

Abstract
It is becoming increasingly clear that herpesviruses can exploit the endocytic pathway to infect cells, yet several important features of this process remain poorly defined. Using herpes simplex virus-1 (HSV-1) as a model, we demonstrate that endocytosis of the virions mimic many features of phagocytosis. During entry, HSV-1 virions associated with plasma membrane protrusions followed by a phagocytosis-like uptake involving rearrangement of actin cytoskeleton and trafficking of the virions in large phagosome-like vesicles. RhoA GTPase was activated during this process and the mode of entry was cell type–specific. Clathrin-coated vesicles had no detectable role in virion trafficking as Eps15 dominant-negative mutants failed to affect HSV-1 uptake. Binding and fusion of the virion envelope with the phagosomal membrane is likely facilitated by clustering of nectin-1 (or HVEM) in phagosomes, which was observed in infected cells. Collectively, our data suggests a novel mode of uptake by which the virus can infect both professional and nonprofessional phagocytes.
Introduction
Mammalian cells have evolved multiple mechanisms for internalizing fluids, surface-bound ligands, and plasma membrane components. Although some are well-characterized mechanisms such as endocytosis in clathrin-coated pits, many others including phagocytosis by nonprofessional phagocytes remain poorly defined. One way to generate new insight into cellular uptake processes is to make use of intracellular microorganisms that exploit endocytic mechanisms to infect mammalian cells. For example, receptor-mediated endocytosis is commonly used by intracellular pathogens including bacteria and viruses for infecting cells (Miller and Hutt-Fletcher, 1992; Akula et al., 2003; Gruenheid and Finlay, 2003; Pelkmans and Helenius, 2003; Dimitrov, 2004). Apart from the convenient transport in vesicles, the acidic environment of endocytic vesicles is used by many viruses to efficiently penetrate into the host cytosol (Marsh and Helenius, 1989). In the case of herpes simplex virus type-1 (HSV-1), endocytosis plays a dominant role in infection of many cell types (Nicola et al., 2003; Nicola and Straus, 2004). This process, however, appears to be unique because it is likely not mediated by formation of clathrin-coated pits or caveolae and it may not always be pH dependent (Gianni et al., 2004; Milne et al., 2005; Nicola et al., 2005).
The other mode of HSV-1 entry is virion fusion at the plasma membrane, which is pH independent (Spear, 1993a) and is facilitated by at least five viral glycoproteins: gB, gC, gD, gH, and gL (Spear, 1993b). Entry via this mode is initiated by interaction of viral gC and/or gB with heparan sulfate (HS) proteoglycan (Shukla and Spear, 2001). This interaction is followed by binding of viral gD to one of its receptors (Spear, 1993b). The gD receptors include HVEM, a member of the tumor necrosis factor receptor family; nectin-1 (CD111), a member of the Ig superfamily; and 3-O-sulfated heparan sulfate or 3-OS HS (Shukla et al., 1999, 2000; Spear et al., 2000; Xia et al., 2002). The interaction between gD and its receptor mobilizes, in an unknown fashion, participation from gB, gH, and gL to trigger the process of membrane fusion (Spear et al., 2000). Noticeably, the gD receptors are also essential for entry via endocytosis (Nicola et al., 2003; Nicola and Straus, 2004).
HSV causes conditions ranging from blisters on mucosal surfaces to deadly brain infections in immunocompromised individuals (Shukla and Spear, 2001). It also causes ocular diseases affecting not only the outer epithelial layer of the cornea but also the stroma and the tissues neighboring the cornea such as the trabecular meshwork (Tiwari et al., 2005) in the eye. Infection of the stroma or herpetic stromal keratitis (HSK) is rare, but is a significant cause of infectious blindness in the developed countries. HSK lasts longer and is more difficult to treat than epithelial keratitis (Russell et al., 1984; Thomas and Rouse, 1997). Development of novel strategies to efficiently treat HSK is hampered by the relatively poor understanding of the infection of the corneal stroma.
To better elucidate the infection of the stroma and to generate specific information on the endocytic mode of entry, the present study compared HSV-1 entry into primary human corneal fibroblasts (CF) cultured from excised tissue of the stroma with that of nectin-1 expressing Chinese hamster ovary (nectin-1-CHO) cells. Both cell types exhibited a pH-dependent mode of viral entry that mimics and retains many features of phagocytosis. Essentially, HSV-1 virions associate with cellular protrusions followed by an actin network–dependent uptake (internalization of the virions into the host cytosol) involving RhoA activation. This is a novel uptake mode that has implications for entry of virions into other professional and nonprofessional phagocytic cells.
Results
Cultured CFs are susceptible to HSV-1 entry and replication
Monolayer cultures of CF and nectin-1-CHO or HVEM-expressing CHO cells (HVEM-CHO) were infected with a recombinant β-galactosidase (β-gal)–expressing reporter virus, HSV-1(KOS)gL86, abbreviated here as KOS-gL86 and interchangeably used in this manuscript for HSV-1. β-gal activity indicates that virus has entered the cell, released its genome into the nucleus, and activated the constitutive promoter for the enzyme production and activity (Shukla et al., 2000). Although no β-gal activity (Fig. 1 A, top) was observed in naturally resistant wild-type CHO-K1 cells (CHO-WT), dosages of input virus sufficient to infect 100% of nectin-1-CHO cells (Fig. 1 A, middle) also caused β-gal expression in 100% of the CF cells (Fig. 1 A, bottom). The viral dose-response curve for CF, performed in the linear range of 0–100 PFU/cell, was similar to that of nectin-1-CHO or HVEM-CHO cells and naturally susceptible HeLa cells (Fig. 1 B). CHO-WT cells transfected with an empty vector pcDNA3 were used as a negative control (Fig. 1 B). As a measure of productive infection, HSV-1 replication was verified by real-time PCR using HSV-1 gD probes and also by syncytial plaque formation. The viral DNA replications were indicated by threshold cycle (CT) values: the lower the value, the higher the viral replication. In CF, the highest replication was seen at 36 h, which was also verified by PCR (Fig. 1, C and D). The HSV-1 virions were also able to form plaques and the numbers of plaques formed (unpublished data) were very similar for CF and nectin-1-CHO cells (Fig. S1, A–C, available at http://www.jcb.org/cgi/content/full/jcb.200509155).
Figure 1.

Corneal fibroblasts (CF) exhibit susceptibility to HSV-1 entry. (A) β-galactosidase staining of viral entry. CHO-WT (top panel), nectin-1-CHO (middle panel) and CF (bottom panel) cells were exposed to a recombinant β-galactosidase-expressing HSV-1 (5 PFU/cell) for 6 h followed by incubation with X-Gal (5-bromo-4-chloro-3-indoyl-β-d-galactoside) to identify infected cells (blue). (B) HSV-1 entry curves. As indicated, CF, HeLa, nectin-1-CHO, HVEM-CHO, or pCDNA3-CHO (control) cells in microwells were exposed to HSV-1 at the dosages and time point indicated followed by incubation with ONPG (O-nitro-phenyl β-d-galactopyranoside) which is cleaved by β-gal to galactose and O-nitro-phenol. The individual OD values were within 10% of the mean and represent the amount of reaction product detected by spectrophotometry (OD 410 nm). (C) Replication of HSV-1 in human CF. Real time PCR shows replication of HSV-1 DNA in nectin-1-CHO and CF at times 12, 24, and 36 h. Mock infected cells were used as controls. Fluorescence curve along with CT (threshold cycle; vertical red lines) values are shown for each sample. The parameter CT is defined as the fractional cycle number at which the fluorescence passes the fixed threshold. (D) Visualization of viral DNA. PCR products electrophoresed on 1.5% agarose gel are shown. The amplified products (HSV-1 gD) were of the predicted size (129 bp*). The molecular size standards are shown in the first lane from left.
Plasma membrane protrusions facilitate viral entry
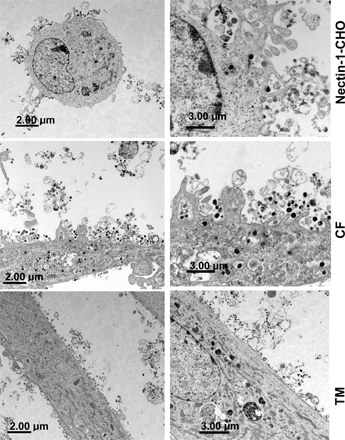
Ultrastructural analyses of HSV-1 entry into CF and nectin-1- CHO cells using EM suggested a role for plasma membrane protrusions and vesicles in entry. Transmission EM (TEM) micrographs demonstrated enveloped virions adjacent to and/or surrounded by protrusions of the plasma membrane in both nectin-1-CHO cells (Fig. 2, A and B, arrows) and CF (Fig. 2, D and E). The internalized virions were enveloped and located in uncoated and relatively large vesicles (Fig. 2, A, B, C, E, and F) not in clathrin-coated pits as seen in typical endocytosis. In contrast, HSV-1–infected primary cells cultured from excised human trabecular meshwork tissues (TM cells) had relatively few protrusions (Fig. 2 K) and the virions were mainly attached to relatively smooth cell surfaces (Fig. 2, G and H, arrows). Intracellular vesicles containing virions were virtually absent in TM cells. The lower magnification views for all three cell types are shown in Fig. S2 (available at http://www.jcb.org/cgi/content/full/jcb.200509155).
Figure 2.

Electron microscopic analysis of HSV-1 entry. TEM (A–H) and SEM (I–J) were performed to analyze HSV-1 entry into cells. Unless otherwise stated, all the images were captured 30 min after infection. (A) Virions (arrows) either bound to a nectin-1-CHO cell surface or loaded in vesicles, bar 0.4 μm. (B) Engulfment of virions (arrow) by protrusions present on the surface of a nectin-1-CHO cell, bar 0.5 μm. (C) Vesicles in nectin-1-CHO cell containing enveloped virions (arrows), bar 0.4 μm. (D–F) CF with membrane protrusions in the vicinity of HSV-1 virions and some enveloped virions in vesicles (arrows), bar 2 μm. (G) Virions (arrows) attaching to a protrusions-less cell surface of a TM cell, bar 0.8 μm. (H) Virions bound and apparently fusing with the plasma membrane of a TM cell, bar 2 μm. (I) Plasma membrane protrusions (arrowheads) from nectin-1-CHO with attached virions (arrows), bar 1 μm. (J) Virions (arrows) attached to a nectin-1-CHO protrusion, bar 100 nm. (K) Determination of percentage of plasma membrane protrusions scored from sampled groups of 250 single cells or clusters (with 5–20 cells), ∼5 μm length of a protrusion and at least 5% of the cell surface covered with protrusions is scored positive.
Scanning EM (SEM) results correlated TEM observations that many HSV-1 virions (Fig. 2, I and J, arrows) were attached to the protrusions seen on nectin-1-CHO cells (arrowheads). The protrusions were ∼5–30 μm long and rarely branched, mainly seen within 30 min of exposure to virus. Immunogold EM of HSV-1 envelope glycoproteins gD or gH–gL further confirmed that enveloped HSV-1 virions associated with the protrusions on nectin-1-CHO cells and localized in the intracellular vesicles (unpublished data). No coated pits or caveolae (“smooth coated” or “non-coated” omega- or flask-shaped invaginations) were observed.
Intracellular vesicles containing HSV-1 virions are not formed from clathrin-coated pits
To completely rule out endocytosis of HSV-1 in clathrin-coated vesicles, dominant-negative EGFR pathway substrate clone 15 (Eps15) mutants were used (Fig. 3). Eps15, a constituent of plasma membrane clathrin-coated pits, is ubiquitously and constitutively associated with AP-2 (Benmerah et al., 1998, 1999). AP-2, clathrin, and dynamin are three major coat proteins. The AP-2 complex plays a principal role in both the organization and function of plasma membrane coated pits; it drives clathrin assembly onto the plasma membrane and interacts with tyrosine based signals of membrane receptors. Clathrin provides an organizing framework to the pit. Dynamin is a GTPase that assembles into rings at the neck of invaginated coated pits and its GTPase activity is necessary for the scission of the vesicle from the plasma membrane (Mellman, 1996; Schmid, 1997; McNiven, 1998). Dominant-negative mutants of Eps15 inhibit clathrin-dependent endocytosis (Carbone et al., 1997; Benmerah et al., 1998). The mutants used were: DIIIΔ2 (control), a C-terminal domain construct of Eps15 lacking all the AP-2–binding sites that does not interfere with endocytosis; DIII, the C-terminal domain construct of Eps15 containing all the AP-2 binding sites (Benmerah et al., 1996; Iannolo et al., 1997), which blocks endocytosis (Benmerah et al., 1998), and EΔ95/295 or EH29, the construct lacking Eps15 homology (EH) domains at the N-terminal that are required for clathrin-coated pit targeting of Eps15 and thus an inhibitor of clathrin-coated pit assembly (Benmerah et al., 1999). These controls and mutants, when transfected into nectin-1-CHO cells, all failed to block virus entry (Fig. 3 A). The transfection efficiency estimated by GFP expression (Fig. 3 B) was at an appropriate level of 60%. In parallel experiments, the dominant-negative mutants DIII and EΔ95/295 expectedly blocked human herpes virus-8 (HHV-8) entry of 293T cells (Fig. 3 C) as HHV-8 is known to use endocytosis via clathrin-coated pits for entry (Akula et al., 2003).
Figure 3.

Eps15 mutants do not inhibit HSV-1 entry. (A) Effect of expression of Eps15 mutants. The nectin-1-CHO cells were transfected with either empty vector pcDNA3 or expression plasmids for Eps15 mutants (as indicated) followed by infection with HSV-1 (5 PFU/cell). The entry was measured by β-galactosidase activity. Entry is presented as a percentage of the entry into untransfected cells. (B) Determination of transfection efficiency of plasmids-expressing GFP tagged Eps15 mutants (indicated) into nectin-1-CHO cells. Shown are the snapshots of GFP expressing cells. (C) Effect of the expression of Eps15 mutants on HHV-8 entry. 293 cells were either infected (+) or mock infected (−) with HHV-8 (5 PFU/cell). Un-internalized virions were removed from cell surface using citrate buffer (pH 3). After permeabilization, a mouse monoclonal antibody against HHV-8 glycoprotein, K8.1A (1:500) along with biotinylated secondary antibody and streptavidin-conjugated horseradish peroxidase were used in an ELISA to measure entry using a spectrophotometer. The reaction products generated by the mutants are shown as percentage of control values obtained in triplicates for HHV-8 infected (+) and pcDNA3 transfected cells. Asterisks indicate significant difference from controls (P < 0.05, t test), error bars represent SD.
HSV-1 induces uptake of an established phagocytic tracer
Absence of any clear evidence supporting endocytosis in coated pits and the development of protrusions in response to HSV-1 entry raised the possibility that virus entered into nectin-1-CHO and CF via a phagocytosis-like uptake. To test this possibility, we used an assay that allows the phagocytosis process to be observed and quantitated in cells. Essentially, the internalization of fluorescently labeled bacteria such as fluorescein-labeled K-12 Escherichia coli (E. coli bioparticles) was monitored. After quenching of the extracelluar fluorescence by trypan blue, the assay measures intracellular fluorescence emitted by the engulfed particles (Sahlin et al., 1983; Wan et al., 1993). The phagocytosis assays (Fig. 4, A and B) were performed using nectin-1-CHO, HVEM-CHO, and a CHO mutant (CHO-745 or pgsA-745) transfected with nectin-1 or HVEM. CHO-745 cells lack glycosaminoglycans including HS (Esko et al., 1985). Because HS is required for HSV-1 attachment to cells (Shukla and Spear, 2001), the purpose of this experiment was to determine if the induction of phagocytosis was dependent on virus attachment to cells. As predicted, exposure of HSV-1 to nectin-1-CHO and HVEM-CHO elicited several hundred-fold induction of phagocytosis activity compared with the unexposed cells (Fig. 4 A). A similar but somewhat muted response was observed with CHO-745 cells transfected with the same receptors, which showed only about a 10-fold increase in the phagocytosis activity (Fig. 4 A). Thus, it appears that HS-mediated attachment of virions (normally mediated by HSPG) to cells is needed for maximal triggering of phagocytosis. Partial, HS-independent attachment of virions to cells does occur in the presence of HSV-1 gD receptors; therefore, complete loss of phagocytosis was not expected (Shukla et al., 1999).
Figure 4.

Induction of phagocytosis during HSV entry and the role of dynamin2. Phagocytosis of E. coli K-12 bioparticles was measured in relative fluorescence units (RFU) using 480 nm excitation and 520 nm emission with (+) or without (−) HSV-1. (A) Effect of HSV-1 on phagocytic activity of nectin-1 (or HVEM)-CHO or CHO-745 cells naturally deficient in HS. (B) Effect of K44A-dyn2 or WT-dyn2 expression on phagocytic activity of nectin-1-CHO cells. (C) Effect of expression of K44A-dyn2 or WT-dyn2 expression on virus entry in nectin-1-CHO cells. The data shown are the means of triplicate measures and are representative of 3 independent experiments. Asterisks indicate significant difference from controls (P < 0.05, t test), error bars represent SD.
To gain further insight into the mechanism, we focused on the ubiquitously expressed dynamin-2 isoform of dynamin, which, as indicated earlier, is crucial for regulation of the actin dynamics at the plasma membrane and for detachment of intracellular vesicles including phagosomes from the plasma membrane (Nichols and Lippincott-Schwartz, 2001; Meier and Greber, 2003; Schafer, 2004). We focused on dynamin also because it is considered to be essential for phagocytosis but not for macropinocytosis (Meier and Greber, 2003), and could potentially be used to differentiate the two processes. Although nectin-1-CHO cells transfected with WT-dyn2 or its dominant-negative mutant, K44A-dyn2, exhibited significant enhancement of phagocytosis activity in the presence of HSV-1 virions (Fig. 4 B), there was a moderate yet significant (∼25%) decrease in the phagocytosis activity by K44A expression. Given a 50–60% transfection efficiency, the net decrease in the phagocytosis activity could be close to 50% if all the cells were expressing the mutant. Thus, dyn2-mediated membrane pinching might play a role in HSV-1 uptake via a phagocytosis-like mechanism. Next, to evaluate how this drop in phagocytosis could be translated into viral entry, we performed an entry assay. Unlike WT-dyn2, which showed the same effect on entry as transfection with an empty plasmid, the expression of K44A-dyn2 resulted in a significant (60% or more) decrease in HSV-1 entry (Fig. 4 C). Again, when extrapolating to 100% transfection efficiency, the data would indicate significant blocking of entry in the presence of the mutant and may implicate dynamin-mediated processes in HSV-1 internalization.
HSV-1 virions and phagocytic tracers are cointernalized in an actin network–dependent process
HSV-1 induction of bioparticle internalization via phagocytosis does not necessarily imply that the virions themselves get internalized using the same route unless a colocalization is demonstrated. To determine whether the virus cointernalized with phagocytic tracers such as red fluorescent latex beads, a series of colocalization experiments was performed. CF or nectin-1-CHO cells were either treated with cytochalasin D (Cyto D), an inhibitor of actin polymerization and phagocytosis (Gottlieb et al., 1993; Rabinovitch, 1995; Schafer, 2004), or mock-treated before coincubation with red fluorescent beads and a recombinant HSV-1 (K26GFP). The recombinant virus contains nucleocapsid tagged with GFP (Desai and Person, 1998) that allows monitoring of the virus internalization into host cytosol (virus entry) using fluorescent confocal microscopy (CM). Non-internalized virions were removed by low pH treatment (citrate buffer pH 3) (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200509155). Using CM, z-stacks were generated and analyzed for internalization of both the virions (green) and the tracer (red). Indeed, in mock-treated cells, virions cointernalized with beads (Fig. 5 A, yellow), although a few clusters of virions were also noted to have entered into cells without the beads. It was not totally unexpected because the virus, in its capacity as an inducer, could potentially invoke its own internalization via phagocytosis-like uptake without the tracer. With Cyto D treatment, internalization of both virions and the tracer was significantly restricted, as very few virions and/or beads were seen (Fig. 5 B). Overall, the finding that the virions and the phagocytic tracers were cointernalized suggests that phagosomes may be the vesicles that transport HSV-1 virions during the entry.
Figure 5.

GFP tagged HSV-1 and red fluorescent beads are cointernalized. Nectin-1-CHO cells were either (A) mock treated or (B) treated with Cyto D, before exposure to green fluorescent HSV-1 (K26GFP) and latex beads (Red-beads). Sequential CM was performed to examine z-stacks for virus and beads internalization and colocalization (Merge). A higher magnification of the merged images is also shown (bottom panels).
Given that Cyto D blocked cointernalization of virions and the beads, we next determined whether the actin inhibitors would also block entry of HSV-1 independent of the beads. Cyto D and another actin inhibitor, latrunculin B (Lat B) (Spector et al., 1989) were used. Although without showing any detectable effect on TM cells (Fig. 6 A), Cyto D blocked in a dose-dependent manner up to 80% of HSV-1 entry into nectin-1- CHO (or HVEM-CHO) and CF (Fig. 6 A). The latter expresses HVEM and 3-OS HS, but not nectin-1 (unpublished data). Likewise, Lat B in a dose-dependent manner blocked up to 60% of HSV-1 entry into nectin-1-CHO and CF, but did not affect entry into TM cells (Fig. 6 B).
Figure 6.

Actin depolymerizers block HSV-1 internalization. Cells were treated with indicated concentrations of Cyto D and Lat B and exposed to HSV-1 (50 PFU/cell). (A) Effects of Cyto D on viral entry into nectin-1-CHO, CF and TM cells. (B) Effects of Lat B on viral entry into nectin-1-CHO, CF and TM cells. The control (Control) represents entry into corresponding mock-treated cells. (C) Effect of Cyto D and Lat B on virus internalization. As indicated cells were treated with Cyto D or Lat B at concentrations indicated for 2 h either after (post-treatment) or before (pre-treatment) exposure to HSV-1 (indicated). The assays were performed either with (+) or without (−) indicated components. Internalization of virions at 37°C after pH 3.0 treatment was quantitated by determining relative fluorescence units (RFU) using a 96-well fluorescence reader. (D) Effect of Cyto D and Lat B on HSV-1 dependent phagocytosis-like uptake of green fluorescent E. coli bioparticles. The extracellular fluorescence was quenched by treatment with trypan blue. The data shown are the means of triplicate measures and are representative of 3 independent experiments. Asterisks indicate significant difference from controls and/or treatments (P < 0.05, t test), error bars represent SD.
To further demonstrate the significance of actin network in HSV-1 entry, cells were treated with Cyto D or Lat B either after (post-treatment) or before (pre-treatment) infection with HSV-1. It was postulated that pre-treatment would have more negative effect on entry than post-treatment, provided the actin-based protrusions (such as filopodia) played a role in the virus attachment to cells as well. After allowing for attachment and internalization of virions in both cases, cell surface virions that failed to infect were removed using low pH (citrate buffer pH 3.0) (Fig. S3). The internalization of the virus HSV-1 (K26GFP) was quantitated by measuring fluorescence intensity. There was ∼60–75% inhibition of internalization of HSV-1 in CF and nectin-1-CHO cells in post-treatment and up to 90% in pre-treatment cells (Fig. 6 C). TM cells did not exhibit significant inhibition in either case. Corresponding effects on phagocytosis-like uptake paralleled that of internalization (Fig. 6 D). It should, however, be noted that experiments using Cyto D and Lat B are suggestive, but not fully conclusive, of the role of actin network and phagocytosis-like uptake of HSV-1 because the drugs may have other effects not fully accounted for by the experiments shown here.
Effect of pH on HSV-1 entry
We also examined the pH dependence of this mode of entry. Nicola et al. (2003) have previously found that HSV-1 entry into nectin-1-CHO or HVEM-CHO is pH dependent. Thus, effects of lysosomotropic agents capable of interfering with vesicular acidification were tested. These included bafilomycin A1 (BFLA-1) (Lukacs et al., 1990; Perez and Carrasco, 1994; Drose and Altendorf, 1997), chloroquine, and ammonium chloride (de Duve et al., 1974). Monolayer cultures of nectin-1-CHO or HVEM-CHO, CF, and TM cells were pretreated with BFLA-1 or chloroquine. In addition, ammonium chloride was applied to CF and TM cells. There was a strong dosage-dependent inhibition of HSV-1 entry by these drugs in nectin-1-CHO (Fig. 7 A) and CF (Fig. 7 B); the inset shows the inhibitory effects of intermediate concentrations of chloroquine. Noticeably, entry into TM cells was inhibited by chloroquine, but not by BFLA-1 or ammonium chloride (Fig. 7 C). Collectively, the inhibition of acidification of vesicles severely retarded viral entry into CF and nectin-1-CHO, but not into TM cells.
Figure 7.
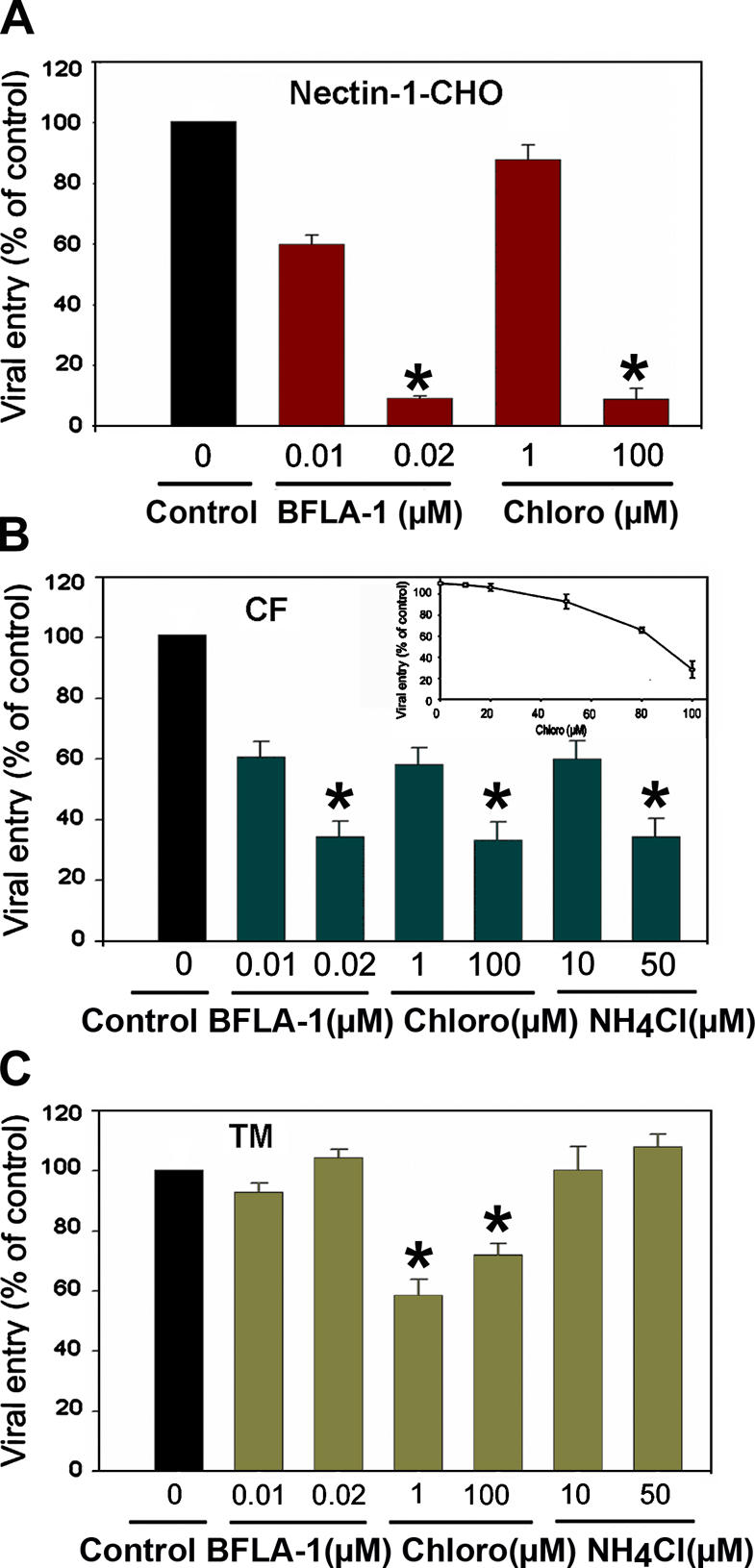
Lysosomotropic agents block HSV-1 entry. Cells were treated with indicated concentrations of bafilomycin A1 (BFLA-1), chloroquine (Chloro), or ammonium chloride (NH4Cl), and exposed to HSV-1 (50 PFU/cell). Viral entry was quantitated at 3 h after infection. The data shown are the means of triplicate measures and are representative of three independent experiments. The control (Control) represents entry into corresponding mock-treated cells. (A) Effects of lysosomotropic agents on entry into nectin-1-CHO cells. (B) Effects of lysosomotropic agents on entry into CF. The inset shows the effect of intermediary concentrations of chloroquine, a commonly used drug for the treatment of malaria. (C) Effects of lysosomotropic agents on entry into TM cells. Asterisks indicate significant differences from corresponding mock-treated controls (P < 0.05, t test), error bars represent SD.
Receptor clustering and the presence of the virions in early stage vesicles
The gD receptor (nectin-1 or HVEM) is essential for HSV-1 entry (Spear et al., 2000). To understand its role in phagocytosis-like uptake mode, we hypothesized that it should be present in the vesicles to facilitate viral capsid penetration into the cytosol, for which it is already known to be required (Pertel et al., 2001). To trace receptor trafficking we used a chimera of full-length nectin-1 and enhanced GFP (nectin-1-EGFP). This chimera is a functional receptor for HSV-1, which maintains full entry and cell-to-cell fusion activities (unpublished data). CHO cells transiently transfected with the chimera (nectin-1-EGFP-CHO) were briefly serum starved to eliminate endogenous transferrin and subsequently replaced with an exogenous Texas red conjugate (Texas red-transferrin), a molecule recycled via early endosomal vesicles including phagosomes (Dautry-Varsat, 1986; Tjelle et al., 2000). Incorporation of nectin-1 into vesicles was traced by sequential CM to eliminate the “cross talk” or leakage between green and red, allowing for a factual colocalization. In mock-infected nectin-1-EGFP-CHO (Fig. 8 A, a–c), nectin-1-EGFP (green) and Texas red-transferrin–positive vesicles (red) were noted throughout the cells, but the nectin-1 and vesicles appeared at distinct diffuse areas with little colocalization. However, when nectin-1-EGFP-CHO cells were infected with purified HSV-1 (Fig. 8 A, d–f), large clusters of nectin-1 (green) and Texas red-transferrin–positive vesicles were observed (red). Their colocalization (yellow) was indicative of the association of nectin-1 with the vesicles.
Figure 8.

HSV-1 induced clustering of receptor and identification of virions containing vesicles. (A) Nectin-1 colocalizes with early stage vesicles in HSV-1 infected cells. CHO cells transfected with a plasmid expressing nectin-1-EGFP were incubated with Texas red-transferrin conjugate (1:250) for 10 min and then either (a–c) mock infected or (d–f) infected with HSV-1 (50 PFU/cell) for 30 min, fixed and examined by confocal microscopy. Nectin-1 expression is seen in green (a and d) and the early stage vesicles appear red (b and e). In mock-infected cells, merger of green and red shows no colocalization (c), but in infected cells there is colocalization (yellow) (f); (B) HSV-1 colocalizes in early stage vesicles. As indicated, nectin-1-CHO or CHO-WT (Control) cells were infected with HSV-1 (K26GFP) (green) for the time points indicated and then treated with citrate buffer (pH 3) and fixed. It was followed by staining with either anti-EEA1 or anti-TfR (red) to identify the vesicles. By sequential CM the z stacks show colocalization of virions with early stage vesicles. Little colocalization with recycling to late endosomes was observed.
To verify the presence of the virions in the vesicles and to evaluate the stage at which virions transit from vesicles to release their capsid into the cytosol, antibodies to early endosome antigen 1 (EEA1) (marker for early stage endosomes and/or phagosomes) (Gillooly et al., 2001, Amer and Swanson, 2002), transferrin receptor (TfR) (marker for recycling endosomes), and LAMP-2 (marker for lysosomes) were independently used to stain infected (or mock-infected control) nectin-1-CHO or CHO-WT cells. Entry was with cold bound K26GFP incubated in media at 37°C over a time course ranging from 0 to 90 min. Significant numbers of virions (green) were found to localize in vesicles that mimic early stage endosomes (red) at time points from 5 to 60 min, with a peak seen at 30 min (Fig. 8 B). This time course corresponded well with the HSV-1 internalization observed while estimating optimal phagocytosis-like uptake (unpublished data). It appears that the virus stabilizes these early stage vesicles to facilitate its transit into the cytosol. Little colocalization of virus with both TfR (Fig. 8 B) and LAMP-2 (unpublished data) was observed, suggesting that neither was significantly involved in virion trafficking.
Cellular signaling supports a phagocytosis-like entry
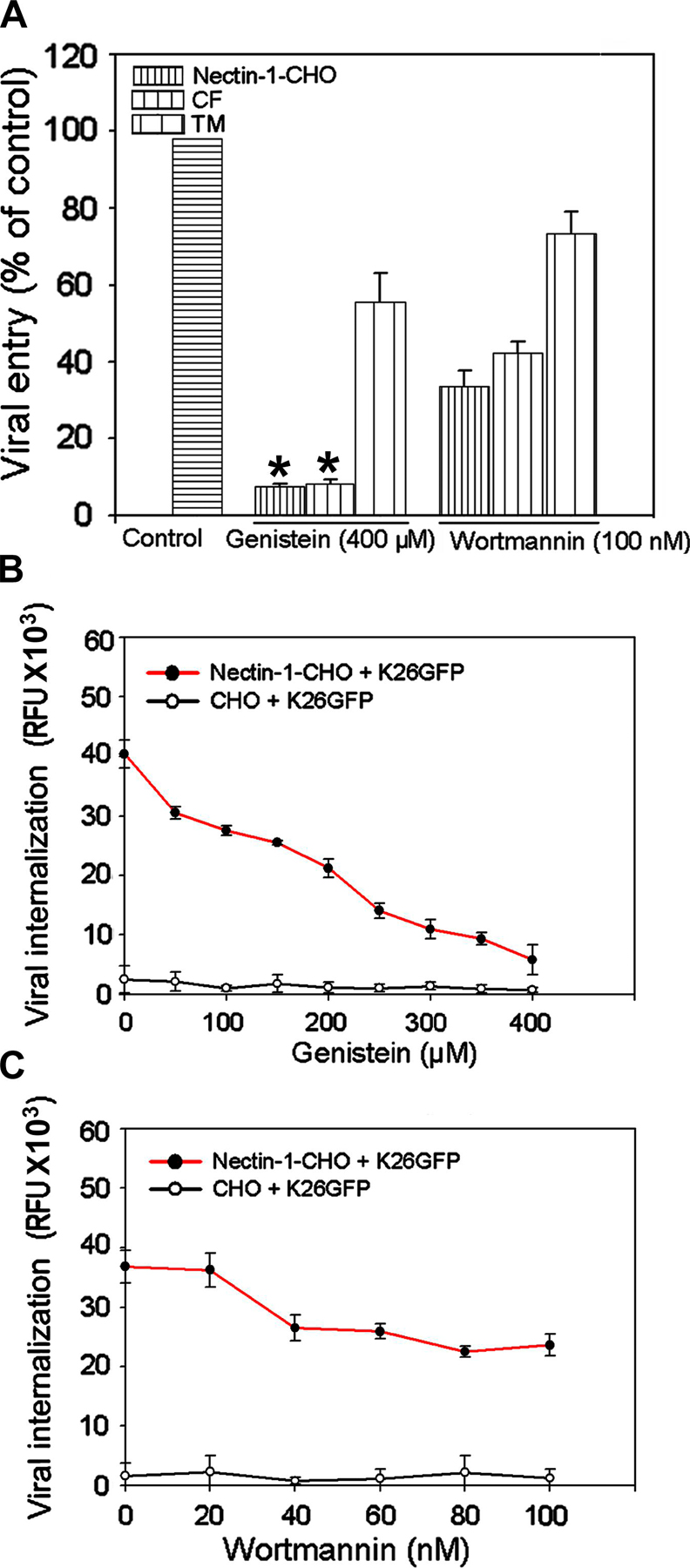
Plasma membrane protrusions that are formed in response to HSV-1 entry may be triggered by upstream signaling. Potential candidates may include tyrosine kinases (Cheshenko et al., 2005) and lipid kinases. Pharmacological agents such as genistein (Nicola et al., 2005), a tyrosine kinase inhibitor, and wortmannin, a specific PI3-kinase inhibitor (Nicola and Straus, 2004), were used to assess the requirements for signaling. Both genistein and wortmannin adversely affected entry of HSV-1 into CF, nectin-1-CHO, and TM cells. However, the inhibitory effects were more pronounced in CF and nectin-1-CHO than in TM cells. Genistein at the higher dosage of 400 μM was more effective (Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200509155). It might be that the less potent effect of wortmannin (∼50–60%) at the higher dosage of 100 nM was because it acted downstream (Fig. S4 A). Therefore, we performed dose-response entry experiments using nectin-1-CHO cells and found that genistein (Fig. S4 B) but not wortmannin (Fig. S4 C) induced a dose-dependent inhibition that significantly blocked HSV-1 entry. In further experiments, genistein blocked cointernalization of K26GFP and red beads but wortmannin failed to do so (unpublished data). Tyrosine kinase activation may play a role in HSV-1 uptake into CF and nectin-1- CHO cells.
We next examined the role of Rho family GTPases that are known to regulate the actin cytoskeleton rearrangement that drives the process of phagocytosis. RhoA is required for complement-mediated phagocytosis and Rac and/or Cdc42 typically are implicated in triggered phagocytosis (Caron and Hall, 1998). Rho alternates between the active GTP-bound and inactive GDP-bound state. Using the substrates GDP (negative control), GTPγS (positive control), and purified HSV-1 (potential activator of Rho) and pull-down assay we found that RhoA was activated in infected CF and nectin-1-CHO cells (Fig. 9 A). A time-course study using nectin-1-CHO cells indicated that the RhoA activation was sustained for 30 min (Fig. 9 B). Incidentally, this time frame matches well with the localization of the virions within early stage vesicles (Fig. 8 B). Cdc42 activation was not sustained. This activation dissipated in nectin-1-CHO cells within roughly 5 min of virus exposure (Fig. 9, C and D). Some of the plasma membrane protrusions were filamentous and rarely branched suggesting transitory filopodia formation, potentially to facilitate virus attachment. Similarly, no activation of Rac1 was detected (Fig. 9, E and F). These data prompted us to look more closely at changes in actin cytoskeleton upon viral exposure. Stress fibers became more prominent with entry of nectin-1-CHO cells, peaking at 30 min (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200509155). This time period, matching with that observed in RhoA activation and virion trafficking experiments, may be critical for virions transit via vesicles.
Figure 9.

Activation of RhoA signaling pathway by HSV-1. Western blot analysis of Rho GTPases. Activation of RhoGTPases (indicated) in the presence of GDP, GTPγS as controls or HSV-1 in cell types (indicated) was determined by affinity precipitation with Rhotekin-RBD-GST or PAK-PBD-GST beads (described in Materials and methods). (A) Activation of RhoA by HSV-1 at 15 min after infection. (B) Time course of HSV-1 induced activation of RhoA from 0 to 90 min in nectin-1-CHO cells. (C) Activation of Cdc42 by HSV-1 at 15 min after infection. (D) Time course of HSV-1 induced activation of Cdc42 from 0 to 10 min in nectin-1-CHO cells. Transient activation of Cdc42 was observed between 1–5 min (E) Activation of Rac1 by HSV-1 at 15 min after infection. (F) Time course of HSV-1 induced activation of Rac1 from 0 to 10 min in nectin-1-CHO cells. As evident, no HSV-1 induced activation of Rac1 was observed.
Discussion
The study presented herein attempts to shed light on many important but yet unclear aspects of a recently described endocytic route of HSV-1 entry (Nicola et al., 2003). Our work began with a goal to examine the ability of cultured human CF to support a productive HSV-1 entry, but it ended up with systematic exploration of the endocytic mode of entry. This cell type–dependent viral uptake was unexpectedly found to share many features of professional phagocytosis, hence the name “phagocytosis-like.” It likely requires actin rearrangement and dynamin assembly and is regulated by signaling pathways that apparently involve action of RhoA GTPase and may also involve tyrosine kinases. This pH-dependent and clathrin-independent viral uptake mechanism is marked by generation of cell surface protrusions in response to HSV-1 entry and clustering of gD receptors in large phagosome-like vesicles. Consistent with the notion that HSV-1 entry could occur by a phagocytosis-like uptake, it was found that the virus can significantly induce phagocytosis of E. coli bioparticles and HSV-1 virions cointernalized with phagocytic tracers.
Induction of phagocytosis by HSV-1 might seem unexpected because it is generally believed that phagocytosis could not be induced by particles smaller than 0.5 μm in diameter (Rabinovitch, 1995; Sansonetti, 2001). The enveloped HSV-1 is only ∼0.2 μm. Yet evidence does exist to the contrary that phagocytosis could ensue for tracers as small as 0.13 μm (Desjardins and Griffiths, 2003). Actually, it has been reported that granulocytes take up HSV via complement-mediated phagocytosis (Van Strijp et al., 1989). Also in the past, phagocytosis has been implicated in entry of viruses including many paramyxoviruses smaller in size than HSV-1 (Silverstein and Marcus, 1964). In addition, certain forms of vaccinia virus use an entry pathway that mimics the initial stages of phagocytosis (Locker et al., 2000). Similarly, the induction of RhoA GTPase during HSV-1 entry (Fig. 9, A and B) but not Cdc42 or Rac1 (Fig. 9, C–F) would support the notion that the mechanism of entry into nectin-1-CHO and CF could be analogous to complement-activated phagocytosis (Caron and Hall, 1998). It is perhaps logical to suggest that the molecular signaling mechanisms, not necessarily size, invokes phagocytosis.
Our findings supported by some additional ultrastructural images allow us to propose a model (Fig. 10). In brief, the infection of nectin-1-CHO and CF cells begins with the virions making initial contact with the cell surface by associating with plasma membrane protrusions (may include filopodia) (Fig. 10, arrows, top left and right panels). As the virions surf to concentrate at the cell body, internalization may occur by phagocytosis-like uptake followed by transport in early stage vesicles that are likely phagosomes, as suggested by their relatively large size and apparent colocalization of HSV-1 with phagocytic tracers (Fig. 5). Presence of nectin-1 (or HVEM) in the vesicles (Fig. 8 A, d–f) could provide the opportunity for the virions to fuse their envelopes with the vesicular membrane (Fig. 10, middle top and bottom panels). Consistent with a possibility that low pH environment found in vesicles can, in fact, enhance the fusion potential of HSV-1 virions, exposure of gB and gD to acidic pH has been shown to improve their membrane fusion potential (Butcher et al., 1990) and similarly, we have also found that low pH shock enhances HSV-1 mediated cell-to-cell fusion (unpublished data). The final stage is the release of the naked nucleocapsid (arrowhead) from the vesicle into the cytosol (cy) proximal to the nucleus (nu) (Fig. 10, bottom) for replication in the nucleus.
Figure 10.

Model for the phagocytic mode of HSV-1 entry. A cartoon (blue) illustrates the major steps (a-e) involved in the phagocytosis-like mode of HSV-1 entry. The panels on the right provide additional ultra-structural evidence to support the model. Top panels: Interaction of HSV-1 (arrows) with nectin-1-CHO cell membrane protrusions. Observations were made by SEM (top left panel, bar 1.0 μm) and by CM (top right panel, bar, 8.0 μm). Middle panels: viral trafficking. TEM images of a virus (arrow) apparently engulfed by the protrusions (top panel) and potentially fusing with the vesicular membrane (bottom panel); bar, 0.6 μm. Bottom panel: viral release. Apparent escape of a nucleocapsid (arrowhead) from a phagosome into the cytoplasm (cy) proximal to the nucleus (nu), bar, 0.6 μm.
Our model raises an obvious question related to the initial molecular events that trigger phagocytosis-like uptake. Specific virus–cell interactions are likely to be important because HS-mediated attachment of virus to cells is needed for maximal induction of phagocytosis-like uptake. Association with HS, however, is likely not the trigger, as cells lacking HSPG can still induce some basal level of phagocytosis (Fig. 4 A). Similarly, gD receptors (nectin-1, HVEM) may not be directly involved because they appear to be essential for entry into all cells, irrespective of the mode of viral entry (Nicola and Straus, 2004). Based on the current theory that RhoA is usually activated in complement mediated phagocytosis (Caron and Hall, 1998), it is possible that either an unknown complement receptor homologue, or a CF and CHO analogue of it, could trigger a similar process. The search for the trigger is further complicated by the existence of a dozen or so different HSV-1 envelope glycoproteins with little known functions. One or perhaps more of these HSV-1 envelope glycoproteins are likely to be needed for the triggering mechanism. Precedent exists, at least for some intracellular bacteria, that interactions with integrins (and potentially HS) could signal internalization by phagocytosis after binding of pathogens (Rabinovitch, 1995; Fukasawa et al., 1997; Tjelle et al., 2000). Interestingly, a glycoprotein (gH) essential for HSV-1 entry has recently been reported to interact in vitro with αvβ3 integrins, although the effect of this interaction on entry is still unclear (Parry et al., 2005). Moreover, new findings including ours indicate that the mode of entry could vary with cell types (Nicola et al., 2005). Conceivably, the functions of various HSV-1 glycoproteins and their corresponding receptors could vary with cell types as well. For instance, gD, the most studied glycoprotein, has been reported to bind multiple cellular molecules including three very diverse cellular receptors (Spear et al., 2000). Given the multi-protein binding ability of HSV-1 envelope proteins and the potential for cell type–dependent interactions, it is evident that extensive amount of work needs to be followed to identify the initial players and dissect their functions in the triggering mechanism(s). In any case, our current study provides new information on the existence of a novel pathway with implications for virus entry into both professional and nonprofessional phagocytes. It also provides new information on the poorly understood infection of the stroma and is likely to help develop new therapies for control of potentially blinding HSK.
Materials and methods
Plasmids
Plasmids used include gD receptors pBG38 (nectin-1) and pBEC10 (HVEM); control vector pcDNA3 (Invitrogen); pNec1-EGFP or pHVEM-EGFP expressing full-length nectin-1 or HVEM fused with EGFP (pEGFP-N1 vector; BD Biosciences) and rat dynamin2 (wild-type, WT-dyn2 and dominant-negative mutant, K44A-dyn2; from Mark McNiven, Mayo Clinic, Rochester, NY) fused with EGFP (CLONTECH Laboratories, Inc.). Eps15 mutants: DIIIΔ2 (control), DIII and EH29 corresponding to EΔ95/295 all subcloned in EGFP-C2 were kind gifts from Alexandre Benmerah (Universitė Paris, Paris, France).
Cell culture and virus preparations
Patricia G. Spear (Northwestern University, Chicago, IL) provided wild-type CHO-WT and CHO-745 cells (Esko et al., 1985). All CHO cell lines were grown in Ham's F12 (Invitrogen) supplemented with 10% fetal bovine serum (FBS). Stable nectin-1 (or HVEM)-CHO or control empty vector pcDNA3 cell lines were selected in medium containing geneticin (500 μg/ml) (Cellgro). For primary cultures of CF and TM cells, human eye tissues were obtained from the Illinos Eye Bank and cultured as described previously (Yue and Baum, 1981). Recombinant viruses HSV-1(KOS) gL86 and K26GFP (gifts from P. Desai, The Johns Hopkins University, Baltimore, MD) were amplified and quantitated as described elsewhere (Desai and Person, 1998; Shukla et al., 2000). HHV-8 virions were a kind gift from J. Vieira (University of Washington, Seattle, WA). A syn mutant strain, HSV-1(KOS) 804 was harvested from Vero cells. HSV-1(KOS) gL86 was purified by a modified sucrose gradient centrifugation method (Pignatti and Cassai, 1980). Experiments discussed in this manuscript were performed using HSV-1 titers within the linear range of 0–100 PFU/cell (Fig. 1 B).
Virus entry assays
Entry assays were based on quantitation of β-gal expressed from HSV-1 genome by either insoluble 5-bromo-4-chloro-3-indoyl-β-d-galactoside (X-Gal; Invitrogen), 1 mg/ml or soluble O-nitro-phenyl β-d-galactopyranoside (ONPG; Pierce Biotechnology), 3 mg/ml. Alternatively, virus entry was determined by fluorescence readout assay.
Stably transfected nectin-1 (or HVEM)-CHO cells, CF, and TM cells were treated with medium containing Cyto D (0.5 or 1.0 μg/ml), Lat B (0.25 or 2.5μM), BFLA-1 (0.01 or 0.1 μM), chloroquine (1.0 or 100 μM), genistein (400 μM) (Sigma-Aldrich), or wortmannin (100 nM; Calbiochem). CF and TM cells were in addition treated with ammonium chloride (10 or 50 mM; Fisher cientific). Infected (HSV-1 at 50 PFU/cell used for all experiments unless otherwise stated) and mock-infected cells were grown for 24 h at 37°C in 96-well culture dishes. Dosages of pharmacological agents for treatments were based on prior reports (Spector et al., 1989; Gottlieb et al., 1993; Fredericksen et al., 2002; Nicola et al., 2003).
Quantitative real time PCR
Real-time quantitative PCR was performed in 25-μl tubes with DNA prepared according to the manufacturer's instruction (QIAamp DNA Mini kit) using a Smart Cycler System (Cepheid). Primers, forward; AAGACCTTCCGGTCCTG and backward; TCCAACACGGCGTAGTA for the HSV-1 (KOS) glycoprotein D target were designed using Clone Manager 6 program (Sci Ed Central). The reaction was performed for 50 cycles under the following conditions: denaturation at 95°C for 30 s, annealing at 58°C for 10 s and extension at 72°C for 16 s. After amplification, one cycle of melting curve from 60 to 95°C by a transition rate of 0.2°C/s with continuous detection of fluorescence, was performed.
Confocal microscopy
Confluent monolayers of cells grown on coverslips were transfected with nectin-1-EGFP plasmid. After 24 h, cells were incubated with Texas red-transferrin conjugate (1:250 dilution; Molecular Probes) for 10 min and washed. Cells were then either infected with purified HSV-1 for 30 min or mock-infected with phosphate-buffered saline–glucose-calf serum (PBS-G-CS). At various times after infection, the cells were rinsed with PBS and fixed with 4% paraformaldehyde. In another set of experiments, HSV-1 was replaced with K26GFP and coincubated with fluospheres sulfate (red beads) (Molecular Probes). Immunofluorescence staining was done with anti EEA1, LAMP-2 (BD Biosciences), anti TfR (Zymed Laboratories) and counterstaining was performed using Cy3 anti–mouse secondary antibody (Jackson ImmunoResearch Laboratories). Confocal microscopy and sequential confocal analyses were performed using a laser-scanning spectrum confocal system (TCS SP2; Leica) linked to a microscope (DMIRE2; Leica). Images were captured using a 100× oil objective (Plan-Apo 1.4) with PMT for GFP and Cy3 at room temperature and using confocal acquisition software, LCS (Leica). The laser intensities for the two channels were separately set by alternately lowering each laser intensity to eliminate cross talk between red and green. The full area was scanned in slow mode, with high resolution and a sampling time of 400 ns (2 s/image). The confocal slit was set at 10 μm in most cases. Images of the cells were collected in focal planes with photomultiplier tubes. Images were processed using the LCS software. Additional sample stainings were analyzed using a fluorescent microscope system (Zeiss Axiovert 100 M; Carl Zeiss MicroImaging, Inc.). Images were captured using a 63× oil objective (Plan-Apo 1.4) at room temperature using FITC, Cy3, and DAPI channels using a Cook camera (SensiCam) and MetaMorph 6.3 software.
For some of the images, adjustments of brightness and contrast were applied to the entire images using Photoshop CS2 (Adobe).
Phagocytosis assay
The vibrant phagocytosis kit (Molecular Probes) developed with an adherent murine macrophage cell line (J774) has been adapted to evaluate phagocytosis in other adherent cell types (Wan et al., 1993; Foukas et al., 1998). Subcultured nectin-1-CHO, HVEM-CHO, and CHO-745 cells were adjusted to 106 cells/ml, aliquoted at 0 (negative control), 100 μl/microwell for positive and test into Microtest 96-well assay plates (BD Falcon) for settlement and adherence overnight at 37°C. The cells were washed and challenged with purified HSV-1 or purified K26GFP for test and PBS-G-CS for negative and positive controls. All cells were incubated at 4°C for 1 h, washed, and with warm medium further incubated at 37°C for 1 h. In experiments using HSV-1, all the microwells were in addition challenged for 2 h with reconstituted suspension of fluorescein-labeled E. coli K-12 bioparticles. For colocalization experiments, K26GFP and red fluorescent beads (FluoSpheres; Molecular Probes) were used. The effect of dynamin was evaluated by transfecting cells with WT-dyn2 or K44A-dyn2. For drug inhibition of internalization and phagocytosis, cells were first cold-bound with purified K26GFP or HSV-1 for 1 h before the 2-h incubation with virus in the presence of Cyto D (1 μg/ml) or Lat B (2.5 μM) at 37°C for post-treatment, or the cells were first incubated for 1 h with the drugs for pre-treatment. Virus that were bound to the cell surface were stripped with citrate buffer pH 3.0 for 1 min. Virus entry or phagocytic activity measured as relative fluorescence units (RFU) per treatment were determined by a fluorescence quenching assay with trypan blue using GENios Pro plate reader (TECAN) at 480-nm excitation and 520-nm emission spectrum. Measurements of five replicates of negative control, positive control, and test samples were performed. Data were expressed as mean ± SD.
Electron microscopy
Nectin-1-CHO, CF, and TM cells cultured in Lab-Tek chamber slides (2.5 × 105 cell/tray) and in anopore wells (Nalge Nunc) (2.5 × 105 cells/well) were infected with purified HSV-1 at 10–100 PFU/cell for 10–90 min. EM was performed using standard methods (Nicola et al., 2003). For TEM (JEM-1220; JEOL USA Inc.), images were captured at 1,000–600,000×, point-0.36 nm (3.6A) and lattice-0.2 nm (2A) at temperature, ACC voltage 40–120 kV using a Gatan camera (Digital CCD; Gatan, Inc.) and using Gatan Digital Micrograph (DM) v2.5 acquisition software. For SEM, Field Emission SEM (JMS-6320F; JEOL Inc. USA) was used to capture images at 25–2,000× in LM and 500–650,000× in HR modes. Images were resolved at 15 kV 1.2 nm (12A) and 1 kV 2.5 nm (25A) at room temperature ACC 0.5–30 kV using camera system ARC64 and Arch software (JEOL Inc. USA). Alternatively, for immunogold the infected cells were processed (Spaur and Moriarty, 1977). Specimens mounted on 150 mesh nickel-formvar–coated grids were incubated in 1% BSA and further incubated at room temperature for 3 h with polyclonal rabbit anti-HSV-1 gD, R7 (1:200), or anti gH-gL, R137 (1:200). Sections were subsequently incubated at room temperature for 1 h with 12-nm colloidal gold-conjugated goat anti–rabbit IgG (H+L) (1:30; Jackson ImmunoResearch Laboratories). The grids were also counterstained with uranyl acetate and examined in TEM as previously described.
Pull-down assays and Western blot analysis
Total cell lysates of equal amount of HSV-1 (50 PFU/cell for 15 min) or GDP (negative control) and GTPγS (positive control) as well as time courses for RhoA, Cdc42, and Rac) were spun in a microfuge at 14,000 rpm for 15 min and proteinase inhibitors were added to the supernatants. Rhotekin- RBD-GST (RhoA) and PAK-PBD-GST (Cdc42/Rac) beads (Cytoskeleton Inc.) were incubated with the supernatants for 1 h at 4°C on a rotator to bind the active proteins. The bound proteins were resolved on SDS-PAGE gels, transferred to nitrocellulose membranes, and immunoblotted with antibodies; RhoA, Cdc42, Rac 1:200 (Santa Cruz Biotechnology) and secondary at 1:10,000 HRP anti–mouse IgG (for RhoA) and HRP anti–rabbit IgG (for Cdc42 and Rac) (Jackson ImmunoResearch Laboratories). Immunoreactive bands were developed by West Pico substrate for enhanced chemiluminescence reactions (Pierce Biotechnology) and imaged on KODAK Biomax Mr film.
Online supplemental material
Fig. S1 shows plaque formation on CF cells after infection with HSV-1(KOS-804). Fig. S2 shows large sections of CF, nectin-1-CHO, and TM cells after infection with HSV-1. Fig. S3 shows inactivation of uninternalized HSV-1 after treatment with citrate buffer (pH 3.0). Fig. S4 shows the effects of tyrosine kinase and PI3K inhibitors on HSV-1 entry. Fig. S5 shows phalloidin staining of actin of nectin-1-CHO and CHO-WT cells demonstrating HSV-1 induced cytoskeletal changes. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200509155.
Supplementary Material
Acknowledgments
The authors thank Drs. Patricia Spear and Thomas Hope (Northwestern University) for helpful comments, and Prashant Desai (Johns Hopkins University) and Alexandre Benmerah (Universitė Paris) for reagents.
This work was supported by National Institutes of Health grants AI053836 (to D. Shukla) and EY03890 and EY05628 (to B.Y. Yue); a core grant EY01792 and a Career Development Award from Research to Prevent Blindness (to D. Shukla). V. Tiwari is supported by an American Heart Association postdoctoral fellowship (AHA0525768Z).
C. Clement and V. Tiwari contributed equally to this work.
Abbreviations used in this paper: CHO, Chinese hamster ovary; CF, corneal fibroblasts; CM, confocal microscopy; Cyto D, cytochalasin D; EEA1, early endosome antigen 1; HS, heparan sulfate; HSK, herpetic stromal keratitis; HSPG, heparan sulfate proteoglycans; HSV-1, herpes simplex virus-1; Lat B, latrunculin B; nectin-1-EGFP, nectin-1 with enhanced green fluorescent protein; TM, trabecular meshwork; TEM, transmission electron microscopy; SEM, scanning electron microscopy.
References
- Akula, S.M., P.P. Naranatt, N.S. Walia, F.Z. Wang, B. Fegley, and B. Chandran. 2003. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 77:7978–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amer, A.O., and M.S. Swanson. 2002. A phagosome of one's own: a microbial guide to life in the macrophage. Curr. Opin. Microbiol. 5:56–61. [DOI] [PubMed] [Google Scholar]
- Benmerah, A., B. Bègue, A. Dautry-Varsat, and N. Cerf-Bensussan. 1996. The ear of alpha-adaptin interacts with the COOH-terminal domain of the Eps15 protein. J. Biol. Chem. 271:12111–12116. [DOI] [PubMed] [Google Scholar]
- Benmerah, A., C. Lamaze, B. Begue, S.L. Schmid, A. Dautry-Varsat, and N. Cerf-Bensussan. 1998. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 140:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmerah, A., M. Bayrou, N. Cerf-Bensussan, and A. Dautry-Varsat. 1999. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J. Cell Sci. 112:1303–1311. [DOI] [PubMed] [Google Scholar]
- Butcher, M., K. Raviprakash, and H.P. Ghosh. 1990. Acid pH-induced fusion of cells by herpes simplex virus glycoproteins gB an gD. J. Biol. Chem. 265:5862–5868. [PubMed] [Google Scholar]
- Carbone, R., S. Fre, G. Iannolo, F. Belleudi, P. Mancini, P.G. Pelicci, M.R. Torrisi, and P.P. Difiore. 1997. Eps15 and Eps15r are essential components of the endocytic pathway. Cancer Res. 57:5498–5504. [PubMed] [Google Scholar]
- Caron, E., and A. Hall. 1998. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 282:1717–1721. [DOI] [PubMed] [Google Scholar]
- Cheshenko, N., W. Liu, L.M. Satlin, and B.C. Herold. 2005. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 280:31116–31125. [DOI] [PubMed] [Google Scholar]
- Dautry-Varsat, A. 1986. Receptor-mediated endocytosis: the intracellular journey of transferrin and its receptor. Biochimie. 68:375–381. [DOI] [PubMed] [Google Scholar]
- de Duve, C., T. de Barsy, B. Poole, A. Trouet, P. Tulkens, and F. Van Hoof. 1974. Lysosomotropic agents. Biochem. Pharmacol. 23:2495–2531. [DOI] [PubMed] [Google Scholar]
- Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins, M., and G. Griffiths. 2003. Phagocytosis: latex leads the way. Curr. Opin. Cell Biol. 15:498–503. [DOI] [PubMed] [Google Scholar]
- Dimitrov, D.S. 2004. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2:109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drose, S., and K. Altendorf. 1997. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 200:1–8. [DOI] [PubMed] [Google Scholar]
- Esko, J.D., T.E. Stewart, and W.H. Taylor. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA. 82:3197–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen, B.L., B.L. Wei, J. Yao, T. Luo, and J.V. Garcia. 2002. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J. Virol. 76:11440–11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foukas, L.C., H.L. Katsoulas, N. Paraskevopoulou, A. Metheniti, M. Lambropoulou, and V.J. Marmaras. 1998. Phagocytosis of Escherichia coli by insect hemocytes requires both activation of the Ras/mitogen-activated protein kinase signal transduction pathway for attachment and beta3 integrin for internalization. J. Biol. Chem. 273:14813–14818. [DOI] [PubMed] [Google Scholar]
- Fukasawa, M., F. Sekine, M. Miura, M. Nishijima, and K. Hanada. 1997. Involvement of heparan sulfate proteoglycans in the binding step for phagocytosis of latex beads by Chinese hamster ovary cells. Exp. Cell Res. 230:154–162. [DOI] [PubMed] [Google Scholar]
- Gianni, T., G. Campadelli-Fiume, and L. Menotti. 2004. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J. Virol. 78:12268–12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly, D.J., A. Simonsen, and H. Stenmark. Phosphoinositides and phagocytosis. 2001. J. Cell Biol. 155:15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb, T.A., I.E. Ivanov, M. Adesnik, and D.D. Sabatini. 1993. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J. Cell Biol. 120:695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid, S., and B.B. Finlay. 2003. Microbial pathogenesis and cytoskeletal function. Nature. 422:775–781. [DOI] [PubMed] [Google Scholar]
- Iannolo, C., A.E. Salcini, I. Gaidarov, O.B. Goodman, J. Baulida, G. Carpenter, P.G. Pelicci, P.P. Di Fiore, and J.H. Keen. 1997. Mapping of the molecular determinants involved in the interaction between eps15 and AP-2. Cancer Res. 57:240–245. [PubMed] [Google Scholar]
- Locker, J.K., A. Kuehn, S. Schleich, G. Rutter, H. Hohenberg, R. Wepf, and G. Griffiths. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol. Biol. Cell. 11:2497–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs, G.L., O.D. Rotstein, and S. Grinstein. 1990. Phagosomal acidification is mediated by a vacuolar-type H(+)-ATPase in murine macrophages. J. Biol. Chem. 265:21099–21107. [PubMed] [Google Scholar]
- Marsh, M., and A. Helenius. 1989. Virus entry into animal cells. Adv. Virus Res. 36:107–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven, M.A. 1998. Dynamin: a molecular motor with pinchase action. Cell. 94:151–154. [DOI] [PubMed] [Google Scholar]
- Meier, O., and U.F. Greber. 2003. Adenovirus endocytosis. J. Gene Med. 5:451–462. [DOI] [PubMed] [Google Scholar]
- Mellman, I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 12:575–625. [DOI] [PubMed] [Google Scholar]
- Miller, N., and L.M. Hutt-Fletcher. 1992. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 66:3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, R.S., A.V. Nicola, J.C. Whitbeck, R.J. Eisenberg, and G.H. Cohen. 2005. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 79:6655–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola, A.V., and S.E. Straus. 2004. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 78:7508–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola, A.V., A.M. McEvoy, and S.E. Straus. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola, A.V., J. Hou, E.O. Major, and S.E. Straus. 2005. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 79:7609–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, B.J., and J. Lippincott-Schwartz. 2001. Endocytosis without clathrin coats. Trends Cell Biol. 11:406–412. [DOI] [PubMed] [Google Scholar]
- Parry, C., S. Bell, T. Minson, and H. Browne. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 86:7–10. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., and A. Helenius. 2003. Insider information: what viruses tell us about endocytosis. Curr. Opin. Cell Biol. 15:414–422. [DOI] [PubMed] [Google Scholar]
- Perez, L., and L. Carrasco. 1994. Involvement of the vacuolar H(+)-ATPase in animal virus entry. J. Gen. Virol. 75:2595–2606. [DOI] [PubMed] [Google Scholar]
- Pertel, P.E., A. Fridberg, M.L. Parish, and P.G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoprotein gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology. 279:313–324. [DOI] [PubMed] [Google Scholar]
- Pignatti, P.F., and E. Cassai. 1980. Analysis of herpes simplex virus nucleoprotein complexes extracted from infected cells. J. Virol. 36:816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch, M. 1995. Professional and non-professional phagocytes: an introduction. Trends Cell Biol. 5:85–87. [DOI] [PubMed] [Google Scholar]
- Russell, R.G., M.P. Nasisse, H.S. Larsen, and B.T. Rouse. 1984. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest. Ophthalmol. Vis. Sci. 25:938–944. [PubMed] [Google Scholar]
- Sahlin, S., J. Hed, and I. Rundquist. 1983. Differentiation between attached and ingested immune complexes by a fluorescence quenching cytofluorometric assay. J. Immunol. Methods. 60:115–124. [DOI] [PubMed] [Google Scholar]
- Sansonetti, P. 2001. Phagocytosis of bacterial pathogens: implications in the host response. Semin. Immunol. 13:381–390. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A. 2004. Regulating actin dynamics at membranes: a focus on dynamin. Traffic. 5:463–469. [DOI] [PubMed] [Google Scholar]
- Schmid, S.L. 1997. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annu. Rev. Biochem. 66:511–548. [DOI] [PubMed] [Google Scholar]
- Shukla, D., and P.G. Spear. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest. 108:503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla, D., J. Liu, P. Blaiklock, N.W. Shworak, X. Bai, J.D. Esko, G.H. Cohen, R.J. Eisenberg, R.D. Rosenberg, and P.G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 99:13–22. [DOI] [PubMed] [Google Scholar]
- Shukla, D., M.C. Dal Canto, C.L. Rowe, and P.G. Spear. 2000. Striking similarity of murine nectin-1alpha to human nectin-1alpha (HveC) in sequence and activity as a glycoprotein D receptor for alphaherpesvirus entry. J. Virol. 74:11773–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein, S.C., and P.I. Marcus. 1964. Early stages of Newcastle disease virus-Hela cell interaction: an electron microscopic study. Virology. 23:370–380. [DOI] [PubMed] [Google Scholar]
- Spaur, R.C., and G. Moriarty. 1977. Improvements of glycol methacrylate I. Its use as an embedding medium for electron microscopy studies. J. Histochem. Cytochem. 25:163–174. [DOI] [PubMed] [Google Scholar]
- Spear, P.G. 1993. a. Entry of alphaherpesviruses into cells. Semin. Virol. 4:167–180. [Google Scholar]
- Spear, P.G. 1993. b. Membrane fusion induced by herpes simplex virus. In Viral Fusion Mechanisms. J. Bentz, editor. CRC Press, Inc., Boca Raton, FL. pp. 201–232.
- Spear, P.G., R.J. Eisenberg, and G.H. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology. 275:1–8. [DOI] [PubMed] [Google Scholar]
- Spector, I., N.R. Shochet, D. Blasberger, and Y. Kashman. 1989. Latrunculins-novel marine macrolides that disrupt mocrofilament organization and affect cell growth: comparison with cytochalasin D. Cell Motil. Cytoskeleton. 13:127–144. [DOI] [PubMed] [Google Scholar]
- Thomas, J., and B.T. Rouse. 1997. Immunopathogenesis of herpetic ocular disease. Immunol. Res. 16:375–386. [DOI] [PubMed] [Google Scholar]
- Tiwari, V., C. Clement, P.M. Scanlan, B.Y.J.T. Yue, and D. Shukla. 2005. A role for HVEM as the receptor for herpes simplex virus-1 entry into primary human trabecular meshwork cells. J. Virol. 79:13173–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjelle, T.E., T. Lovdal, and T. Berg. 2000. Phagosome dynamics and function. Bioessays. 22:255–263. [DOI] [PubMed] [Google Scholar]
- Van Strijp, J.A., K.P. Van Kessel, M.E. van der Tol, and J. Verhoef. 1989. Complement-mediated phagocytosis of herpes simplex virus by granulocytes. Binding or ingestion. J. Clin. Invest. 84:107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, C.P., C.S. Park, and B.H. Lau. 1993. A rapid and simple microfluorometric phagocytosis assay. J. Immunol. Methods. 162:1–7. [DOI] [PubMed] [Google Scholar]
- Xia, G., J. Chen, V. Tiwari, W. Ju, J.P. Li, A. Malmstrom, D. Shukla, and J. Liu. 2002. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 277:37912–37919. [DOI] [PubMed] [Google Scholar]
- Yue, B.Y.J.T., and J.L. Baum. 1981. Studies of corneas in vivo and in vitro. Vision Res. 21:41–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}