

Abstract
In this study, the mechanisms of actin-bundling in filopodia were examined. Analysis of cellular localization of known actin cross-linking proteins in mouse melanoma B16F1 cells revealed that fascin was specifically localized along the entire length of all filopodia, whereas other actin cross-linkers were not. RNA interference of fascin reduced the number of filopodia, and remaining filopodia had abnormal morphology with wavy and loosely bundled actin organization. Dephosphorylation of serine 39 likely determined cellular filopodia frequency. The constitutively active fascin mutant S39A increased the number and length of filopodia, whereas the inactive fascin mutant S39E reduced filopodia frequency. Fluorescence recovery after photobleaching of GFP-tagged wild-type and S39A fascin showed that dephosphorylated fascin underwent rapid cycles of association to and dissociation from actin filaments in filopodia, with t 1/2 < 10 s. We propose that fascin is a key specific actin cross-linker, providing stiffness for filopodial bundles, and that its dynamic behavior allows for efficient coordination between elongation and bundling of filopodial actin filaments.
Introduction
Cells generate two major types of actin-based protrusive organelles, lamellipodia and filopodia. Sheetlike lamellipodia contain a branched actin network (Svitkina and Borisy, 1999) and are thought to be the major engine for cell locomotion. Spikelike filopodia are thought to be the cell's sensory and guiding organelles, which function to explore the local environment and form cell–substratum or cell–cell interactions (Ridley et al., 2003).
Filopodia contain actin filaments that are organized into parallel bundles (Small, 1988; Lewis and Bridgman, 1992; Small et al., 2002). The proximal part of the bundle is usually embedded in the lamellipodial network, whereas the distal part of the bundle may or may not protrude beyond the leading edge. Nonprotruding filopodia are also called microspikes (or ribs), but, as we documented, microspikes and protruding filopodia are mechanistically related (Svitkina et al., 2003). Therefore, throughout this paper, we do not distinguish protruding and nonprotruding structures and collectively call them filopodia. Based on our recent kinetic and structural investigation, we proposed the convergent elongation mechanism for filopodial initiation by reorganization of the lamellipodial dendritic network (Svitkina et al., 2003). The first step in the process is the association of processively elongating actin filaments at their barbed ends that leads to the formation of the so-called Λ-precursors. Subsequently, these self-segregated filaments are bundled to make mature filopodia. Formation of filopodia-like bundles by a similar mechanism was reconstituted in vitro using cytoplasmic extracts or pure proteins (Vignjevic et al., 2003). Cross-linking of actin filaments is proposed to be a critical step in filopodia protrusion because individual long actin filaments lack the stiffness required to efficiently push the membrane (Mogilner and Oster, 1996; Mogilner and Rubinstein, 2005).
The leading candidate for filament bundling in filopodia is fascin, which is a 55-kD monomeric protein that cross-links actin filaments in vitro into unipolar and tightly packed bundles (Bryan and Kane, 1978; Yamashiro-Matsumura and Matsumura, 1985). In various cells, fascin localizes to filopodial bundles (Yamashiro-Matsumura and Matsumura, 1986; Sasaki et al., 1996; Yamashiro et al., 1998; Cohan et al., 2001; Kureishy et al., 2002; Svitkina et al., 2003) and is highly expressed in specialized cells that are particularly rich in filopodia, such as neurons and mature dendritic cells, as well as in many transformed cells (Kureishy et al., 2002; Hashimoto et al., 2005). Formation of filopodia-like bundles in vitro was also dependent on fascin (Vignjevic et al., 2003).
However, additional actin cross-linking proteins, including α-actinin, espin, fimbrin, and villin, are involved in the formation of certain parallel actin bundles, such as those in microvilli, bristles, or stereocilia (Bartles, 2000; DeRosier and Tilney, 2000). A recent in vitro study (Brieher et al., 2004) showed that fimbrin, α-actinin, and filamin, as well as fascin, can support transient Listeria monocytogenes motility via actin bundling. Therefore, it is still uncertain whether fascin is sufficient for filopodia formation or multiple bundlers share the role.
During the dynamic process of the filopodial life cycle, filament bundling should be coordinated with actin polymerization. Biochemical experiments predict very tight bundling of actin filaments by fascin. The actin-bundling activity of fascin (Yamakita et al., 1996; Ono et al., 1997) and fascin localization in cells (Adams et al., 1999) are regulated by phosphorylation of serine 39 within the N-terminal actin-binding domain, suggesting that phosphorylation–dephosphorylation cycles of fascin may be coupled with filopodial dynamics. This hypothesis needs to be experimentally tested, as the dynamics of fascin in filopodia has not yet been closely investigated.
In this work, we investigated the function and dynamics of fascin during filopodia formation by a combination of RNAi and the expression of phosphomimetic mutants. Our results demonstrate that fascin plays critical roles in the initiation and protrusion of filopodia by providing them the necessary stiffness, and that fascin-mediated cross-linking of actin filaments in filopodial bundles is unexpectedly highly dynamic.
Results
Fascin is the major actin cross-linker localized in filopodia
We evaluated the presence of known actin cross-linkers in filopodia of B16F1 mouse melanoma cells. Expression of YFP-fascin (Fig. 1) and immunostaining with fascin antibody (not depicted) demonstrated that fascin localized throughout the length of all filopodia. Espin, villin, fascin 3, and L-fimbrin were not expressed in these cells, as revealed by microarray analysis (see Materials and methods). These results are consistent with previous reports on tissue-specific expression (DeRosier and Tilney, 2000), whereas α-actinin and T-fimbrin (the ubiquitous isoform) were expressed.
Figure 1.

Localization of actin cross-linking proteins in B16F1 melanoma cells. (A) YFP-fascin and CFP–α-actinin expression patterns. Fascin is enriched in filopodia, whereas α-actinin localizes to actin “spots” (arrow) and focal contacts (arrowhead), and, slightly, to filopodial roots, but not to protruding parts (bottom row). (B) Localization of GFP-fimbrin. Actin filaments are labeled with the Texas red–X phalloidin. Fimbrin is enriched both in lamellipodia and filopodia. (C) Localization of ectopically expressed GFP-espin. Actin filaments are labeled with the Texas red–X phalloidin. Espin is present only in proximal regions of filopodial bundles. Bars, 10 μm.
Coexpression of YFP-fascin and CFP–α-actinin showed different distribution of these two cross-linkers (Fig. 1 A). As expected, α-actinin was found in stress fibers and focal contacts (Lazarides and Burridge, 1975; Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200603013/DC1), in actin “spots” (Schafer et al., 1998), probably corresponding to invadopodia or podosomes (Buccione et al., 2004), and weakly in lamellipodia and, occasionally, in the internal, but not the protruding, parts of filopodial bundles (Svitkina et al., 2003).
Fimbrin and espin are actin cross-linkers in microvilli of epithelial cells and in hair cell stereocilia (Tilney et al., 1989, 1992; Bartles et al., 1998; Zheng et al., 2000; Loomis et al., 2003). In B16F1 cells, expressed GFP–T-fimbrin localized to lamellipodia and filopodia, whereas its localization to stress fibers was very weak (Fig. 1 B). Similar localization of the endogenous protein was detected by immunostaining (Fig. S1 B). Ectopically expressed GFP-espin was targeted to many, although not all, filopodia, but predominantly to their proximal parts (Fig. 1 C), whereas mCherry-fascin always localized throughout the entire length of filopodia (Fig. S1 C). Thus, molecular marker analysis showed that endogenously expressed actin cross-linkers, other than fascin, did not exhibit specific filopodial targeting, but also localized to other actin structures, lamellipodia, and/or stress fibers. These results suggest that fascin is a major specific bundling protein in filopodia of B16F1 cells, although the contribution of other cross-linkers is not necessarily excluded.
Fascin depletion inhibits filopodia formation
The roles of fascin in filopodia formation were investigated by RNAi using pG-SUPER plasmid, which coexpresses small hairpin RNA (shRNA) and GFP simultaneously (Kojima et al., 2004). The following three target sequences were used (Fig. 2 A): Tc, which is common to mouse and human fascin; mouse-specific Tm; and human-specific Th, as a control.
Figure 2.

Expression of shRNA depletes fascin in B16F1 cells. (A) Target sequences for RNAi. Tc (left) is common for mouse and human fascin; Th and Tm (right) are common for human and mouse fascin mRNA, respectively, and differ in two base pairs. (B) Immunoblot analysis. Lysates prepared from FACS-purified cell populations 5 d after transfection were probed with fascin antibody. Tubulin served as a loading control. (C) Microscopic analysis. GFP fluorescence serves as a marker for cells transfected with the Tm, Th, or Tc constructs. Fascin is visualized by immunofluorescence and actin is stained with fluorescent phalloidin. Merged images show GFP (green) and fascin staining (red) simultaneously. Intensity line scans (right) through one transfected and one untransfected cell (lines in merged images) show fascin and GFP intensities in red and green, respectively. Bar, 20 μm.
Fascin silencing in B16F1 cells was assayed by immunoblotting and immunostaining. For immunoblotting, GFP-expressing cells were collected by FACS 1 d after transfection and cultured for an additional 4 d. Blots (Fig. 2 B) were analyzed by densitometry (two independent experiments), and normalized to a loading control and to the percentage of GFP-positive cells (∼75% on day four). The average reduction in fascin was 90% for the Tc and 85% for the Tm shRNAs. The control Th shRNA did not decrease fascin. Immunostaining showed that expression of Tm or Tc, but not of Th, shRNAs significantly decreased levels of fascin in mouse B16F1 cells (Fig. 2 C). Conversely, Th, but not Tm, was effective in fascin knockdown in human HeLa cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200603013/DC1).
Phalloidin staining of fascin-depleted cells showed a 4–5-fold decrease in the number of filopodial bundles, whereas lamellipodia were apparently unaffected (Fig. 3, A and B). Remaining filopodia did not contain detectable amounts of fascin as determined by immunostaining (not depicted) and were often wavy and running parallel to the leading edge (Fig. 3 A, right), as if they were buckled because of the compromised stiffness. EM analysis (see the following paragraph) confirmed that these lateral actin-rich structures were bundles of long filaments, but not ruffles.
Figure 3.

Inhibition of filopodia formation by fascin knockdown. (A) Distribution of actin revealed by phalloidin staining. Fewer filopodia can be seen in cells transfected with knockdown constructs (Tm and Tc) compared with the control (Th). The remaining filopodia are bent and buckled in Tm- and Tc-transfected cells. Asterisk-labeled filopodia are enlarged on the right. (B) Quantification of filopodia. Number of filopodia per 20 μm of cell leading edge was counted in cells untransfected or transfected with the indicated constructs. WT*-fascin stands for YFP-fascin refractory to Tc shRNA. Inset illustrates an example of the cell perimeter area selected for the analysis. Frequency of filopodia is reduced significantly by Tm and Tc shRNA (P < 0.002). (C) Rescue of the knockdown phenotype by fascin. A cell expressing both CFP-Tc shRNA (CFP) and YFP-WT*-fascin refractory to siRNA (fascin-WT*) displays numerous filopodia. (D) Rescue of the knockdown phenotype by α-actinin or T-fimbrin. Filopodia formation in CFP-Tc shRNA–expressing cells (CFP) is not rescued by YFP–α-actinin (left column), and only partially rescued by GFP-fimbrin (right column). Bars, 10 μm.
By platinum replica EM, the remaining filopodia of fascin-depleted cells did not contain tightly packed straight actin filament bundles as in control cells (Fig. 4). Instead, they consisted of rather loosely arranged actin filaments that were wavy and ran along the cell edge, which is consistent with light microscopic data. The internal parts of these bundles were remarkably long (9.5 ± 3.9 μm versus 3.0 ± 1.7 μm in control cells; P < 0.001; n Tc = 6 cells, 25 filopodia; n control = 8 cells, 49 filopodia). In contrast, the structural organization of the lamellipodial network looked indistinguishable from normal B16F1 cells. These results establish that fascin is required for filopodia formation in B16F1 cells and suggest that it participates in filopodial protrusion by cross-linking actin filament bundles and, thus, providing them the necessary stiffness.
Figure 4.

EM of filopodia in fascin knockdown cells. Filopodium in a control Th-shRNA–transfected cell (left) is tightly bundled and runs almost perpendicular to the leading edge. The remaining filopodium in fascin knockdown Tc-shRNA–transfected cell (right) has very long internal bundles, which are bent and buckled behind the leading edge and consist of loosely organized filaments, whereas neighboring lamellipodium has normal dendritic organization. Boxed region is enlarged at bottom right. Bar, 100 nm.
The specificity of fascin knockdown phenotype was confirmed by rescue experiments. YFP-tagged human fascin refractory to RNAi (WT*-fascin) restored filopodia formation in cells expressing Tc-shRNA together with CFP marker (Fig. 3 C). Almost all cells (90%; n = 80) expressing both CFP-Tc and YFP-WT*-fascin had numerous filopodia (5.5 ± 2.5 per 20 μm of the perimeter) containing fascin along their length, which is similar to control cells (6.8 ± 2.6). Rescue also established that fascin tagged with derivatives of GFP is functional.
Furthermore, we tested whether different actin filament cross-linkers, i.e., α-actinin or fimbrin, could rescue filopodia formation in the absence of fascin. Cells cotransfected with CFP-Tc and YFP–α-actinin showed α-actinin localizing to focal adhesions and stress fibers, but not to filopodia; phase-contrast images of such cells did not show restoration of filopodia (Fig. 3 C). In similar conditions, GFP–T-fimbrin was able to partially rescue filopodia formation in ∼50% of cells (n = 31), which showed on average 2.4 ± 0.6 filopodia per 20 μm of the cell perimeter, whereas other cells did not show any signs of rescue. This variability might depend on the level of GFP–T-fimbrin overexpression. Thus, other actin filament cross-linkers cannot fully substitute for fascin function in vivo.
Role of fascin phosphorylation in filopodia formation
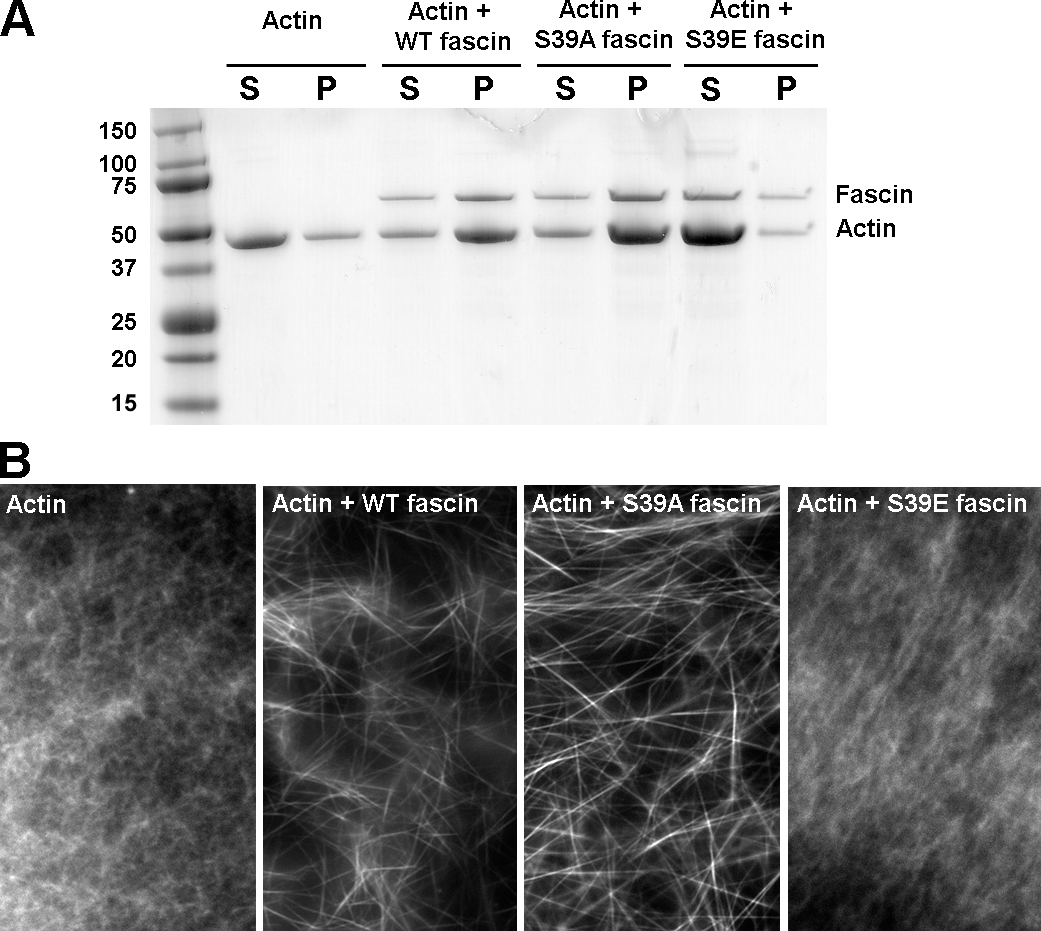
Phosphorylation of fascin at serine 39 is important for its actin bundling activity in vitro (Yamakita et al., 1996; Ono et al., 1997) and proper localization in vivo (Adams et al., 1999). To examine the roles of serine39 phosphorylation in filopodia formation, we produced point mutants mimicking the active dephosphorylated (S39A) or inactive phosphorylated (S39E) states of fascin. In vitro bundling assays with F-actin and bacterially expressed and purified wild-type (WT), S39A, or S39E fascins confirmed the actin-bundling activity of WT and S39A fascins, but not of S39E fascin (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200603013/DC1), which is thus similar to the previously characterized S39D mutant (Adams et al., 1999). WT and mutant fascin proteins were also assayed for their ability to associate with filopodial bundles in detergent-extracted and buffer-incubated cytoskeletons. Long incubation of lysed cells in a stabilizing buffer results in complete dissociation of endogenous fascin, whereas the actin cytoskeleton remains almost intact (Svitkina et al., 2003). When applied to fascin-depleted cytoskeletons, WT and S39A, but not S39E, fascin associated with filopodia along their length (unpublished data).
Expression of GFP-tagged S39A or S39E fascin mutants in B16F1 cells had opposite effects on filopodia formation. The S39A mutant induced long, overabundant filopodia extending from the cell edges, as well as from the dorsal surface (Fig. 5 A). Quantification of lateral filopodia revealed an ∼1.6-fold increase in filopodia frequency compared with control cells (Fig. 5 B). The actual degree of filopodia stimulation was even greater because dorsal filopodia were not scored. Expression of the S39A mutant also led to elongation of the protruding parts of filopodial bundles, whereas their internal parts remained of the same length (Fig. 5 C). In contrast, expression of the S39E mutant reduced the number of filopodia by ∼2.5-fold and had no effect on the length of remaining filopodia (Fig. 5, B and C). The dominant-negative effect of S39E was also observed in 3T3 cells, where S39E expression significantly reduced the number of filopodia from 4.3 ± 2.9 to 1.9 ± 1.9 (n = 31; P < 0.0002). 3T3 cells expressing S39A mutant had a similar frequency of filopodia as control cells (3.9 ± 5.1; n = 30).
Figure 5.

Effects of fascin mutants on filopodia number and length. (A) Phalloidin staining of untransfected cells and cells expressing indicated GFP-fascins. S39A- or S39E-expressing cells show increased or decreased filopodia formation, respectively. (B) Number of filopodia per 20 μm of cell leading edge in cells transfected with indicated constructs. Asterisks indicate statistically significant changes (P < 0.003) compared with untransfected cells. GFP stands for empty GFP vector. Error bars represent the mean ± the SEM. (C) Lengths of the filopodia protruding beyond leading edge (dark gray bars) and internal (light gray bars) parts of filopodia. Expression of S39A fascin increases the length of the protruding parts of filopodia. (D) EM of filopodia in cells transfected with fascin mutants. A filopodium in S39A-expressing cell (left) is long, thin, straight, and tightly bundled. In S39E-expressing cell (right), a filopodium is loosely bundled (top). Many long filaments converge at the leading edge, forming an abnormally large Λ-precursor (bottom). Bars: (A) 10 μm; (D) 100 nm.
The structural organization of filopodia in B16F1 cells expressing the fascin mutants was analyzed by EM. Similar to normal ones (Fig. 5 D), filopodia induced by the S39A mutant were straight, orthogonal to the edge, and composed of tightly bundled actin filaments. However, these bundles were typically thinner than in control cells (Fig. 4), correlating with the lower fluorescence intensity of phalloidin staining in S39A-induced filopodia (Fig. 5 A). In S39E-transfected cells, filopodial bundles had normal thickness, but were often loosely bundled, especially farther away from the tip (Fig. 5 D, right). In contrast to siRNA-treated cells, the length of the internal parts of filopodial bundles (2.9 ± 1.1 μm; n = 7 cells, 33 filopodia) was not significantly different from control cells (3.0 ± 1.7 μm; n = 8 cells, 49 filopodia), which is consistent with light microscopic measurements. However, the surrounding lamellipodia frequently contained multiple series of long actin filaments that converged to a common tip at the leading edge (Fig. 5 D, bottom right). Such structures were similar to Λ-precursors, which are the intermediates during filopodia initiation (Svitkina et al., 2003), except that they were about fourfold larger than normal Λ-precursors (0.48 ± 0.66 μm2 versus 0.10 ± 0.17 μm2 in control cells, P < 0.001; n control = 28 and n S39E = 56 in 5 cells each). These results suggest that the S39E fascin mutant acted as a dominant negative by inhibiting filopodia formation at the stage of initiation of bundling.
Dynamics of fascin in filopodia
In live B16F1 cells, WT and active S39A fascins essentially colocalized with actin in filopodia (Fig. 7, A and B), except for the proximal regions of filopodial bundles, where the fascin/actin fluorescence ratio was slightly lower, as previously reported (Cohan et al., 2001). In contrast, S39E fascin was present only in the distal (∼65%) regions of filopodial bundles visualized by phalloidin staining. Approximately 38% of filopodia also displayed prominent enrichment of S39E fascin at filopodial tips (Fig. 7 B). The dynamics of fascins were analyzed by three different approaches.
Figure 7.

Distribution of fascin mutants in filopodial bundles. (A) Fluorescence images of cells expressing GFP-tagged WT, S39A, and S39E fascins. S39E fascin is enriched at filopodial tips. Bar, 10 μm. (B) Individual filopodia (top) and corresponding fluorescent intensity line scans (bottom) from GFP-fascin–transfected cells costained with phalloidin. Red, actin; green, fascin. Inactive S39E mutant is significantly enriched at filopodial tips, whereas distribution of WT and S39A fascins matches that of actin. (C, left) Fascin dissociation from filopodia after detergent extraction. Cells transfected with indicated GFP-tagged fascins are shown live immediately before extraction (time 0) and after detergent extraction for indicated periods of time. WT and S39A fascin remain associated with filopodia for a long time after cell lysis, whereas S39E fascin dissociates within the first 30 s from the tip of filopodia. (right) Line-scan analysis of kinetics of fascin dissociation. Fluorescence intensity profile of GFP-fascin is shown along the length of the filopodia indicated by asterisks in A (filopodial tips are at left). Individual lines in each plot represent different time points after lysis, as indicated.
FRAP of GFP-fascin was used to analyze fascin turnover in filopodia. Surprisingly, FRAP of GFP-WT fascin was rather rapid in B16F1 cells with half time of recovery t 1/2 = 9 ± 6 s (n = 41; Fig. 6 A). Furthermore, 96 ± 37% of fluorescence was recovered, indicating that the majority of fascin population in filopodia was dynamic. In contrast, FRAP of GFP-actin in B16F1 cells resulted in no detectable recovery (Fig. 6, B and C). Instead, bleached zones displayed retrograde flow, as shown previously (Mallavarapu and Mitchison, 1999). Next, we examined whether fascin dynamics depends on Ser39 phosphorylation. FRAP of GFP-S39A fascin (Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200603013/DC1) was similar to that of GFP-WT fascin both for the half-life time (t 1/2 = 6 ± 4 s) and the final recovery level (101 ± 31%; n =39), indicating that active dephosphorylated fascin also undergoes rapid association/dissociation cycles. The similar fast dynamics of WT and S39A fascins were also observed in mouse neuroblastoma Neuro2A cell line (unpublished data). We tried FRAP of GFP-S39E fascin in B16F1 cells, but a small number of filopodia remained in S39E-expressing cells, and their lateral movement, rather than persistent extension, precluded obtaining enough data for meaningful curve fitting.
Figure 6.

FRAP of GFP-fascin and actin in filopodia. (A) FRAP of GFP-fascin. Fluorescence and phase-contrast images of a GFP-fascin–expressing B16F1 cell show an overview of the cell (left) and a time-lapse sequence of the boxed region before and after photobleaching. Fluorescence of GFP-fascin recovered within 30 s. (B) FRAP of GFP-actin. Fluorescence time-lapse sequence of B16F1 cell expressing GFP-actin. There was no fluorescence recovery of GFP-actin over time. Time after bleaching in A and B is given in seconds. Fluorescence images are presented after correction of photofading during image acquisition (see Materials and methods). (C) Kymograph analysis of the bleached filopodium. The bleached region moved toward the base of the filopodium at the rate of 2 μm/min. Bars, 1 μm.
As an alternative approach to evaluate the dynamics of fascins, we monitored the dissociation of GFP-fascins from filopodia after cell lysis during incubation in the stabilization buffer. Cell lysis almost immediately removed cytoplasmic fluorescence of all GFP-fascins, suggesting that this pool represented soluble and weakly bound proteins. Filopodia-associated WT and S39A fascins dissociated slowly, with half-life times of 62 ± 40 min and 25 ± 6 min, respectively (Fig. 7 C). Faster dissociation of S39A may be related to lower thickness of S39A-induced filopodia, where internal fascin molecules can reach the surface more rapidly. In contrast, S39E fascin was lost within 1 min upon cell lysis. Importantly, the brighter tip fluorescence seemed to dissociate more quickly than the dimmer shaft fluorescence, such that the fluorescence profile of filopodia at intermediate stages of extraction lost the characteristic peak at the tip (Fig. 7 C). These results show that S39E fascin is very weakly bound to actin bundles and that its enrichment at the tips may be independent of actin.
Next, the dynamics of mutant fascins, on the background of the silenced endogenous fascin, were analyzed in living cells during filopodia initiation. Refractory to silencing YFP-fascins (S39A* and S39E*) were coexpressed with CFP-Tc plasmid (Fig. 8 A). In these conditions, the active YFP-S39A* fascin enhanced filopodia formation (7.1 ± 1.4 filopodia per 20 μm of cell perimeter; n = 18), as in normal cells (Fig. 3 B). Coexpression of the inactive YFP-S39E* and Tc shRNA did not exhibit additive inhibition of filopodia formation (1.0 ± 0.9; n = 33) as compared with YFP-S39E alone (Fig. 3 B). Time-lapse imaging showed that at the initial stages of filopodia formation (Fig. 8 B), WT, S39E, and S39A fascins similarly appeared as dots at the leading edges, suggesting that recruitment of fascin to the tips is not critically dependent on the phosphorylation state of fascin. In contrast to WT and S39A that are continuously incorporated into the shaft of the elongating filopodium, S39E fascin seemed to dissociate from the filopodia soon after it appeared at the tip. Consistent with fast dissociation of S39E mutant from filopodia in lysed cells, the lifetime of S39E in filopodia in live cells was very short, estimated as a few tens of seconds, whereas WT and S39A filopodia lived for minutes. Thus, incorporation of fascin into filopodial shafts is likely dependent on Ser39 dephosphorylation.
Figure 8.

Dynamics of WT and mutant fascins in living cells. (A) Fluorescence images of cells expressing both CFP-Tc-shRNA (left) and one of YFP-tagged fascins refractory to shRNA (right). Active S39A fascin mutant (S39A*) induces numerous filopodia and shows localization along their length, whereas inactive S39E fascin mutant (S39E*) fails to increase the frequency of filopodia even at high levels of expression and shows mostly diffuse localization throughout the cytoplasm with slight enrichment in occasional filopodia. Bar, 10 μm. (B) Filopodial dynamics of WT or mutant fascins. Asterisks mark specific filopodia over time. Time is shown in seconds.
Discussion
We previously formulated the convergent elongation model based on analysis of filopodia initiation in B16 cells. This model suggests that a special structure at filopodial tips, the filopodial tip complex, drives reorganization of the lamellipodial dendritic network into filopodial bundles (Svitkina et al., 2003). According to this model, filaments nucleated within the lamellipodial network gain protection against capping and, thus, can elongate persistently at their barbed ends (Mejillano et al., 2004). Collisions and clustering of barbed ends during elongation leads to formation of filopodial precursors, called Λ-precursors. The Λ-precursors consist of long actin filaments converging at the cell edge; they are subsequently reorganized to filopodia by bundling at their vertices. In this work, we investigated the molecular mechanisms of this last step of filopodia formation and the functional contribution of an actin cross-linker, fascin, to this process.
Fascin is essential for filopodia formation
Several previous studies, using antibodies, membrane-permeable peptides, and fascin mutants, indicated that fascin is an excellent candidate for actin filament bundling in filopodia (Adams et al., 1999; Al-Alwan et al., 2001; Anilkumar et al., 2003), but this hypothesis was not directly and definitively tested. In this study, using several complementary functional approaches—by evaluating other potential candidates, by suppressing fascin expression by RNAi, and by analyzing the phenotypes of the Ser39 mutants—we demonstrated that fascin is indeed a key filopodial bundler that plays a critical role in formation and protrusion of filopodia by providing them with the necessary stiffness to push against the membrane.
In the first approach, analysis of potential cross-linkers by gene microarray, immunofluorescence, and ectopic protein expression revealed that among known endogenously expressed actin cross-linkers, only fascin specifically localized to filopodia. Although T-fimbrin was also detected in filopodia, it bound lamellipodial filaments equally well, suggesting that T-fimbrin may not be primarily responsible for initiation of filopodial bundles. These results allowed us to focus on fascin while investigating the molecular mechanisms of filopodia initiation.
Depletion of fascin by RNAi led to significant reduction of filopodia frequency and drastically compromised the mechanical properties of remaining filopodia. Because the combination of siRNA and dominant-negative approaches did not show additive effects, other cross-linkers likely contributed to formation of the remaining bundles. One candidate is T-fimbrin, because its overexpression could partially restore filopodia formation, although the endogenous level of T-fimbrin was not sufficient. Interestingly, many other specialized bundles usually require two cross-linkers to be fully functional, and fascin is commonly the major one (Tilney and DeRosier, 2005). The inability of α-actinin to rescue filopodia suggests that tight bundling with short interfilament spacing, like that mediated by fascin or fimbrin, is required to provide filopodial bundles with suitable mechanical properties. Interestingly, the remaining filopodial bundles in fascin-depleted cells not only buckled under the membrane but were also extremely long, suggesting that filopodial bundles may depolymerize in more internal cell regions, but not in the leading edge vicinity.
The importance of fascin for filopodia formation can be explained based on physical properties of actin filaments. Individual filaments are too flexible to resist compressive forces, and therefore elongation of individual filaments, per se, is predicted to be insufficient for filopodial protrusion. Bundling provides enough rigidity to actin filaments for protrusion against the compressive force (Mogilner and Rubinstein, 2005). This prediction was proven by in vitro assays using deformation of large liposomes (Honda et al., 1999) and L. monocytogenes motility (Brieher et al., 2004). Our observation is a proof of this idea in vivo. The remaining filopodia in fascin-depleted B16F1 cells were usually bent and buckled under the cell membrane, as if they were not able to resist compression. Structurally, individual filaments in these filopodia were not tightly packed, suggesting a correlation between bundling and rigidity.
Role of fascin phosphorylation in filopodia formation
Phosphorylation at serine39 regulates actin-binding and bundling abilities of fascin. Phosphorylation of fascin by protein kinase Cα in vitro reduced its actin-binding constant from 3.5 × 106 M−1 to <0.5 × 105 M−1 (Ono et al., 1997). In vivo, C2C12 cells expressing S39D fascin were able to attach to thrombospondin-1, but did not form microspikes (Adams et al., 1999). In our study, expression of a similar S39E mutant inhibited formation of filopodia and microspikes. This dominant-negative activity of inactive fascin mutants could be explained by competition with endogenous fascin for actin filaments. Indeed, the S39E mutation compromised only the one actin-binding site, while another site located in the C-terminal half (Ono et al., 1997) is presumably intact. However, our EM analysis suggests additional mechanism of the S39E dominant-negative effect because S39E-expressing cells displayed even more severe phenotype compared with knockdown cells. Although shRNA-expressing cells formed long, although morphologically unusual, bundles, S39E-expressing cells had abundant oversized Λ-precursors at their leading edges. This observation suggests that in S39E-expressing cells the process of filopodia initiation occurred normally up to the stage of formation of Λ-precursors, but then failed because of the cell's inability to bundle actin filaments. A possible explanation for this phenomenon is that S39E, for some reason, may compete not only with endogenous fascin, but also with other cross-linkers, potentially T-fimbrin. In contrast to the inactive S39E mutant, the active S39A fascin positively regulated filopodia and increased their length and number. Interestingly, targeting of protein phosphatase I to the actin cytoskeleton in neurons by neurabin I promoted filopodia formation (Oliver et al., 2002). It is tempting to speculate that this effect may be caused by fascin dephosphorylation, although it remains unknown whether fascin is a substrate of this phosphatase. S39A fascin may increase the stiffness per diameter of filopodia. If this is the case, newly formed filopodia will be able to start protrusion with fewer actin filaments, as was observed by EM. Such filopodia need fewer actin monomers per length of filopodial extension. That would explain excessive elongation of the external part of S39A-induced filopodia, where the availability of actin monomers is likely a limiting factor (Mogilner and Rubinstein, 2005). Considering the opposite phenotype produced by S39E and S39A fascins, we propose that the level of active fascin in cells determines the number and thickness of filopodia by converting Λ-precursors to genuine filopodia through initiation of bundling at different stages of precursor maturation.
Fascin dynamics in filopodia
An important and unexpected finding in this work comes from the FRAP experiments, which demonstrate that fascin in filopodial bundles undergoes frequent cycles of association and dissociation rather than remaining stably bound to actin filaments. Importantly, fast dynamics was also observed for the constitutively active S39A mutant, suggesting that fast fascin turnover is not coupled to cycles of phosphorylation and dephosphorylation. Such behavior of active dephosphorylated fascin may seem counterintuitive if one assumes that rapid dynamics is not consistent with the rigidity of actin bundles. However, a high dissociation rate does not necessarily imply a weak binding affinity. If the association constant is fast as well, the affinity can be sufficiently strong. Also, actin filaments in filopodia are very densely packed and many fascin molecules participate in the formation of actin bundles.
The fast dynamics of fascin in vivo seems to conflict with the results of cell lysis experiments that gave much slower dissociation rates for S39A and WT fascin. However, the in vitro dissociation experiments were performed after detergent extraction in stabilizing buffer containing polyethyleneglycol, which significantly slows down the dissociation of fascin. This delayed dissociation allowed us to perform more reliable comparison of WT, S39A, and S39E fascins. In the absence of polyethyleneglycol, fascin dissociation from actin filaments was faster (unpublished data).
What might be an advantage of the dynamic cross-links compared with stable ones? We propose that dynamic binding would enhance the availability of fascin molecules for newly polymerized actin filaments at the filopodial tips by decreasing the diffusion distance. This would ensure timely filament bundling during filopodia elongation. If fascin was stably bound to actin filaments in filopodia and released only with filament disassembly at the rear, free fascin would need to diffuse through the whole filopodial length, which is on the order of micrometers, to reach sites of growth. Another possible advantage of dynamic cross-links is to allow for transport of other molecules. For instance, myosin X is proposed to deliver its cargo toward the tip of filopodia (Berg and Cheney, 2002). The local debundling and rebundling may transiently generate space for passage of such cargos. Finally, local stresses constantly generated inside filopodial bundles during motility, such as uneven tension, torque, bends, etc., can be more easily relieved by dynamic cross-links. Therefore, although dynamic association/dissociation cycles of fascin seem counterintuitive, such a property may be advantageous for rapid growth and integrity of filopodia.
The mechanism of fascin targeting to filopodia
As a potent actin filament cross-linker, expressed fascin might be able to associate with actin filaments throughout a cell. Yet, it specifically localized to filopodia, but not, for example, to stress fibers. This targeting specificity was also displayed by the constitutively active S39A fascin, suggesting that dephosphorylation and phosphorylation of fascin are not necessary for regulating its specific targeting. One possible mechanism of fascin targeting to filopodia is that it specifically recognizes closely opposed parallel actin filaments. During normal filopodia formation, accumulation of closely spaced parallel filaments at the tips of Λ-precursors may serve as sites for fascin recruitment. Additional mechanisms may contribute to fascin exclusion from other bundles, for example, through competition with tropomyosin (Ishikawa et al., 1998; DesMarais et al., 2002).
Even if parallel-aligned filaments are sufficient to recruit fascin, our data suggest an additional mechanism that coordinates filament elongation and bundling in growing filopodia in B16F1 cells. We found that the inactive S39E fascin was frequently enriched at the filopodia tips, suggesting a recruitment mechanism independent of actin bundling. As we previously showed, the filopodial tips are associated with a large protein complex. The composition of the tip complex is poorly characterized, but some components are known, including Ena/VASP proteins (Lanier et al., 1999; Rottner et al., 1999), myosin X (Berg and Cheney, 2002), Vav 1 (Kranewitter et al., 2001), Abi (Stradal et al., 2001), and formins (Peng et al., 2003; Pellegrin and Mellor, 2005; Schirenbeck et al., 2005). Some of these or unknown components of the filopodial tip complex might be responsible for recruiting inactive fascin to filopodial tips. Interestingly, it was shown that neurotrophin receptor p75 specifically binds fascin in human melanoma cells (Shonukan et al., 2003) and thus is a candidate for recruiting fascin to the membrane. However, it is not known whether this protein is specifically enriched at filopodial tips. Another possibility for fascin targeting to filopodial tips is that fascin could preferably bind ATP-actin near the growing barbed ends. Concentration of fascin at the filopodia tips, which may also be coupled to activation by dephosphorylation, would enhance the availability of the cross-linker for actin bundling. Although we were unable to detect enrichment of S39E fascin at the filopodial tips in all tested cell lines, such a mechanism of fascin targeting may be important for some specialized cells.
In summary, we propose the following model for the role of fascin in filopodia formation (Fig. 9). Fascin is present in the cytoplasm as a mixture of inactive phosphorylated and active dephosphorylated forms. Active fascin is recruited to sites where actin filaments are parallel and nearly aligned, like at the tips of Λ-precursors. Concentration of active fascin in the cytoplasm determines the frequency of transformation of Λ-precursors to filopodia. Recruitment of fascin to filopodia tips may be enhanced by some component(s) of the filopodial tip complex. When bound, fascin increases the stiffness of filopodial bundles and, thus, promotes extension of filopodia beyond the leading edge. Fast dynamics of fascin, while bound to actin filaments, is important for extension and maintenance of filopodia.
Figure 9.

Model of fascin role in filopodia formation. (1) Uncapped actin filaments elongate and form Λ-precursors. (2) Fascin becomes recruited to the tip complex, through low-affinity interaction. (3) Elongation of filaments is closely followed by fascin recruitment and cross-linking, which allows the bundling process to keep up with elongation and guarantees efficient pushing. (4) Filopodia is formed. (5 and 6) Fascin molecules undergo fast dissociation/association from the shaft of filopodia, ensuring its concentration is high enough, which is necessary for coordination of actin elongation and bundling at the filopodia tips. Different shades of fascin are used to illustrate the dynamic character of the bundling process.
Materials and methods
Plasmids
The pEGFP-fascin construct was provided by J. Adams (Cleveland Clinic Foundation, Cleveland, OH). A site-directed mutagenesis kit (QuikChangeII; Stratagene) was used to create point mutations. YFP/CFP/mCherry-fascin constructs (WT and mutants) were obtained by transferring the fascin cDNA from pEGFP-fascin to pEYFP/pECFP-C1 (CLONTECH Laboratories, Inc.) and pmCherry-C1 with BsrGI–BamHI sites. pmCherry-C1 was constructed by replacing EGFP cDNA in pEGFP-C1 with a PCR-amplified mCherry sequence. The original mCherry construct was provided by R. Tsien (University of California, San Diego, CA). An expression construct of EGFP–α-actinin was obtained from C. Otey (University of North Carolina, Chapel Hill, NC). The α-actinin–coding sequence was transferred to HindIII gaps of pEYFP/pECFP-N1 to prepare pEYFP/pECFP–α-actinin. pEGFP–T-fimbrin and pEGFP-espin2B were provided by J. Bartles (Northwestern University Medical School, Chicago, IL). pG-SUPER was previously described elsewhere (Kojima et al., 2004). pG-SUPER-Fascin-Th, -Tm, and -Tc were constructed as previously described (Brummelkamp et al., 2002). The selected target sequences were as follows: for Th, nt 741–759 of human fascin1 (NM_003088); for Tm, nt 741–759 of mouse fascin1 (NM_007984); and for Tc, nt 393–411 of human/mouse fascin1 (conserved). The selected sequences did not have significant homology to any other known genes in the mouse database, as determined using the Basic Local Alignment Search Tool (National Center for Biotechnology Information). Th and Tm had two base mismatches, in that Th served as a negative control of Tm (Fig. 2 A). pC-SUPER-Fascin-Tm and -Tc were constructed by replacing EGFP cDNA of pG-SUPER with ECFP cDNA from pECFP-N1 (CLONTECH Laboratories, Inc.) using SacII–NotI. The rescue construct, pEYFP-fascin* contained three silent point mutations in two codons, S133S (AGC–TCC) and A137A (GCC–GCT), and was refractory to Tc-siRNA.
Cell culture and microscopy
B16F1 mouse melanoma and Neuro2a mouse neuroblastoma lines were provided by C. Ballestrem (Weizmann Institute of Science, Rehovot, Israel) and A. Ferreira (Northwestern University, Chicago, IL), respectively, and maintained in MEM supplemented with 10% FBS at 37°C. NIH 3T3 and HeLa lines were obtained from American Type Culture Collection and cultured according to their instruction. For microscopy experiments, cells were plated onto coverslips, which were coated with 20 μg/ml mouse laminin (Invitrogen or Sigma-Aldrich) for 1 h, and then incubated with 1% bovine serum albumin for 20 min. Transfection was performed by FuGENE6 (Roche) according to the manufacturer's instructions.
Light microscopy was performed using a microscope (Eclipse TE2000; Nikon) equipped with a Plan 100×, 1.3 NA, objective (Nikon) and a back-illuminated cooled charge-coupled device camera (model CH250; Roper Scientific). The MetaMorph Imaging software (Universal Imaging Corp.) was used for image acquisition and analysis. For live cell imaging, cells were kept in a CO2-independent culture medium (L-15; Invitrogen) at 37°C.
Immunofluorescence
For actin staining of FACS-purified cells, the procedures are described elsewhere (Mejillano et al., 2004). For actin staining of cells expressing GFP fusion proteins, cells were fixed with 4% formaldehyde for 20 min, extracted with 1% Triton X-100 for 2 min, and stained with 33 nM Texas red–X phalloidin (Invitrogen) for 10–30 min. For T-fimbrin staining, cells were fixed with 4% formaldehyde, extracted with 1% Triton X-100, and reacted to rabbit anti-fimbrin antibodies (a gift from M. Arpain, Institut Curie, Paris, France). For simultaneous fascin and actin staining, the formaldehyde-fixed samples were stained for F-actin with Texas red–X phalloidin and the first set of images was taken. The specimens were treated with methanol at –20°C for 5 min, incubated by mouse monoclonal anti-fascin antibody (clone 55K-2; DakoCytomation) for 30 min, and subsequently reacted with Cy5-conjugated anti–mouse IgG (Jackson ImmunoResearch Laboratories). The second set of images was aligned with the first set of actin staining by locators on etched grid coverslips (Bellco Glass, Inc.). Secondary antibodies were obtained from Jackson ImmunoResearch Laboratories and Invitrogen.
EM
Transfectants of pG-SUPER-fascin and pEGFP-fascin were collected by FACS. The procedures of platinum replica EM were previously described (Svitkina and Borisy, 1998, 2006). Quantification of the projected area of Λ-precursors was performed on scanned negatives by measuring the base and the height of triangles approximating the shape of Λ-precursors.
Fascin dissociation assay
Cells expressing EGFP-fascin were incubated on a microscope stage with the extraction solution (1% Triton X-100, 4% polyethylene glycol [40,000 kD], 100 mM PIPES, pH 6.9, 1 mM MgCl2, and 1 mM EGTA) supplemented with 2 μM Texas red–X phalloidin. GFP images were taken immediately before and after addition of the extraction solution (within 15–30 s), and then at 5-min intervals. 10–20 cells were analyzed for each group (WT, S39A, or S39E). The total GFP intensity of each filopodium was measured at every time point. After subtraction of the background level, the relative fluorescence intensities were calculated by normalization to the initial intensity in the first image after adding the extraction solution. The decay of the relative intensity was plotted to a single exponential function, as follows: I0exp(−koff t), where koff is the dissociation rate constant, t is time, and I0 is the initial fluorescence immediately after extraction. t 1/2 was calculated as t 1/2 = ln2/koff.
Quantification of filopodia frequency and length
Transfectants of pG-SUPER-fascin and pEGFP-fascin were collected by FACS. Spread cells containing prominent lamella with convex-shaped leading edges were chosen from phalloidin-stained samples. The contour lengths of the lamellipodia were measured by the MetaMorph Imaging software. The number of filopodial bundles touching or crossing the edge was counted. Only bundles having fluorescence intensity of at least 1.2 times above the background were considered. For each filopodium, the lengths of the internal and protruding parts were also measured. Two researchers performed quantification independently. Statistical analysis was done using SigmaPlot software (Systat Software, Inc.). 50–150 cells were quantified for each sample. Analysis of variance, the Holm-Sidak method, was used as a significance test, with P ≤ 0.001 and an overall significance level of 0.05. For the rescue experiments, the number of filopodia analyzed was from cells expressing YFP-fascin.
Immunoblotting
12 h after transfection with pG-SUPER-Tm, -Th, or -Tc, GFP-positive cells were collected by FACS and cultured for an additional 3–4 d in the culture medium. The detailed procedures of immunoblotting were previously described elsewhere (Kojima et al., 2004). The protein samples (40 μg per lane) were assayed with mouse anti-fascin (clone 55K-2). The blots were analyzed with Image 1.6 software (National Institutes of Health). The linearity of the signals was confirmed by dilution series of purified fascin.
FRAP
FRAP experiments were performed on a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) with a 110×, 1.3 NA, PlanApochromat oil objective (Carl Zeiss MicroImaging, Inc.). Cells were maintained at 37°C with a heated stage. GFP-tagged proteins in the midregions of protruding filopodia were bleached with a rectangular region of area ranging from 6 to 11 μm2 for ∼1 s using the 488-nm laser line at 100% laser power (25 mW). Thereafter, fluorescence recovery within the bleached region was monitored every 0.5–1 s over a period of 30–60 s. For quantification, MetaMorph software was used.
The fluorescence recovery was analyzed as follows: the average intensity over the bleached zone at each time of imaging was measured. To calculate the loss of fluorescence attributed to photofading during image acquisition, the fluorescence intensity of a nonbleached protruding filopodium was determined over time. The photofading constant, kfade, was obtained by curve-fitting the intensity of a nonbleached filopodium over time, t, to the single exponential decay function, exp(−kfade t). The intensity of the bleached zone was corrected for photofading by multiplying a correction factor, exp(kfade t), at each time point in the experiment. The equation I(t) = If + (IPB−If)exp(−krec t) was used to curve fit the corrected recovery intensities, where I(t) = intensity at a given time (t), If = intensity at final time, IPB = intensity at photobleaching event, and krec = fluorescence recovery rate constant. Half recovery time was calculated as ln2/krec. Final recovery was calculated by dividing If by the average intensity over the flanking regions next to the bleached zone. Examples of fluorescence recovery curves are presented in Fig. S4 B, with normalization to the flanking regions.
Microarray
RNA was purified from B16F1 cells using RNeasy kit (QIAGEN) and analyzed to Affymetrix MOE430A chips in triplicate. The microarray data can be viewed in the National Center for Biotechnology Information's Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under the accession no. GSE4819.
Online supplemental material
Fig. S1 shows the localization of actin cross-linking proteins in B16F1 melanoma cells. Fig. S2 shows fascin depletion by shRNA in HeLa cells. Fig. S3 shows actin-bundling activity of fascin WT and mutant proteins. Fig. S4 shows FRAP analysis of GFP-fascin. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200603013/DC1.
Supplementary Material
Acknowledgments
We thank Drs. J. Adams, C. Otey, J. Bartles, R. Tsien, M. Arpain, A. Ferreira, and C. Ballestrom for generous gifts of reagents; Drs. V. Gelfand and E. Taylor for stimulating discussions; and Drs. A. Biyasheva, T. Chew, and J. Peloquin for supporting experiments.
This work was supported by United States Army Medical Research and Material Command grant DAMD 17-00-1-0386 to D. Vignjevic and National Institutes of Health grants GM 62431 to G.G. Borisy and GM 70898 to T. Svitkina.
D. Vignjevic and S.I. Kojima contributed equally to this paper.
D. Vignjevic's present address is Equipe de Morphogenese et Signalisation Cellulaires, UMR 144, Centre National de la Recherche Scientifique/Institut Curie, Institut Curie, 75248 Paris Cedex 05, France.
Abbreviations used in this paper: shRNA, small hairpin RNA; WT, wild-type.
References
- Adams, J.C., J.D. Clelland, G.D. Collett, F. Matsumura, S. Yamashiro, and L. Zhang. 1999. Cell-matrix adhesions differentially regulate fascin phosphorylation. Mol. Biol. Cell. 10:4177–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Alwan, M.M., G. Rowden, T.D. Lee, and K.A. West. 2001. Fascin is involved in the antigen presentation activity of mature dendritic cells. J. Immunol. 166:338–345. [DOI] [PubMed] [Google Scholar]
- Anilkumar, N., M. Parsons, R. Monk, T. Ng, and J.C. Adams. 2003. Interaction of fascin and protein kinase Calpha: a novel intersection in cell adhesion and motility. EMBO J. 22:5390–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartles, J.R. 2000. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 12:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartles, J.R., L. Zheng, A. Li, A. Wierda, and B. Chen. 1998. Small espin: a third actin-bundling protein and potential forked protein orthologue in brush border microvilli. J. Cell Biol. 143:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J.S., and R.E. Cheney. 2002. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 4:246–250. [DOI] [PubMed] [Google Scholar]
- Brieher, W.M., M. Coughlin, and T.J. Mitchison. 2004. Fascin-mediated propulsion of Listeria monocytogenes independent of frequent nucleation by the Arp2/3 complex. J. Cell Biol. 165:233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp, T.R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296:550–553. [DOI] [PubMed] [Google Scholar]
- Bryan, J., and R.E. Kane. 1978. Separation and interaction of the major components of sea urchin actin gel. J. Mol. Biol. 125:207–224. [DOI] [PubMed] [Google Scholar]
- Buccione, R., J.D. Orth, and M.A. McNiven. 2004. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell Biol. 5:647–657. [DOI] [PubMed] [Google Scholar]
- Cohan, C.S., E.A. Welnhofer, L. Zhao, F. Matsumura, and S. Yamashiro. 2001. Role of the actin bundling protein fascin in growth cone morphogenesis: localization in filopodia and lamellipodia. Cell Motil. Cytoskeleton. 48:109–120. [DOI] [PubMed] [Google Scholar]
- DeRosier, D.J., and L.G. Tilney. 2000. F-actin bundles are derivatives of microvilli: what does this tell us about how bundles might form? J. Cell Biol. 148:1–6. [PMC free article] [PubMed] [Google Scholar]
- DesMarais, V., I. Ichetovkin, J. Condeelis, and S.E. Hitchcock-DeGregori. 2002. Spatial regulation of actin dynamics: a tropomyosin-free, actin-rich compartment at the leading edge. J. Cell Sci. 115:4649–4660. [DOI] [PubMed] [Google Scholar]
- Hashimoto, Y., M. Skacel, and J.C. Adams. 2005. Roles of fascin in human carcinoma motility and signaling: prospects for a novel biomarker? Int. J. Biochem. Cell Biol. 37:1787–1804. [DOI] [PubMed] [Google Scholar]
- Honda, M., K. Takiguchi, S. Ishikawa, and H. Hotani. 1999. Morphogenesis of liposomes encapsulating actin depends on the type of actin-crosslinking. J. Mol. Biol. 287:293–300. [DOI] [PubMed] [Google Scholar]
- Ishikawa, R., S. Yamashiro, K. Kohama, and F. Matsumura. 1998. Regulation of actin binding and actin bundling activities of fascin by caldesmon coupled with tropomyosin. J. Biol. Chem. 273:26991–26997. [DOI] [PubMed] [Google Scholar]
- Kojima, S., D. Vignjevic, and G.G. Borisy. 2004. Improved silencing vector co-expressing GFP and small hairpin RNA. Biotechniques. 36:74–79. [DOI] [PubMed] [Google Scholar]
- Kranewitter, W.J., C. Danninger, and M. Gimona. 2001. GEF at work: Vav in protruding filopodia. Cell Motil. Cytoskeleton. 49:154–160. [DOI] [PubMed] [Google Scholar]
- Kureishy, N., V. Sapountzi, S. Prag, N. Anilkumar, and J.C. Adams. 2002. Fascins, and their roles in cell structure and function. Bioessays. 24:350–361. [DOI] [PubMed] [Google Scholar]
- Lanier, L.M., M.A. Gates, W. Witke, A.S. Menzies, A.M. Wehman, J.D. Macklis, D. Kwiatkowski, P. Soriano, and F.B. Gertler. 1999. Mena is required for neurulation and commissure formation. Neuron. 22:313–325. [DOI] [PubMed] [Google Scholar]
- Lazarides, E., and K. Burridge. 1975. Alpha-actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell. 6:289–298. [DOI] [PubMed] [Google Scholar]
- Lewis, A.K., and P.C. Bridgman. 1992. Nerve growth cone lamellipodia contain two populations of actin filaments that differ in organization and polarity. J. Cell Biol. 119:1219–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis, P.A., L. Zheng, G. Sekerkova, B. Changyaleket, E. Mugnaini, and J.R. Bartles. 2003. Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J. Cell Biol. 163:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallavarapu, A., and T. Mitchison. 1999. Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J. Cell Biol. 146:1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejillano, M.R., S. Kojima, D.A. Applewhite, F.B. Gertler, T.M. Svitkina, and G.G. Borisy. 2004. Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell. 118:363–373. [DOI] [PubMed] [Google Scholar]
- Mogilner, A., and G. Oster. 1996. Cell motility driven by actin polymerization. Biophys. J. 71:3030–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilner, A., and B. Rubinstein. 2005. The physics of filopodial protrusion. Biophys. J. 89:782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver, C.J., R.T. Terry-Lorenzo, E. Elliott, W.A. Bloomer, S. Li, D.L. Brautigan, R.J. Colbran, and S. Shenolikar. 2002. Targeting protein phosphatase 1 (PP1) to the actin cytoskeleton: the neurabin I/PP1 complex regulates cell morphology. Mol. Cell. Biol. 22:4690–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, S., Y. Yamakita, S. Yamashiro, P.T. Matsudaira, J.R. Gnarra, T. Obinata, and F. Matsumura. 1997. Identification of an actin binding region and a protein kinase C phosphorylation site on human fascin. J. Biol. Chem. 272:2527–2533. [DOI] [PubMed] [Google Scholar]
- Pellegrin, S., and H. Mellor. 2005. The Rho family GTPase Rif induces filopodia through mDia2. Curr. Biol. 15:129–133. [DOI] [PubMed] [Google Scholar]
- Peng, J., B.J. Wallar, A. Flanders, P.J. Swiatek, and A.S. Alberts. 2003. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 13:534–545. [DOI] [PubMed] [Google Scholar]
- Ridley, A.J., M.A. Schwartz, K. Burridge, R.A. Firtel, M.H. Ginsberg, G. Borisy, J.T. Parsons, and A.R. Horwitz. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709. [DOI] [PubMed] [Google Scholar]
- Rottner, K., B. Behrendt, J.V. Small, and J. Wehland. 1999. VASP dynamics during lamellipodia protrusion. Nat. Cell Biol. 1:321–322. [DOI] [PubMed] [Google Scholar]
- Sasaki, Y., K. Hayashi, T. Shirao, R. Ishikawa, and K. Kohama. 1996. Inhibition by drebrin of the actin-bundling activity of brain fascin, a protein localized in filopodia of growth cones. J. Neurochem. 66:980–988. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A., M.D. Welch, L.M. Machesky, P.C. Bridgman, S.M. Meyer, and J.A. Cooper. 1998. Visualization and molecular analysis of actin assembly in living cells. J. Cell Biol. 143:1919–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirenbeck, A., T. Bretschneider, R. Arasada, M. Schleicher, and J. Faix. 2005. The Diaphanous-related formin dDia2 is required for the formation and maintenance of filopodia. Nat. Cell Biol. 7:619–625. [DOI] [PubMed] [Google Scholar]
- Shonukan, O., I. Bagayogo, P. McCrea, M. Chao, and B. Hempstead. 2003. Neurotrophin-induced melanoma cell migration is mediated through the actin-bundling protein fascin. Oncogene. 22:3616–3623. [DOI] [PubMed] [Google Scholar]
- Small, J.V. 1988. The actin cytoskeleton. Electron Microsc. Rev. 1:155–174. [DOI] [PubMed] [Google Scholar]
- Small, J.V., T. Stradal, E. Vignal, and K. Rottner. 2002. The lamellipodium: where motility begins. Trends Cell Biol. 12:112–120. [DOI] [PubMed] [Google Scholar]
- Stradal, T., K.D. Courtney, K. Rottner, P. Hahne, J.V. Small, and A.M. Pendergast. 2001. The Abl interactor proteins localize to sites of actin polymerization at the tips of lamellipodia and filopodia. Curr. Biol. 11:891–895. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1998. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 298:570–592. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 1999. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J. Cell Biol. 145:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina, T.M., and G.G. Borisy. 2006. Correlative light and electron microscopy studies of cytoskeletal dynamics. In Cell Biology: A Laboratory Handbook. Third Edition. Vol. 3. J. Celis, editor. Elsevier, San Diego. 277–285.
- Svitkina, T.M., E.A. Bulanova, O.Y. Chaga, D.M. Vignjevic, S. Kojima, J.M. Vasiliev, and G.G. Borisy. 2003. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 160:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney, L.G., and D.J. DeRosier. 2005. How to make a curved Drosophila bristle using straight actin bundles. Proc. Natl. Acad. Sci. USA. 102:18785–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney, M.S., L.G. Tilney, R.E. Stephens, C. Merte, D. Drenckhahn, D.A. Cotanche, and A. Bretscher. 1989. Preliminary biochemical characterization of the stereocilia and cuticular plate of hair cells of the chick cochlea. J. Cell Biol. 109:1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney, L.G., M.S. Tilney, and D.J. DeRosier. 1992. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu. Rev. Cell Biol. 8:257–274. [DOI] [PubMed] [Google Scholar]
- Vignjevic, D., D. Yarar, M.D. Welch, J. Peloquin, T. Svitkina, and G.G. Borisy. 2003. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 160:951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignjevic, D., J. Peloquin, and G.G. Borisy. 2006. In vitro assembly of filopodia-like bundles. Methods Enzymol. 406:727–739. [DOI] [PubMed] [Google Scholar]
- Yamakita, Y., S. Ono, F. Matsumura, and S. Yamashiro. 1996. Phosphorylation of human fascin inhibits its actin binding and bundling activities. J. Biol. Chem. 271:12632–12638. [DOI] [PubMed] [Google Scholar]
- Yamashiro, S., Y. Yamakita, S. Ono, and F. Matsumura. 1998. Fascin, an actin-bundling protein, induces membrane protrusions and increases cell motility of epithelial cells. Mol. Biol. Cell. 9:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro-Matsumura, S., and F. Matsumura. 1985. Purification and characterization of an F-actin-bundling 55-kilodalton protein from HeLa cells. J. Biol. Chem. 260:5087–5097. [PubMed] [Google Scholar]
- Yamashiro-Matsumura, S., and F. Matsumura. 1986. Intracellular localization of the 55-kD actin-bundling protein in cultured cells: spatial relationships with actin, alpha-actinin, tropomyosin, and fimbrin. J. Cell Biol. 103:631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, L., G. Sekerkova, K. Vranich, L.G. Tilney, E. Mugnaini, and J.R. Bartles. 2000. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell. 102:377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}