Aplysia two-hybrid system (Choi et al., 2003). We measured β-galactosidase activity in Aplysia kurodai neurons as an indication of the interactions among ApAF, ApCREB2, and ApC/EBP. We found the following types of interaction: (a) those between full-length ApAF and the basic leucine zipper (bZIP) domain of ApAF; (b) those between full-length ApAF and the bZIP domain of ApC/EBP; and (c) those between full-length ApAF and the bZIP domain of ApCREB2 (Fig. 1). The ApAF–ApC/EBP or ApAF–ApCREB2 heterodimers showed higher β-galactosidase activities than ApAF or ApC/EBP homodimers, indicating that ApAF may heterodimerize better with ApC/EBP or ApCREB2 than it homodimerizes with itself. These were consistent with both yeast two-hybrid and in vitro binding assay data (Bartsch et al., 2000) showing that ApAF formed stable heterodimers with both ApC/EBP and ApCREB2 proteins, although ApAF formed homodimers inefficiently. In contrast, neither the pair alone nor the Gal4DB-bait (bZIP of ApC/EBP or ApCREB2) on its own activated the reporter gene (Fig. 1).
Figure 1.

Interaction between the nuclear protein ApAF and ApC/EBP or ApCREB2 in an A. kurodai neuron. The histogram represents the degree of interaction in terms of normalized β-galactosidase activity (β-galactosidase/luciferase activity). ApAF or ApC/EBP can form homodimers through bZIP (n = 9 and n = 6, respectively). ApAF can also form heterodimers with ApC/EBP (n = 4) or with ApCREB2 (n = 4). Heterodimerization seems to be more favorable than homodimerization (*, P < 0.05; one-way analysis of variance (ANOVA) and Newman-Keuls multiple comparison test for ApAF(FL)-ApAFbz, C/EBP(FL)-C/EBPbz, ApAF(FL)-C/EBPbz, and ApAF(FL)-CREB2bz). ApAF (S266A) has a similar binding affinity with ApC/EBP (n = 3) and ApCREB2 (n = 3) as ApAF(WT). In contrast, neither the pair alone (n = 6) nor the Gal4DB-bait (bZIP of ApC/EBP [n = 6], ApCREB2 [n = 7], or ApAF [n = 3]) on its own could activate reporter genes. Gal4 is used as a positive control (n = 5). The bars correspond to normalized mean β-galactosidase activity ± the SEM. BD and AD represent the DNA-binding domain and transcription activation domain, respectively. bz represents the bZIP domain of the transcription factors. AD, BD, ApAF (FL), C/EBP (FL), C/EBPbz, CREB2bz, ApAFbz, and S266A represent Gal4AD, Gal4BD, Gal4AD-ApAF (full-length), Gal4AD-ApC/EBP (full-length), Gal4BD-ApC/EBPbz, Gal4BD-ApCREB2bz, Gal4BD-ApAFbz, and ApAF (S266A), respectively.
These results indicate that nuclear protein ApAF actually interacts with both a transcriptional activator ApC/EBP and a repressor ApCREB2 in the nucleus, implying that ApAF can function in the sensory cells as a transcriptional cofactor during LTF in A. kurodai.
ApAF is a transactivator with ApC/EBP acting on ERE in A. kurodai sensory neurons
Because ApAF interacts both with ApC/EBP and with ApCREB2, it raises the question: what is the functional role of the interaction of ApAF with ApC/EBP in the consolidation of LTF? Because the consensus-binding sequences of ApAF from Aplysia californica were previously reported to contain CAAT box sequences (Bartsch et al., 2000), we investigated whether ApAF regulates reporter gene expression through enhancer response element (ERE) sequences, which contain the CAAT core box sequence, and whether 5-HT can activate the reporter gene expression in Aplysia sensory neurons (Alberini et al., 1994; Lee et al., 2001). Accordingly, we microinjected pNEXδ-ApAF together with ERE-luciferase reporter into sensory neurons. 1 d after this microinjection, we found that reporter gene expression was increased by approximately fivefold by the ApAF construct, as compared with the ERE-luciferase reporter that was introduced as a control (Fig. 2 A). We next asked whether reporter gene expression induced by ApAF introduction is activated by 5-HT. To address this question, we microinjected pNEXδ-ApAF with the ERE-luciferase reporter into sensory neurons. 1 d later, we exposed sensory neurons to one pulse of 5-HT (10 μM), which by itself only produces short-term facilitation (STF) and does not induce gene expression (Fig. 2 A; Montarolo et al., 1986). We found that 1 d after this one pulse of 5-HT, reporter gene expression in neurons into which ApAF had been introduced was increased by approximately twofold compared with untreated cells (Fig. 2 A). ApC/EBP overexpression combined with one pulse of 5-HT also activated ERE reporter gene expression (Fig. 2 A). These results show that ApAF as well as ApC/EBP can activate ERE-driven gene expression by 5-HT in A. kurodai sensory neurons.
Figure 2.

ApAF induced ERE-mediated gene expression and LTF by interacting with ApC/EBP. (A) Transcriptional activity of ApAF, ApC/EBP, or ApAF–ApC/EBP via the ERE sequence in A. kurodai sensory neurons. +, DNA constructs injected with ERE-luciferase reporter into sensory cells in the pleural ganglia. Either one (1×) or five pulses (5×) of 5-HT were applied to sensory cells. ERE-driven reporter gene expression was increased by ApAF (n = 4), ApC/EBP (n = 4), or ApAF–ApC/EBP (n = 4) overexpression in comparison to nonexpressing control cells (n = 4; ***, P < 0.001; one-way ANOVA and Newman-Keuls multiple comparison test for groups indicated by open bars). The reporter gene expression was further enhanced by 5-HT treatment in ApAF- (n = 4), ApC/EBP- (n = 5), and ApAF–ApC/EBP-introduced (n = 8) cells (*, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed unpaired t test). Each bar corresponds to normalized mean luciferase activity ± the SEM. Five pulses of 5-HT treatment significantly increased ERE-mediated gene expression (n = 4), whereas one pulse of 5-HT treatment did not (n = 5). (B) The effect and specificity of ApAF dsRNA. The expression level of ApAF mRNA was examined by in situ hybridization. The expression level of ApAF was significantly lower in ApAF dsRNA–injected neurons (n = 5) than in luciferase dsRNA–injected neurons (n = 9; *, P < 0.05; two-tailed unpaired t test), whereas the expression level of ApCREB1a was not affected by ApAF dsRNA injection (n = 6). The percentage of change in mRNA is calculated as the relative expression level of ApAF or ApCREB1a mRNA in ApAF dsRNA–injected neurons compared with luciferase dsRNA–injected neurons. The bar represents the mean ± the SEM. Bar, 25 μm. (C) The effect of ApAF–ApC/EBP heterodimer on LTF. +, DNA constructs injected into sensory cells. EPSPs were measured before and 24 h after one or five pulses of 5-HT were applied to the sensory-to-motor synapse. For shaded bars, injection of ApAF dsRNA completely blocked both the LTF induced by five pulses of 5-HT (n = 11) and the LTF induced by ApC/EBP overexpression combined with one pulse of 5-HT (n = 8), whereas the injection of luciferase dsRNA did not block either type of LTF (5×5-HT, n = 8; 1×5-HT, n = 4; **, P <0.01; one way ANOVA and Newman-Keuls multiple comparison test). For open bars, ApAF overexpression combined with one pulse of 5-HT did not produce LTF (n = 8), whereas ApC/EBP overexpression combined with 5-HT did (n = 13). However, ApAF overexpression enhanced the LTF induced by ApC/EBP and one pulse of 5-HT (n = 17; *, P < 0.05). The noninjected control cells produced normal LTF by five pulses of 5-HT (n = 10). Representative examples of recording traces are shown on the right. (D) STF was not affected by ApAF dsRNA microinjection. The mean percentage of changes in EPSP of ApAF dsRNA–injected cells (n = 4) was not significantly different from that of noninjected control cells (n = 4) or luciferase dsRNA–injected cells (n = 4; P > 0.05; one way ANOVA and Newman-Keuls multiple comparison test). Bars correspond to mean percentage changes ± the SEM in EPSP amplitudes.
We next investigated the consequences of the ApAF–ApC/EBP interaction on ERE-mediated gene expression. For this purpose, pNEXδ-ApAF and pNEXδ-ApC/EBP with ERE-luciferase reporter were microinjected into A. kurodai sensory neurons. 1 d after DNA microinjection, sensory neurons were treated with one pulse of 5-HT. We found that 24 h after 5-HT treatment, ERE-mediated reporter gene expression by ApAF–ApC/EBP overexpression was further increased by approximately twofold compared with that by either ApC/EBP or ApAF alone (Fig. 2 A). Furthermore, we also found that reporter gene expression by ApAF–ApC/EBP was increased by approximately fourfold in 5-HT–treated cells versus non–5-HT–treated cells (Fig. 2 A). These results indicate that ApAF can enhance the ERE-mediated gene expression by cooperating with ApC/EBP and that ApAF can be activated by 5-HT treatment.
Functional cooperation between ApAF and ApC/EBP is essential for the consolidation of LTF
Because ApAF could activate reporter gene expression through ERE by interacting with ApC/EBP, we next investigated the actual role in long-term synaptic facilitation of ApAF heterodimerized to ApC/EBP. We previously reported that ApC/EBP is essential for the consolidation of LTF by showing that blocking ApC/EBP blocks the facilitation (Alberini et al., 1994), and that overexpression of ApC/EBP combined with only one pulse of 5-HT converted STF to LTF (Lee et al., 2001). We also showed that LTF produced by ApC/EBP overexpression and one pulse of 5-HT was comparable to that produced by five pulses of 5-HT (Lee et al., 2001; Fig. 2 C). Therefore, we considered the possibility that the ApAF–ApC/EBP heterodimer may be essential for the consolidation of LTF because overexpressed ApC/EBP interacts with constitutively expressing endogenous ApAF.
To test this idea by blocking the ApAF expression, we used the RNAi technique (Lee et al., 2001). First, to confirm the effectiveness and the specificity of ApAF double-stranded (ds) RNA, we injected ApAF dsRNA into sensory neurons. 1 d later, we examined the expression level of ApAF mRNA by in situ hybridization. We injected the luciferase dsRNA as a control. The expression of ApAF mRNA was significantly knocked down in ApAF dsRNA–injected neurons compared with luciferase dsRNA–injected neurons (Fig. 2 B). In contrast, ApAF dsRNA did not affect the expression level of ApCREB1 mRNA compared with luciferase dsRNA (Fig. 2 B). These results show that ApAF dsRNA specifically impairs the expression of ApAF.
Given these results, we examined the effect of the inhibition of ApAF synthesis on LTF induced by five pulses of 5-HT. The cells injected with dsApAF failed to produce LTF induced by five pulses of 5-HT without effecting STF and the basal synaptic strength (Fig. 2, C and D, and not depicted). These results are consistent with the earlier finding (Bartsch et al., 2000) that ApAF is required for LTF in A. californica sensory-to-motor synapses.
Next, we coinjected pNEXδ-ApC/EBP with ApAF dsRNA into a sensory neuron. Cells injected with ApAF dsRNA also failed to produce the LTF induced by pairing one pulse of 5-HT with ApC/EBP overexpression, whereas cells injected with luciferase dsRNA showed normal long-term synaptic facilitation (Fig. 2 C). These results suggest that ApAF–ApC/EBP heterodimer is essentially required for the consolidation of LTF, and indicate that the ApC/EBP homodimer is insufficient to convert STF to LTF.
If ApAF–ApC/EBP heterodimer is important for LTF, can overexpression of ApAF enhance the LTF induced by ApC/EBP overexpression when combined with one pulse of 5-HT? To address this question, we overexpressed ApAF, ApC/EBP, or ApAF + ApC/EBP in the sensory neurons of sensory-to-motor synapses, and then exposed cocultures to one pulse of 5-HT. We found that ApAF overexpression enhanced LTF induced by ApC/EBP overexpression combined with one pulse of 5-HT (Fig. 2 C), without affecting basal synaptic transmission (unpublished data). However, ApAF overexpression alone did not produce LTF in response to one pulse of 5-HT (Fig. 2 C), suggesting that ApC/EBP is also critical in LTF.
Collectively, these data indicate that ApAF is a key player in the induction of LTF by cooperating with ApC/EBP.
The Ser-266 in ApAF needs to be phosphorylated by PKA
If ApAF plays an important role in LTF by cooperating with ApC/EBP, how is the functional activation of ApAF regulated by 5-HT to induce LTF? Our reporter assays and electrophysiological recordings showed that a signaling pathway activated by 5-HT is required to activate ERE-mediated gene expression and to induce ApAF-mediated LTF. However, as is the case with ApCREB2, ApAF is a constitutive protein that is not induced by 5-HT, whereas ApC/EBP is rapidly induced (Alberini et al., 1994; Bartsch et al., 1995, 2000; unpublished data). Therefore, we considered the possibility that ApAF might be regulated by kinases activated by 5-HT. Because 5-HT can activate the cAMP–PKA pathway and ApAF has two possible PKA phosphorylation sites at Ser-175 and -266 (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200512066/DC1), we explored whether a PKA-signaling pathway is involved in ApAF-mediated LTF by blocking the PKA pathway using the PKA inhibitor KT5720. Incubation with 10 μM KT5720 was found to completely block LTF consolidated by ApC/EBP overexpression and 5-HT. Moreover, the ApAF-mediated enhancement of LTF was completely impaired by KT5720 (Fig. 3 A). These results suggest that PKA signaling is essentially involved in ApAF–ApC/EBP–mediated LTF.
Figure 3.

ApAF phosphorylation by PKA is involved in ApAF-mediated LTF. (A) The effect of PKA activation on ApAF-mediated LTF. +, DNA constructs injected into sensory cells of sensory-to-motor synapses. The cultures were exposed to one (1×) or five pulses (5×) of 5-HT. Incubation with 10 μM KT5720 for 2 h completely blocked the LTF induced by one pulse of 5-HT with ApC/EBP overexpression (n = 4) or with overexpression of ApAF and ApC/EBP (n = 4), as well as the LTF induced by five pulses of 5-HT (n = 5; *, P < 0.05; **, P < 0.01; ***, P < 0.001; two-tailed Mann-Whitney test). Bars correspond to mean percentage changes ± the SEM in EPSP amplitudes. The data without inhibitor treatment are the same as in Fig. 2 C. (B) Phosphorylation of ApAF. (top) Purified ApAF was incubated with Aplysia cell lysate as a source of kinases. PKA inhibitor KT5720 dramatically blocked the phosphorylation of ApAF, whereas the MEK inhibitor PD98059 or the PKC inhibitor chelerythrine did not. (bottom) ApAF proteins were purified from E. coli as GST fusion proteins and were incubated with catalytic subunits of PKA. WT and S175A mutant were directly phosphorylated by PKA, whereas S266A mutant and S175/266A double mutant were not. This phosphorylation was completely blocked by KT5720.
Because 5-HT can also activate MAPK, we examined whether MAPK activity is required for the activation of the ApAF–ApC/EBP complex using PD98059, which is an inhibitor of the MAPK kinase MEK. PD98059 did not block LTF consolidated by ApAF and ApC/EBP overexpression (Fig. 3 A). Although PD98059 treatment slightly attenuated ApAF–ApC/EBP–mediated LTF, the degree of facilitation was not significantly different from that without drug treatment (P > 0.05; two-tailed Mann-Whitney test; Fig. 3 A). This result suggests that MAPK signaling is not critically involved in ApAF–ApC/EBP–mediated LTF.
To clarify whether ApAF is an endogenous substrate of Aplysia PKA, we examined ApAF phosphorylation by using A. kurodai cell lysate as a source of the kinases. As shown in Fig. 3 B, KT5720 dramatically blocked the phosphorylation of ApAF, whereas either PD98059 or chelerythrine (PKC inhibitor) did not (Fig. 3 B). Moreover, to examine whether ApAF is directly phosphorylated by PKA at Ser-175 or -266, we incubated the proteins with the catalytic subunit of bovine PKA. We found that PKA phosphorylation was detected in wild-type (WT) ApAF and in ApAF (S175A), but not in ApAF (S266A) or ApAF (S175/266A; Fig. 3 B). Thus, these results clearly suggest that PKA specifically phosphorylates ApAF at Ser-266.
Ser-266 phosphorylation by PKA is required for the functional activation of ApAF
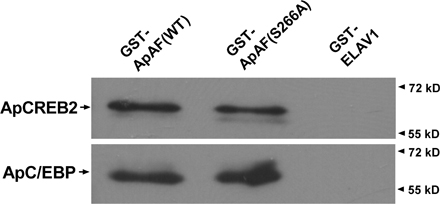
Next, we examined whether PKA is required for transcriptional activation by ApAF–ApC/EBP. To address this issue, ERE-mediated gene expression regulation by ApAF mutants (S175A, S266A, or S175/266A) was first tested by reporter gene assay. ApAF (WT, S175A, S266A, or S175/266A) and ApC/EBP constructs were injected together with the ERE-reporter construct into sensory neurons. The day after microinjection, sensory cells were treated with one pulse of 5-HT, and the following day luciferase assays were performed. Consistent with previous results (Fig. 3, A and B), reporter gene expression regulated by ApAF–ApC/EBP (S266A) or by ApC/EBP–ApAF (S175/266A) was found to be attenuated by ∼4.6- and ∼7-fold compared with that regulated by ApAF–ApC/EBP (WT; Fig. 4 A). However, reporter gene expression regulated by ApAF–ApC/EBP (S175A) was not reduced compared with that regulated by ApAF–ApC/EBP (WT; Fig. 4 A). In addition, we found that both WT ApAF and ApAF (S266A) form a heterodimer with ApC/EBP with similar affinity in GST pull-down assay and Aplysia two-hybrid assay (Fig.1 and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200512066/DC1). These data suggest that ApAF (S266A) is likely to act as a dominant-negative mutant through interfering with the transcriptional machinery, such as RNA polymerase II complex, that may recognize only a phosphorylated ApAF–ApC/EBP heterodimer. Collectively, our results suggest that ApAF phosphorylation by PKA at Ser-266 essentially regulates the ERE-mediated gene expression via cooperation between ApAF and ApC/EBP.
Figure 4.

ApAF phosphorylation by PKA on Ser-266 is essential for ERE-mediated gene expression and LTF. (A) The effect of ApAF phosphorylation by PKA at Ser-266 on ERE-mediated gene expression. +, DNA constructs injected with ERE-luciferase into sensory cells in the pleural ganglia. 24 h after microinjection, the cells were treated with one pulse (1×) of 5-HT, which does not induce gene expression. ERE-driven reporter gene expressions by the introduction of ApAF(S266A) and ApAF(S175/266A) were completely blocked (both n = 4; *, P < 0.001; one-way ANOVA and Newman-Keuls multiple comparison test). Each bar corresponds to the mean normalized luciferase activity ± the SEM. The experimental data for WT ApAF are the same as in Fig. 2 A. (B) The effect of ApAF phosphorylation by PKA at Ser-266 on LTF. +, DNA constructs injected into the sensory cell of sensory-to-motor synapses. The cultures were exposed to one (1×) or five pulses (5×) of 5-HT. Overexpression of ApAF mutants (S266A and S175/266A) blocked ApAF–ApC/EBP–mediated LTF (n = 5 and n = 8, respectively), whereas overexpression of ApAF(S175A) did not block LTF (n = 9; *, P < 0.05, **, P < 0.01; one-way ANOVA and Newman-Keuls multiple comparison test for one-pulse 5-HT groups indicated by shaded bars). Moreover, ApAF(S266A) impaired the LTF induced by five pulses of 5-HT (n = 5; **, P < 0.01; two-tailed unpaired t test for five-pulse 5-HT groups indicated by open bar). Bars correspond to mean percentage changes ± the SEM in EPSP amplitudes. The data for WT ApAF and the data without injection are the same as in Fig. 2 C.
Because Ser-266 phosphorylation is important for ERE-mediated gene expression (Fig. 4 A), we examined whether phosphorylation at Ser-266 is also required for LTF. We injected both pNEXδ-ApAF constructs (WT, S175A, S266A, or S175/266A) and pNEXδ-ApC/EBP into sensory neurons and treated them with one pulse of 5-HT the next day. ApC/EBP-mediated LTF was completely blocked in cells expressing pNEXδ-ApAF mutants (S266A or S175/266A), which cannot be phosphorylated, without affecting basal synaptic strength (Fig. 4 B). However, cells injected with pNEXδ-ApAF (WT) or pNEXδ-ApAF (S175A) showed enhanced ApC/EBP-mediated LTF (Fig. 4 B). To further confirm whether ApAF phosphorylation at Ser-266 is required for LTF produced by five pulses of 5-HT, we overexpressed ApAF (S266A) in sensory neurons. We found that ApAF (S266A), functioning as a dominant-negative mutant, completely blocked 5-HT–induced LTF (Fig. 4 B). Collectively, these results suggest that ApAF phosphorylation by PKA at Ser-266 is essential for the LTF induced by cooperation between ApAF and ApC/EBP.
Ser-266 phosphorylation of ApAF by PKA is not required for the relief of ApCREB2 repression
We next asked whether PKA is required for the function of ApAF–ApCREB2 heterodimer in CRE-driven gene expression and LTF.
First, we examined whether ApAF could regulate CRE-driven gene expression by interacting with ApCREB2 using a transcriptional reporter gene assay. ApCREB1 binds to the CRE sequence and activates CRE-mediated gene expression, whereas ApCREB2 inhibits CRE-mediated gene expression by repressing ApCREB1 (Bartsch et al., 1995, 1998; Lee et al., 2003). Therefore, we microinjected into A. kurodai neurons pNEXδ-ApCREB1, pNEXδ-ApCREB2, and pNEXδ-ApAF with CRE-luciferase reporter and found that ApCREB2 overexpression completely blocked the CRE-driven gene expression activated by ApCREB1 (Fig. 5 A). But remarkably, both ApAF mutants and ApAF WT relieved the repression by ApCREB2 of CRE-driven reporter gene expression (Fig. 5 A). In addition, we found that both ApAF WT and ApAF mutant (S266A) form a heterodimer with ApCREB2 with similar affinity in GST pull-down and Aplysia two-hybrid assays (Fig.1 and Fig. S2). Collectively, these results indicate that ApAF can activate ApCREB1 indirectly by direct interaction with ApCREB2, and that ApAF phosphorylation by PKA at Ser-266 is not required to relieve the repression of ApCREB2.
Figure 5.

ApAF phosphorylation by PKA on Ser-266 is not required to relieve repression by ApCREB2. (A) The effect of ApAF phosphorylation by PKA at Ser-266 on ApCREB2-mediated repression. +, DNA constructs injected with CRE-luciferase reporter into cells in the abdominal ganglia. ApCREB2 overexpression (n = 6) blocked CRE-driven reporter gene expression (n = 5) activated by ApCREB1. ApAF(S175A) (n = 3), ApAF(S266A) (n = 3), and ApAF(S175/266A) (n = 4), as well as ApAF WT (n = 3) restored CRE-driven gene expression repressed by ApCREB2 (*, P < 0.001; one-way ANOVA and Newman-Keuls multiple comparison test). Luciferase activity was normalized to β-galactosidase activity. Each bar corresponds to normalized mean luciferase activity ± the SEM. CRE-luciferase alone (n = 5); CRE-luciferase with ApAF (n = 8); CRE-luciferase with ApAF and ApCREB2 (n = 4). (B) The effect of ApAF phosphorylation by PKA at Ser-266 on the blockage of LTF by ApCREB2 repressor. +, DNA constructs injected into sensory cells of sensory-to-motor synapse. The cultures were treated with five pulses of 5-HT. ApCREB2 overexpression significantly impaired the LTF induced by five pulses of 5-HT (n = 12). ApAF overexpression combined with five pulses of 5-HT relieved the blockage of LTF by ApCREB2 (n = 13), whereas ApAF overexpression alone did not affect LTF (n = 9; *, P < 0.05; **, P < 0.01; one-way ANOVA and Newman-Keuls multiple comparison test for the groups represented by a hatched bar and shaded bars). Moreover, ApAF mutants (S175A [n = 5], S266A [n = 4], and S175/266A [n = 4]) also relieved the blockage of LTF by ApCREB2 (**, P < 0.01; one-way ANOVA and Newman-Keuls multiple comparison test for the groups represented by a hatched bar and open bars). For five pulses of 5-HT, control is n = 17. For ApAF and ApCREB2 overexpression without the 5-HT treatment n = 5. Bars correspond to mean percentage changes ± the SEM in EPSP amplitudes.
If this is so, then is Ser-266 phosphorylation by PKA essential for the relief of LTF repressed by ApCREB2? To address this question, ApAF constructs (WT, S175A, S266A, or S175/266A) were coinjected with ApCREB2 into a sensory neuron in a sensory-to-motor coculture. One day later, the cultures were exposed to five pulses of 5-HT. We found that ApAF mutants, which cannot be phosphorylated by PKA, and the ApAF (WT) restored the LTF repressed by ApCREB2 (Fig. 5 B) without affecting basal synaptic transmission (not depicted). The apparent loss of the dominant-negative effect of ApAF (S266A) on ApC/EBP is likely to be caused by a large quantity of the overexpressed ApCREB2. ApAF (S266A) and ApCREB2 can inhibit each other by direct interaction. Thus, overexpressed ApCREB2 may prevent ApAF (S266A) from interfering with ApC/EBP in a dominant-negative fashion. Control cells and cells injected with pNEXδ-ApAF produced normal 5-HT–induced LTF (Fig. 5 B). Collectively, these data suggest that ApAF relieves the repression of LTF by ApCREB2 through a direct binding with ApCREB2, and indicate that ApAF phosphorylation on Ser-266 is not necessary to relieve LTF repressed by ApCREB2.
PKA-activated ApAF–ApC/EBP is sufficient for induction for LTF
If Ser-266 phosphorylation in ApAF is essential for induction of LTF, is PKA-activated ApAF–ApC/EBP sufficient for LTF? To test whether PKA-activated ApAF induces LTF by interacting with ApC/EBP without CRE- and CREB-mediated gene expression, we blocked CRE- and CREB-mediated gene expression by microinjecting CRE oligonucleotide (Alberini et al., 1994) and by overexpressing dominant-negative “killer” CREB (K-CREB ; Walton et al., 1992). The injection of CRE oligonucleotide blocked the LTF produced by five pulses of 5-HT (Fig. 6). However, CRE injection did not affect the LTF produced by ApAF–ApC/EBP heterodimer in the presence of one pulse of 5-HT (Fig. 6). Moreover, the injection of CRE mutant containing the randomly scrambled CRE sequences did not affect the LTF produced by 5 pulses of 5-HT or the LTF produced by ApAF–ApC/EBP heterodimer in the presence of one pulse of 5-HT (unpublished data). To further demonstrate whether PKA-activated ApAF–ApC/EBP heterodimer is sufficient for the consolidation of LTF, we next blocked CREB-mediated gene expression using dominant-negative K-CREB. The cells injected with pNEXδ-K-CREB failed to produce a normal 5-HT–induced LTF. However, the cells overexpressing K-CREB did not block the LTF produced by ApAF and ApC/EBP in the presence of one pulse of 5-HT. Collectively, these results suggest that the PKA-activated ApAF–ApC/EBP heterodimer is sufficient to induce LTF, independent of CREB activity, and imply that ApAF–ApC/EBP is a downstream effector of ApCREB.
Figure 6.

LTF produced by PKA-activated ApAF and ApC/EBP was not blocked by inhibiting CRE- and CREB-mediated gene expression, but was still blocked by inhibition of ERE-mediated gene expression. +, DNA constructs or oligonucleotides (CRE and ERE) injected into sensory cells. The cultures were exposed to one (1×) or five pulses (5×) of 5-HT. Injection of CRE oligonucleotides (oligo; n = 7), as well as K-CREB (n = 6), did not block the LTF (n = 13) produced by ApAF and ApC/EBP in the presence of one-pulse 5-HT treatment. However, injection of CRE (n = 4) or pNEXδ–K-CREB (n = 8) blocked the LTF produced by five pulses (5×) of 5-HT (*, P < 0.05; **, P < 0.01; one-way ANOVA and Newman-Keuls multiple comparison test). In contrast, injection of ERE oligonucleotides completely blocked the LTF induced by ApAF–ApC/EBP in the presence of one-pulse 5-HT (n = 6; **, P < 0.01). ERE oligonucleotides also blocked the LTF induced by five pulses of 5-HT (n = 7; *, P < 0.05). Bars correspond to mean percentage changes ± the SEM in EPSP amplitudes. 5-HT controls were 1×, n = 7; 5×, n = 13.
To examine whether the PKA-activated ApAF–ApC/EBP heterodimer produces LTF through ERE-mediated gene expression, we injected ERE oligonucleotides into the sensory neurons overexpressing ApAF and ApC/EBP. As shown in Fig. 6, ERE oligonucleotide injection impaired the LTF induced by ApAF–ApC/EBP overexpression combined with one pulse of 5-HT. ERE oligonucleotide injection also blocked the LTF induced by five pulses of 5-HT (Fig. 6). In contrast, cells injected with ERE mutant oligonucleotides affected neither the ApAF–ApC/EBP-mediated LTF nor the 5-HT induced LTF (unpublished data). Collectively, these data suggest that the ApAF–ApC/EBP heterodimer, once activated, induces LTF through ERE-mediated gene expression.
Discussion
In this study, we demonstrated a novel, functional cooperation between ApAF and ApC/EBP (an activator) or ApCREB2 (a repressor) for LTF. Moreover, we found that the methods of ApAF interaction with these two factors were quite different. Our study suggests that PKA-activated ApAF–ApC/EBP is a key transcriptional component that is necessary and sufficient for the consolidation of LTF.
The functional interaction between ApAF and ApC/EBP
In our earlier study, ApC/EBP overexpression combined with one pulse of 5-HT, but not ApC/EBP overexpression alone, consolidated STF to LTF (Lee et al., 2001). The failure of ApC/EBP alone to induce LTF indicates that certain factors activated by one pulse of 5-HT may be required to induce LTF. ApAF seems to be such a factor; it can work with ApC/EBP to induce LTF in the presence of one pulse of 5-HT. We also found that when combined with one pulse of 5-HT ApAF overexpression did not induce LTF. Moreover, in the previous experiments, phosphorylated ApAF alone did not induce LTF (Bartsch et al., 2000). We believe that this is because ApC/EBP is not induced by one pulse of 5-HT (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200512066/DC1; Kaang et al., 1993; Kim et al., 2006) and that both ApAF and ApC/EBP, which interact with each other, are critical for LTF. In support of this idea, our RNAi study demonstrated that blocking ApAF by dsRNA injection impaired not only LTF induced by five pulses of 5-HT, but also LTF induced by ApC/EBP overexpression combined with one pulse of 5-HT (Fig. 2 C). Bartsch et al. (2000) showed that ApAF protein injection could convert 5-HT induced-STF to LTF. In our study, however, ApAF DNA overexpression combined with one pulse of 5-HT could not induce LTF (Fig. 2 C). We believe that this discrepancy probably came from the different experimental systems. A larger quantity of proteins could be introduced by a direct protein injection than by DNA expression system. A large quantity of ApAF could lower the threshold of LTF effectively by titrating ApCREB2 out. Similarly, anti-ApCREB2 antibody injection produced LTF with one pulse of 5-HT (Bartsch et al., 2000).
How, then, is the heterodimer ApAF–ApC/EBP involved in the consolidation of LTF? We found that the functional cooperation between ApAF and ApC/EBP in the presence of one pulse of 5-HT enhanced ERE-mediated gene expression and LTF (Fig. 2, A and C). Although we could not exclude the possibility that ApAF homodimer or ApC/EBP homodimer were involved in gene expression, ApAF and ApC/EBP were relatively poor at forming homodimer (Fig. 1; Bartsch et al., 2000), and their homodimers had a lower transcriptional activity in response to one pulse of 5-HT treatment than ApAF–ApC/EBP heterodimer (Fig. 2 A).
The role of PKA in functional activation of ApAF in the consolidation of LTF
The failure of ApAF–ApC/EBP heterodimer to enhance transcriptional activity or LTF in the absence of 5-HT, suggests that a combination of other components or signal cascades activated by 5-HT may be required for memory enhancement. Our biochemical and pharmacological data showed that ApAF was directly phosphorylated at Ser-266 by PKA, but not by either MAPK or PKC (Fig. 3, A and B). Moreover, we demonstrated that ApAF mutants, which cannot be phosphorylated at Ser-266, completely blocked ERE-mediated gene expression and LTF. ApAF mutants may function as dominant-negative proteins, for they bind ApC/EBP equally as well as the WT does, but they cannot recruit and/or activate RNA polymerase II complex, thus, interfering with the interaction between phospho-ApAF–ApC/EBP heterodimer and transcriptional machinery. Collectively, our results strongly suggest that Ser-266 phosphorylation by PKA in ApAF is required for 5-HT–induced LTF.
The functional interaction between ApAF and ApCREB2
ApCREB2 is known as a gatekeeper of LTF because it represses ApCREB1-mediated transcription (Bartsch et al., 1995; Lee et al., 2003). Thus, the initiation of the ApCREB1-activated gene cascade by repressing ApCREB2 is essential for the induction of LTF. If this is so, then how is the repression of ApCREB1 by ApCREB2 relieved? This relief may require covalent modifications via MAPK or PKC, both of which phosphorylate ApCREB2 (Bartsch et al., 1995; Abel and Kandel, 1998). In addition, Bartsch et al. (2000) and our reporter assay and electrophysiological studies showed that the expression level of ApAF may also be important in regulating ApCREB2 activity (Fig. 5). Because ApAF interacts with ApCREB2, ApAF can recruit ApCREB2 and inhibit the repressive function of ApCREB2. This suggests that ApAF may contribute to lowering the threshold required for LTF by repressing ApCREB2.
However, ApAF phosphorylation at Ser-266 did not seem to be required to repress ApCREB2 because each ApAF mutant relieved the ApCREB2-mediated repression of LTF. In support of this idea, we found interactions of ApAF mutant with ApCREB2 or ApC/EBP were similar to those of the ApAF WT (Fig. 1).
Can the interaction between ApAF and ApCREB2 be regulated? The phosphorylation of ApCREB2 by MAPK or PKC could be involved in the relief of ApCREB2-mediated repression (Bartsch et al., 1995). It is also possible that the phosphorylation of ApCREB2 by the kinases may lower its affinity for ApAF as well as ApCREB1, thus, allowing more ApAF to interact with ApC/EBP. Further work will be needed to examine the role of ApCREB2 phosphorylation in ApAF–ApCREB2 heterodimerization.
ApAF–ApC/EBP heterodimer, which is a core downstream effector of ApCREB1, is sufficient for LTF
Phosphorylated ApCREB1 is necessary and sufficient for LTF (Bartsch et al., 1998). This suggests that gene expression induced by activated ApCREB1 is required and is sufficient for the induction of LTF. Of the genes regulated by ApCREB1, ApC/EBP is known to be a consolidating factor for LTF (Alberini et al., 1994). In this study, we found that the phospho-ApAF–ApC/EBP heterodimer was necessary and sufficient for LTF, acting as a key downstream factor from CREB (Fig. 6).
If so, how are ApAF and ApC/EBP activated by phosphorylated ApCREB1? Phosphorylated ApCREB1 could induce not only ApC/EBP but also ubiquitin carboxyl-terminal hydrolase (Ap-uch), which activates PKA persistently (Hegde et al., 1997). This persistent PKA activity may result in ApAF phosphorylation. Phosphorylated ApCREB1 can also bind to ApCREB2 (Bartsch et al., 1995, 1998). Thus, it is possible that ApAF released from ApCREB2 is allowed to form a heterodimer with ApC/EBP induced by phospho-ApCREB1.
In addition, we showed that PKA (not MAPK) was critically involved in ApAF–ApC/EBP–mediated LTF (Fig. 3 A). Thus, we suggest that PKA-, not MAPK-, activated ApAF–ApC/EBP heterodimer is a core downstream effector of ApCREB, and that it is a powerful player essential for the consolidation and stabilization of LTF. Yamamoto et al. (1999) suggested that ApC/EBP phosphorylation by MAPK could regulate DNA binding activity of ApC/EBP homodimer. In Fig. 3 A, LTF induced by ApAF–ApC/EBP overexpression combined with one pulse of 5-HT was slightly attenuated by MEK inhibitor PD98059. This attenuation might represent the contribution of ApC/EBP homodimer although it was not statistically significant.
Transcriptional regulation for memory consolidation
Based on our study, we ascribe a dual role to ApAF in LTF; in the basal state, ApAF lowers the threshold of LTF by repressing ApCREB2, and in the 5-HT–induced activated state, ApAF induces LTF by interacting with ApC/EBP. Of the transcriptional complexes ApAF–ApC/EBP, which is a heterodimer activated by 5-HT, is a downstream effector of ApCREB1 and a core component during the consolidation of STF to LTF. We propose a mechanism of transcriptional regulation for memory consolidation (Fig. 7). Several transcription factors, including positive regulators, such as ApCREB1 or ApAF, and negative regulators, such as ApCREB2, exist in sensory neurons. ApCREB2 can form heterodimers with ApCREB1 or with ApAF. In basal states, ApCREB2 represses CRE-mediated gene expression by ApCREB1. ApCREB2 and ApAF also repress each other by forming a heterodimer. Thus, a balance between the expression levels of positive regulators and negative regulators can determine the threshold of how easily LTF is induced by 5-HT. Repeated pulses of 5-HT activate the cAMP–PKA–MAPK pathway. Activated PKA and MAPK then translocate to the nucleus and phosphorylate various transcription factors, including ApCREB1, ApCREB2, and ApAF. MAPK phosphorylation leads to a reduction in the affinity of ApCREB2 for ApCREB1 and probably reduces the affinity for ApAF. As a result, ApCREB1 has a higher probability to form a homodimer and to bind to the CRE enhancer element of the target genes. PKA now activates transcriptional activators ApCREB1 (Kaang et al., 1993; Martin et al., 1997) and ApAF. Phosphorylated ApCREB1 initiates a gene activation cascade required for memory consolidation (Bartsch et al., 1995; Lee et al., 2003), which leads to the induction of ApC/EBP as an immediately early gene. Once induced, ApC/EBP interacts with ApAF that is activated by PKA and free from ApCREB2. Phospho–ApAF–ApC/EBP heterodimer constitutes the core downstream effector of ApCREB1 and activates the expression of genes that have ERE promoter and are essential for the consolidation and stabilization of LTF.
Figure 7.

The transcriptional regulation by ApAF in consolidation of LTF. In basal state without stimulation by 5-HT, ApCREB2 represses CRE-mediated gene expression by ApCREB1. ApAF binds to ApCREB2, allowing ApCREB1 to form a homodimer, which can bind to the CRE element. Thus, ApAF can lower the threshold of LTF, producing a “primed” state for LTF. The multiple pulses of 5-HT up-regulate cAMP levels within sensory cells via G-protein–coupled receptors, and activate PKA, which then recruits another kinase, MAPK. Both PKA and MAPK translocate to the nucleus, where they phosphorylate the transcriptional factors ApCREB1/2 and ApAF. Phosphorylated ApCREB2 may dissociate easily from ApCREB1 and ApAF. Activation of ApCREB1 by PKA induces an immediate-early gene such as ApC/EBP, and then phosphorylated ApAF can form a heterodimer with newly synthesized ApC/EBP. Functional cooperation of PKA-activated ApAF–ApC/EBP heterodimer is essential for gene induction involved in the consolidation of LTF. Therefore, ApAF may serve as a powerful cofactor during consolidation of LTF.
Materials and methods
Cloning of ApAF from A. kurodai
To clone ApAF from A. kurodai, we used the ∼850-bp N-terminal fragment, which was obtained using NdeI–BbsI–digested pNEX-ACT-ApAF (A. californica) as a probe. We screened ∼1.5 × 105 clones from an A. kurodai cDNA library, and five positive signals were obtained after the second screening. Five clones were analyzed by DNA sequencing. One of them had an open reading frame of 1,083 bp encoding a polypeptide of 360 amino acids, and it also contained a bZIP domain (Fig. S1). Moreover, the amino acid sequence of ApAF from A. kurodai shares ∼80% similarities with that of A. californica. A search for potential phosphorylation sites revealed a common consensus sequence for the phosphorylation by PKA (Fig. S1), PKC, and CaMKII, indicating that ApAF may be regulated by these kinases.
DNA constructs
The ApAF obtained by PCR using specific primers (sense primer, 5′-CCCAAGCTTGCCACCACCATGATATCCAGCATTTCC-3′; antisense primer, 5′-CGGGATCTTTAGGCTGTACCTGCCAT-3′) was separately subcloned into HindIII–BamHI–digested pNEXδ and SalI–BamHI–digested pNEXδ-EGFP (Han et al., 2004) to create pNEXδ-ApAF or pNEXδ-EGFP-ApAF, respectively. The mutant fragments of ApAF (S175A, S266A or S175/266A) were generated by recombinant PCR using specific sense or antisense primers (sense primer containing S175A, 5′-CAAGAAAGCTGCCAAATCACCTG-3′; antisense primer containing S175A, 5′-CAGGTGATTTGGCAGCTTTCTTGG; sense primer containing S266A, 5′-CAAGAGGATTGCTTCCACAGCTTCTG-3′; and antisense primer containing S266A, 5′-GCTGTGGAAGCAATCCTCTTG-3′). The PCR products containing mutations (S175A, S266A, or S175/266A) in ApAF amino acid residues were subcloned into HindIII–KpnI–digested pNEXδ vector. The full-length of ApAF was subcloned into BamHI–SacI–digested pNEX-ACT (Choi et al., 2003) to create pNEX-ACT-ApAF (Gal4-AD-ApAF). The bZIP domain of ApAF and the bZIP domain of ApCREB2 were subcloned into BamHI–SacI–digested pNEX-AS (Choi et al., 2003) to create pNEX-AS-ApAF (bZIP) (Gal4-DB-ApAF [bZIP]) and pNEX-AS-ApCREB2 (bZIP) (Gal4-DB-ApCREB2 [bZIP]), respectively. To generate pLitmis28i-luciferase, luciferase was subcloned into BamHI–KpnI–digested pLitmis28i vector (New England Biolabs, Inc.). To create pLitmus28i-ApAF, ApAF was subcloned into StuI–BamHI–digested pLitmus28i. The full length of ApC/EBP was inserted into pNEXδ-EGFP (Han et al., 2004) to examine its cellular localization. The construction of pNEXδ-EGFP-CREB2 was previously described (Lee et al., 2003).
GST pull-down assay
WT GST-ApAF, its mutant GST fusion protein (GST-ApAF S266A), and GST-ELAV1 (an RNA-binding protein with a protein size similar to ApAF) as a negative control were purified from Escherichia coli using a general purification method (GE Healthcare). ApCREB2-HA and ApC/EBP-HA fusion proteins were translated in the TNT rabbit reticulocyte lysate (Promega). 5 μl of the lysates were mixed with 20 μl of saturated GST protein–bound beads in 200 μl of PBS containing 0.5% NP-40; these binding mixtures were incubated for 3 h at RT. The bound complexes were washed in 600 μl of TNET (25 mM Tris, pH 7.8, 150 mM NaCl, 0.2 mM EDTA, and 0.2% Triton X-100) and 300 μl of NET (25 mM Tris, pH 7.8, 150 mM NaCl, and 0.2 mM EDTA) serially, and then eluted by 2× LDS sample buffer (Invitrogen). The eluted samples were loaded in 12% SDS-PAGE gel and detected with anti-HA mouse monoclonal antibody (Sigma-Aldrich).
In situ hybridization
In situ hybridization experiments were basically performed as previously described (Giustetto et al., 2003). In brief, cultured A. kurodai sensory neurons were fixed with cold 4% paraformaldehyde in PBS and then permeabilized using 0.1% Triton X-100. After prehybridization with a hybridization solution (50% formamide, 5× SSC, 5× Denhardt's reagent, 0.25 g/ml yeast tRNA, and 0.5 g/ml salmon sperm DNA), hybridization was performed using 1 ng/μl of ApAF- or ApCREB1-specific, DIG-labeled antisense probe at 58°C for 12–18 h in a humidified chamber. After washing with 5× SSC at 58°C for 1 h, the sample was incubated with 10% heat-inactivated goat serum in PBS at RT for 1 h. After overnight incubation with the anti-DIG antibody (Roche), the sample was washed three times with PBS at RT for 30 min, followed by 10 mM Tris-Cl, pH 9.5, containing 0.5 mM MgCl2 for 5 min at RT. Development was performed using NBT/BCIP (Roche) for 24–36 h. The resulting cell images were acquired using a microscope (Diaphot; Nikon) attached to a digital camera system (Coolpix 995; Nikon) with a 20× objective at 18 ± 1°C. The hybridization signal in each cell was measured by outlining the cell body using the histogram function of Photoshop software (Adobe). The mean pixel intensity in the cell bodies was calculated by subtracting the background intensity from the cell body intensity.
Recombinant protein purification
WT and mutant ApAF (S175A, S266A, and S175/266A) coding sequences were amplified by PCR with primers containing restriction enzyme sites (sense primer, CGGGATCCATGATATCCAGCATTTCC; antisense primer, GCTCTAGATTAGGCTGTACCTGCCAT; restriction enzyme sites are underlined). Amplified coding sequences were serially digested with BamHI and XbaI and ligated into pGEX-KG vector (Pharmacia, Inc.) for induction in E. coli. The mutation of each construct was confirmed by sequencing. Each construct was transformed into E. coli, and protein expression was induced by 0.2 mM IPTG for 3 h at 37°C. Protein induction was confirmed by SDS-PAGE and purified by a GST purification kit (M-Biotech) as recommended by the manufacturer.
Kinase assays
To examine whether ApAF is an endogenous substrate of Aplysia PKA, the crude tissue extract preparation from A. kurodai pedal-pleural ganglia was performed as previously described (Yamamoto et al., 1999). The reaction was performed at 18°C for 20 min, containing 1 μg GST bead–binding GST-ApAF, 10 μg tissue extract, and 1 mCi γ [32P]ATP in extraction buffer. To confirm the specificity of phosphorylation, the crude tissue extracts were previously incubated for 10 min with inhibitors of specific kinases, 40 μM KT5720 (PKA inhibitor; Khabour et al., 2004), 20 μM PD98059 (MEK inhibitor), and 10 μM chelerythrine (PKC inhibitor). GST pull-down assay was performed as previously described (Sander et al., 1998). The [32P]phosphate incorporation was analyzed by SDS-PAGE and phosphoimager analysis.
To examine whether ApAF is directly phosphorylated by PKA at Ser-175 or -266, PKA kinase assay was performed at 30°C for 30 min in a final volume of 25 μl, containing 1 μg substrate, 200 μM ATP, 1 mCi γ[32P]ATP, and 5 U PKA catalytic subunit (New England Biolabs, Inc.). The reaction solution contained 50 mM Tris-Cl and 10 mM MgCl2, pH 7.5. Reactions were stopped by adding SDS sample buffer and boiling at 100°C for 5 min. [32P]phosphate incorporation was analyzed by SDS-PAGE and phosphoimager analysis. To confirm the specificity of phosphorylation by PKA, either 40 μM KT5720 (A.G. Scientific, Inc.) or DMSO was added.
Microinjection, dsRNA in vitro transcription, and reporter gene assay
Microinjection into A. kurodai neurons was performed using air pressure, as previously described (Kaang et al., 1992).
To investigate the interaction of ApAF with other transcription factors in A. kurodai neurons, such as ApC/EBP or ApCREB2, ∼20 giant cells of the abdominal ganglion were injected with 70 μg/ml of each activator DNA construct (Gal4AD-ApAF [full], Gal4AD-ApC/EBP [full], Gal4BD-ApAFbz, Gal4BD-ApC/EBPbz, and Gal4BD-ApCREB2bz), 760 μg/ml 4×gal4-lacZ, and 30 μg/ml pNEX2-luciferase, as previously described (Choi et al., 2003). Luciferase and β-galactosidase assays were carried out as previously described (Choi et al., 2003).
To investigate the effect of ApAF on CRE-driven gene expression, pNEXδ-ApAF (WT or mutants), pNEXδ-ApCREB1, or pNEXδ-ApCREB2 was coinjected with CRE-luciferase (Lee et al., 2001) into ∼30 neurons of abdominal ganglion. To examine transcriptional activity of ApAF (WT or mutants) or ApC/EBP, pNEXδ-ApAF (WT, S175A, S266A, or S175/266A) or pNEXδ-ApC/EBP was coinjected with ERE-luciferase (Lee et al., 2001) into ∼30 sensory cells of pleural ganglion. ERE (Metz and Ziff, 1991; Alberini et al., 1994) and CRE sequences were inserted into the BglII site of the pGL3-promotor vector (Promega), producing ERE- and CRE-luciferase, respectively. The inserted sequences are 5′-CGCGCGCATATTAGGACATGCGGGGCCCAGATC -3′_ (ERE) and 5′-TGGGCCCTGACGTCACGCGCGGATC-3′_ (CRE) (core enhancer sequences are underlined; Lee et al., 2001). ERE sequences in this reporter construct contain a core CAAT motif, which is homologous to the binding sequences for ApAF homodimer and ApAF–ApC/EBP heterodimer (Bartsch et al., 2000). As an internal control for gene expression, pNEX2-lacZ was included in the injection solution (Kaang, 1996). The DNA concentrations (mg/ml) in the injection solution of activators (pNEXδ-ApAF [WT or mutant], pNEXδ-ApC/EBP, pNEXδ-ApCREB1, or pNEXδ-ApCREB2), reporter (CRE- or ERE-luciferase), and pNEX2-lacZ were 0.1, 0.7, and 0.05, respectively. 24 h after microinjection, the injected ganglia were exposed to one pulse (5 min) of 10 μM 5-HT to examine the effect of ApAF regulated by 5-HT on transcriptional regulation. Ganglion (sensory cluster of pleural ganglion) cell extracts were made 48 h after microinjection using 30 μl of reporter lysis buffer per ganglion (Promega) and luciferase, and β-galactosidase assays were performed as previously described (Lee et al., 2001).
To examine the effect of dsRNA of ApAF on long-term synaptic facilitation, the sensory neurons of sensory-to-motor synapses were injected with the injection buffer containing 500 μg/ml of pNEXδ-EGFP and 500 μg/ml of dsRNA (ApAF or luciferase as a control). To make dsRNA of ApAF or luciferase, each cDNA template was linearized by HindIII–BamHI digestion of pLitmus28i-ApAF or BamHI–KpnI digestion of pLitmus28i-luciferase. dsRNAs were generated using a MEGAscript RNAi kit (Ambion) as previously described (Lee et al., 2003).
To investigate the effect of ApAF (WT or mutants) related to ApC/EBP or ApCREB2 on synaptic facilitation, sensory cells of sensory-to-motor synapses were injected with 500 μg/ml pNEXδ-ApAF (WT or mutants), 500 μg/ml pNEXδ-ApC/EBP, and 500 μg/ml pNEXδ-ApCREB2. 500 μg/ml pNEXδ-hrGFP (Lee et al., 2003) was used as an injection marker. When only either pNEXδ-ApAF or pNEXδ-ApC/EBP was injected, each plasmid was injected at concentration of 1 mg/ml instead of 500 μg/ml. To inhibit CRE-mediated gene expression using CRE oligonucleotide (Alberini et al., 1994) or K-CREB (Walton et al., 1992), the sensory neurons of sensory- to-motor synapses were injected with the injection buffer containing 50 μg/ml of CRE (or ERE) or 500 μg/ml of K-CREB before 5-HT treatment. The oligonucleotides used were as follows: CRE, TGGCATCTACGTCAAGGCTT; ERE, GATCATATTAGGACATGCGG (Alberini et al., 1994).
Cell cultures and electrophysiology
Culture dishes and media were prepared as previously described (Montarolo et al., 1986). Cultures of sensory neurons and sensory-to-motor cocultures were made as previously described (Montarolo et al., 1986; Lee et al., 2001, 2003). The motor cell was then impaled intracellularly with a glass microelectrode filled with 2 M K-acetate, 0.5 M KCl, and 10 mM K-Hepes (10–15 MΩ), and the membrane potential was held at 40 mV below its resting value. The excitatory postsynaptic potential (EPSP) was evoked in an LFS motor cell by stimulating the sensory neurons with a brief depolarizing stimulus using an extracellular electrode. The initial EPSP value was measured 24 h after microinjection. The cultures then received one or five pulses of 5-HT (10 μM) for 5 min at 15-min intervals to induce LTF. We applied one pulse of 5-HT to cells overexpressing ApAF and/or ApC/EBP to examine whether ApAF and/or ApC/EBP overexpression could convert STF to LTF. Some cultures were exposed for 2 h starting 30 min before the first application of 5-HT to membrane-permeable PKA inhibitor (10 μM KT5720; Calbiochem; Hu et al., 2003). In the MAPK experiments, 30 μM PD98059 was applied to the cultures 1 h before the application of 5-HT and during the 5-HT treatment.
The amount of synaptic facilitation was calculated as a percentage change in EPSP amplitude recorded after the 5-HT treatment versus its initial value before treatment. All data are presented as the mean percentage change ± the SEM in EPSP amplitude.
Online supplemental material
Fig. S1 shows the amino acid sequence of ApAF in A. kurodai. Fig. S2 shows the interaction of ApAF with ApC/EBP and ApCREB2. Fig. S3 shows the lack of ApC/EBP mRNA induction by one pulse of 5-HT. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200512066/DC1.
Supplementary Material
Acknowledgments
This work was supported by the Korea Ministry of Science and Technology under the Brain Research Center Frontier Program (M103KV010008-06K2201-00810), by the Korea Ministry of Marine Affairs and Fisheries under the Marine and Extreme Genome Research Center Program, and by the Korea Research Foundation grant (KRF-2005-005-J16001).
J.-A. Lee and S.-H. Lee contributed equally to this paper.
Abbreviations used in this paper: ANOVA, analysis of variance; ApAF, Aplysia activating factor; CRE, cAMP response element; ds, double stranded; EPSP, excitatory postsynaptic potential; ERE, enhancer response element; HT, hydroxytryptamine; LTF, long-term facilitation; PKA, protein kinase A; STF, short-term facilitation; WT, wild-type.
References
- Abel, T., and E. Kandel. 1998. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res. Brain Res. Rev. 26:360–378. [DOI] [PubMed] [Google Scholar]
- Alberini, C.M., M. Ghirardi, R. Metz, and E.R. Kandel. 1994. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 76:1099–1114. [DOI] [PubMed] [Google Scholar]
- Barondes, S.H., and H.D. Cohen. 1968. Memory impairment after subcutaneous injection of acetoxycycloheximide. Science. 160:556–557. [DOI] [PubMed] [Google Scholar]
- Bartsch, D., M. Ghirardi, P.A. Skehel, K.A. Karl, S.P. Herder, M. Chen, C.H. Bailey, and E.R. Kandel. 1995. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 83:979–992. [DOI] [PubMed] [Google Scholar]
- Bartsch, D., A. Casadio, K.A. Karl, P. Serodio, and E.R. Kandel. 1998. CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell. 95:211–223. [DOI] [PubMed] [Google Scholar]
- Bartsch, D., M. Ghirardi, A. Casadio, M. Giustetto, K.A. Karl, H. Zhu, and E.R. Kandel. 2000. Enhancement of memory-related long-term facilitation by ApAF, a novel transcription factor that acts downstream from both CREB1 and CREB2. Cell. 103:595–608. [DOI] [PubMed] [Google Scholar]
- Brunelli, M., V. Castellucci, and E.R. Kandel. 1976. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science. 194:1178–1181. [DOI] [PubMed] [Google Scholar]
- Byrne, J.H., and E.R. Kandel. 1996. Presynaptic facilitation revisited: state and time dependence. J. Neurosci. 16:425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellucci, V.F., E.R. Kandel, J.H. Schwartz, F.D. Wilson, A.C. Nairn, and P. Greengard. 1980. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase simulates facilitation of transmitter release underlying behavioral sensitization in Aplysia. Proc. Natl. Acad. Sci. USA. 77:7492–7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J.H., J.A. Lee, S.W. Yim, C.S. Lim, C.H. Lee, Y.D. Lee, D. Bartsch, E.R. Kandel, and B.K. Kaang. 2003. Using an Aplysia two-hybrid system to examine the interactions between transcription factors involved in long-term facilitation in the nervous system of Aplysia. Learn. Mem. 10:40–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer, J.R., F. Manseau, V.F. Castellucci, and W.S. Sossin. 2003. Serotonin persistently activates the extracellular signal-related kinase in sensory neurons of Aplysia independently of cAMP or protein kinase C. Neuroscience. 116:13–17. [DOI] [PubMed] [Google Scholar]
- Giustetto, M., A.N. Hegde, K. Si, A. Casadio, K. Inokuchi, W. Pei, E.R. Kandel, and J.H. Schwartz. 2003. Axonal transport of eukaryotic translation elongation factor 1alpha mRNA couples transcription in the nucleus to long-term facilitation at the synapse. Proc. Natl. Acad. Sci. USA. 100:13680–13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J.H., C.S. Lim, Y.S. Lee, E.R. Kandel, and B.K. Kaang. 2004. Role of Aplysia cell adhesion molecules during 5-HT-induced long-term functional and structural changes. Learn. Mem. 11:421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde, A.N., K. Inokuchi, W. Pei, A. Casadio, M. Ghirardi, D.G. Chain, K.C. Martin, E.R. Kandel, and J.H. Schwartz. 1997. Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell. 89:115–126. [DOI] [PubMed] [Google Scholar]
- Herdegen, T., and J.D. Leah. 1998. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Brain Res. Rev. 28:370–490. [DOI] [PubMed] [Google Scholar]
- Hinoi, E., V.J. Balcar, N. Kuramoto, N. Nakamichi, and Y. Yoneda. 2002. Nuclear transcription factors in the hippocampus. Prog. Neurobiol. 68:145–165. [DOI] [PubMed] [Google Scholar]
- Hu, J.Y., X. Meng, and S. Schacher. 2003. Redistribution of syntaxin mRNA in neuronal cell bodies regulates protein expression and transport during synapse formation and long-term synaptic plasticity. J. Neurosci. 23:1804–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaang, B.K. 1996. Parameters influencing ectopic gene expression in Aplysia neurons. Neurosci. Lett. 221:29–32. [DOI] [PubMed] [Google Scholar]
- Kaang, B.K., P.J. Pfaffinger, S.G. Grant, E.R. Kandel, and Y. Furukawa. 1992. Overexpression of an Aplysia shaker K+ channel gene modifies the electrical properties and synaptic efficacy of identified Aplysia neurons. Proc. Natl. Acad. Sci. USA. 89:1133–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaang, B.K., E.R. Kandel, and S.G. Grant. 1993. Activation of cAMP-responsive genes by stimuli that produce long-term facilitation in Aplysia sensory neurons. Neuron. 10:427–435. [DOI] [PubMed] [Google Scholar]
- Kandel, E.R. 2001. The molecular biology of memory storage: a dialog between genes and synapses. Biosci. Rep. 21:565–611. [DOI] [PubMed] [Google Scholar]
- Kandel, E.R., M. Brunelli, J. Byrne, and V. Castellucci. 1976. A common presynaptic locus for the synaptic changes underlying short-term habituation and sensitization of the gill-withdrawal reflex in Aplysia. Cold Spring Harb. Symp. Quant. Biol. 40:465–482. [DOI] [PubMed] [Google Scholar]
- Khabour, O., J. Levenson, L.C. Lyons, L.S. Kategaya, J. Chin, J.H. Byrne, and A. Eskin. 2004. Coregulation of glutamate uptake and long-term sensitization in Aplysia. J. Neurosci. 24:8829–8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H., S.H. Lee, J.H. Han, J.A. Lee, Y.H. Cheang, D.J. Chang, Y.S. Lee, and B.K. Kaang. 2006. A nucleolar protein ApLLP induces ApC/EBP expression required for long-term synaptic facilitation in Aplysia neurons. Neuron. 49:707–718. [DOI] [PubMed] [Google Scholar]
- Lee, J.A., H.K. Kim, K.H. Kim, J.H. Han, Y.S. Lee, C.S. Lim, D.J. Chang, T. Kubo, and B.K. Kaang. 2001. Overexpression of and RNA interference with the CCAAT enhancer-binding protein on long-term facilitation of Aplysia sensory to motor synapses. Learn. Mem. 8:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.A., H. Kim, Y.S. Lee, and B.K. Kaang. 2003. Overexpression and RNA interference of Ap-cyclic AMP-response element binding protein-2, a repressor of long-term facilitation, in Aplysia kurodai sensory-to-motor synapses. Neurosci. Lett. 337:9–12. [DOI] [PubMed] [Google Scholar]
- Martin, K.C., D. Michael, J.C. Rose, M. Barad, A. Casadio, H. Zhu, and E.R. Kandel. 1997. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 18:899–912. [DOI] [PubMed] [Google Scholar]
- Mayford, M., and E.R. Kandel. 1999. Genetic approaches to memory storage. Trends Genet. 15:463–470. [DOI] [PubMed] [Google Scholar]
- Metz, R., and E. Ziff. 1991. The helix-loop-helix protein rE12 and the C/EBP-related factor rNFIL-6 bind to neighboring sites within the c-fos serum response element. Oncogene. 6:2165–2178. [PubMed] [Google Scholar]
- Montarolo, P.G., P. Goelet, V.F. Castellucci, J. Morgan, E.R. Kandel, and S. Schacher. 1986. A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 234:1249–1254. [DOI] [PubMed] [Google Scholar]
- Ormond, J., J. Hislop, Y. Zhao, N. Webb, F. Vaillaincourt, J.R. Dyer, G. Ferraro, P. Barker, K.C. Martin, and W.S. Sossin. 2004. ApTrkl, a Trk-like receptor, mediates serotonin- dependent ERK activation and long-term facilitation in Aplysia sensory neurons. Neuron. 44:715–728. [DOI] [PubMed] [Google Scholar]
- Patterson, T.A., S.P. Rose, and P.M. Bradley. 1989. Anisomycin and amnesia in the chick: state-dependent effects are not present with intracranial injections. Brain Res. Dev. Brain Res. 49:173–178. [DOI] [PubMed] [Google Scholar]
- Pedreira, M.E., B. Dimant, D. Tomsic, L.A. Quesada-Allue, and H. Maldonado. 1995. Cycloheximide inhibits context memory and long-term habituation in the crab Chasmagnathus. Pharmacol. Biochem. Behav. 52:385–395. [DOI] [PubMed] [Google Scholar]
- Pittenger, C., Y.Y. Huang, R.F. Paletzki, R. Bourtchouladze, H. Scanlin, S. Vronskaya, and E.R. Kandel. 2002. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 34:447–462. [DOI] [PubMed] [Google Scholar]
- Rainbow, T.C. 1979. Role of RNA and protein synthesis in memory formation. Neurochem. Res. 4:297–312. [DOI] [PubMed] [Google Scholar]
- Sander, E.E., S. van Delft, J.P. ten Klooster, T. Reid, R.A. van der Kammen, F. Michiels, and J.G. Collard. 1998. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt, J.D., and E.R. Kandel. 1989. Persistent and transcriptionally-dependent increase in protein phosphorylation in long-term facilitation of Aplysia sensory neurons. Nature. 339:51–54. [DOI] [PubMed] [Google Scholar]
- Walton, K.M., R.P. Rehfuss, J.C. Chrivia, J.E. Lochner, and R.H. Goodman. 1992. A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol. Endocrinol. 6:647–655. [DOI] [PubMed] [Google Scholar]
- West, A.E., W.G. Chen, M.B. Dalva, R.E. Dolmetsch, J.M. Kornhauser, A.J. Shaywitz, M.A. Takasu, X. Tao, and M.E. Greenberg. 2001. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA. 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, N., A.N. Hegde, D.G. Chain, and J.H. Schwartz. 1999. Activation and degradation of the transcription factor C/EBP during long-term facilitation in Aplysia. J. Neurochem. 73:2415–2423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}