

Abstract
Apicomplexan pathogens are obligate intracellular parasites. To enter cells, they must bind with high affinity to host cell receptors and then uncouple these interactions to complete invasion. Merozoites of Plasmodium falciparum, the parasite responsible for the most dangerous form of malaria, invade erythrocytes using a family of adhesins called Duffy binding ligand-erythrocyte binding proteins (DBL-EBPs). The best-characterized P. falciparum DBL-EBP is erythrocyte binding antigen 175 (EBA-175), which binds erythrocyte surface glycophorin A. We report that EBA-175 is shed from the merozoite at around the point of invasion. Shedding occurs by proteolytic cleavage within the transmembrane domain (TMD) at a site that is conserved across the DBL-EBP family. We show that EBA-175 is cleaved by PfROM4, a rhomboid protease that localizes to the merozoite plasma membrane, but not by other rhomboids tested. Mutations within the EBA-175 TMD that abolish cleavage by PfROM4 prevent parasite growth. Our results identify a crucial role for intramembrane proteolysis in the life cycle of this pathogen.
Introduction
Malaria is caused by species of Plasmodium, a protozoan parasite belonging to the phylum Apicomplexa. The parasite grows within erythrocytes, which it invades in a rapid, multistep process. Initial attachment of the merozoite is followed by reorientation, formation of an electron-dense junction between its apical prominence and the erythrocyte surface, and entry through this “moving junction” into a parasitophorous vacuole. Invasion is facilitated by the discharge of apical secretory organelles called micronemes.
The various steps in invasion involve different receptor–ligand interactions. Primary binding is low affinity and is probably mediated by a glycosyl phosphatidylinositol–anchored protein complex mostly composed of fragments of merozoite surface protein 1 (MSP1), which is expressed around the merozoite circumference (Li et al., 2004), or by other surface-resident glycosyl phosphatidylinositol–anchored proteins (Sanders et al., 2005). Reorientation requires a microneme protein called apical membrane antigen 1 (AMA1), which is released onto the merozoite surface before attachment (Mitchell et al., 2004). Junction formation constitutes an essentially irreversible, high-affinity binding step and involves members of a family of adhesive type I integral membrane microneme proteins known as the Duffy binding ligand-erythrocyte binding proteins (DBL-EBPs). The DBL-EBPs share a similar overall structure, with one or two erythrocyte binding DBL domains situated near their N terminus, a juxtamembrane cysteine-rich domain of unknown function called region VI, a transmembrane domain (TMD), and short cytoplasmic domain (Adams et al., 2001). Despite this overall similarity, the DBL-EBP exhibit extensive sequence diversity and interact with a range of receptors.
The best-characterized P. falciparum DBL-EBP is erythrocyte binding antigen 175 (EBA-175), which binds to a sialic acid–containing structure on glycophorin A (Dolan et al., 1994; Sim et al., 1994; Tolia et al., 2005). Although some P. falciparum strains use predominantly this interaction for invasion, others (including many field isolates) do not, invading through alternative pathways (Dolan et al., 1990; Okoyeh et al., 1999) that use other members of the DBL-EBP family (Duraisingh et al., 2003; Gilberger et al., 2003b). There is extensive redundancy in invasion pathways, and the variety of DBL-EBP paralogues expressed by P. falciparum together with polymorphism within them may allow the parasite to evade immune responses and to invade a range of host cells despite widespread polymorphism in human erythrocyte surface molecules (Maier et al., 2003; Mayer et al., 2004). The structural and antigenic diversity across the DBL-EBP family poses hurdles for the development of vaccines or drugs that target these important ligands.
Invasion by all Apicomplexa is accompanied by extensive proteolysis of surface and microneme proteins. During invasion by P. falciparum merozoites, both the MSP1 complex and AMA1 are quantitatively shed by cleavage at juxtamembrane sites. The enzyme responsible is a subtilisin-like “sheddase” called PfSUB2 that translocates across the parasite surface (Howell et al., 2003; Harris et al., 2005). A distinct mode of shedding has been observed in the related apicomplexan Toxoplasma gondii. Here, several transmembrane microneme proteins that translocate across the parasite through interactions with a subplasmalemmal actinomyosin motor are shed in the final stages of invasion by cleavage within their TMD (Opitz et al., 2002; Zhou et al., 2004). This “capping proteolysis” is mediated by an enzyme, initially dubbed microneme processing protease 1 (MPP1; Carruthers et al., 2000), that is now thought to be a rhomboid, a class of polytopic membrane serine proteases that cleave within the TMD of their substrates (Urban et al., 2001; Urban and Freeman, 2003; Zhou et al., 2004; Brossier et al., 2005; Dowse et al., 2005). All the proteins shed by PfSUB2 and MPP1 have demonstrated or putative binding activity, and the fact that interventions that prevent shedding also inhibit invasion suggests that shedding is required to disengage binding interactions between the parasite and host cell surface, enabling rapid entry into the parasitophorous vacuole (Carruthers and Blackman, 2005). It has not been established whether intramembrane proteolysis plays a role in the release of Plasmodium adhesins.
EBA-175 and all other known DBL-EBPs accumulate in soluble form in culture supernatants (Orlandi et al., 1990; Reed et al., 2000; Gilberger et al., 2003b), suggesting that their role at invasion may culminate in their being shed in a manner similar to the adhesins described above. The significance of DBL-EBP shedding has not been investigated. Here, we address this issue. We show that EBA-175 is secreted onto the surface of P. falciparum merozoites and is shed at or around the point of invasion. Shedding occurs by cleavage within the TMD at a site that is conserved in all DBL-EBPs. Cleavage is mediated by a rhomboid called PfROM4 that is expressed at the merozoite plasma membrane but not by other rhomboids tested. Importantly, parasite lines carrying mutations that prevent PfROM4-mediated cleavage could not be established, suggesting that shedding of EBA-175 is important for successful invasion. Our results show that intramembrane proteolysis by PfROM4 is critical to maintenance of the parasite life cycle and suggest that targeting PfROM4 activity may provide a means to interfere with the function of the entire DBL-EBP family.
Results
EBA-175 is shed from the merozoite at or around the point of erythrocyte invasion
Antibodies raised against a folded, recombinant form of EBA- 175 region VI (Fig. 1 A) were specific for schizont and merozoite-derived EBA-175 on Western blots and also reacted with the shed form in culture supernatants (Fig. 1 B). The shed protein therefore retains some or all of region VI, as previously suggested (Kaneko et al., 2000; Duraisingh et al., 2003). The antibodies produced a strong punctate immunofluorescence assay (IFA) pattern in acetone-fixed schizonts and free merozoites, consistent with the micronemal location of EBA-175 (Fig. 1 C). They also reacted specifically with the apical surface of many nonpermeabilized free merozoites, suggesting that EBA-175 is secreted onto the merozoite surface just before invasion (Fig. 1 D). In contrast, the anti–region VI antibodies did not react at all with most newly invaded rings of either the W2mef or 3D7 P. falciparum clones, labeling only a small fraction with a weak, punctate pattern (Fig. 1 E and Table I, second column). To examine this in more detail, we took advantage of the fact that parasite lines vary in their degree of dependence on EBA-175 as an invasion ligand. W2mef relies predominantly on sialic acid–dependent pathways for invasion and uses EBA-175 as its dominant invasion ligand, whereas 3D7 is capable of invading by both sialic acid–dependent and –independent routes (Reed et al., 2000; Duraisingh et al., 2003). Pretreatment of host erythrocytes with neuraminidase removes the sialic acids required for EBA-175 binding to glycophorin A (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1), precluding EBA-175–mediated invasion and forcing invasion to proceed by alternative pathways. As shown in Table I, invasion into neuraminidase-treated cells did not alter the proportion of 3D7 rings reactive with the anti–region VI antibodies. Collectively, these results imply that at around the time of invasion, EBA-175 is predominantly shed from the merozoite and that this occurs to the same degree regardless of whether EBA-175 is used as a primary ligand mediating invasion. As EBA-175 plays its role in invasion at the parasite–host interface, shedding presumably takes place from the merozoite surface. The truncated form of EBA-175 that appears in culture supernatants is the product of this.
Figure 1.

EBA-175 is shed at or around the point of erythrocyte invasion and retains region VI. (A) Schematic of EBA-175 structure, indicating signal sequence (SS), F1 and F2 DBL domains, TMD, and cytoplasmic domain (CYT). The position of region VI is shown, with a Coomassie blue–stained SDS-PAGE gel of purified recombinant region VI (inset). (B) Western blot of 3D7 schizonts, merozoites, and culture supernatant, run on nonreducing 7.5% SDS-PAGE and probed with anti–region VI antibodies. Shed EBA-175 (arrow) migrates more rapidly than the parasite forms. Molecular mass markers are indicated. (C) IFA images of an acetone-fixed segmented 3D7 schizont and free merozoites dual labeled after fixation with mAb 1E1 (anti-MSP119; red) and anti–region VI antibodies (green). The punctate, apical pattern obtained with the latter is typical of microneme staining. (D) EBA-175 is discharged onto the apical surface of free merozoites. Merozoites released from 3D7 schizonts in the presence of anti–region VI antibodies were washed, fixed, and probed with FITC anti-mouse IgG to detect bound anti–region VI antibodies (green), followed by mAb 1E1 (anti-MSP119; red). Many free merozoites exhibit a strong apical FITC signal (thin arrows), whereas residual intact schizonts containing merozoites that were not accessible to the anti–region VI antibodies show only background FITC labeling (thick arrows). No signal was associated with merozoites released in the presence of preimmune mouse antisera (unpublished data). (E) IFA images of newly invaded (≤3-h-old) ring-stage 3D7 parasites probed with anti–region VI antibodies after acetone fixation. Most rings did not react at all with the antibodies. Nuclei were stained with DAPI (blue). Identical results were obtained with the W2mef clone (unpublished data). Bars, 5 μm.
Table I.
Shedding of EBA-175 at erythrocyte invasion occurs to the same extent whether or not it is used as the dominant invasion ligand
|
|
Proportion of rings reactive with anti–region VI antibodies in IFAa |
||
|---|---|---|---|
|
P. falciparum line
|
Erythrocyte pretreatment
|
||
| untreated | mock | neuraminidaseb | |
| % | % | % | |
| W2mef | 13.4 ± 3.8 | NDc | NDc |
| 3D7 | 16.7 ± 3.5 | 14.9 ± 3.2 | 15.4 ± 5.1d |
Figures shown are derived from three experiments. At least 1,000 rings were examined in each case. 95% confidence levels are indicated.
Erythrocytes were neuraminidase treated or mock treated as described in Materials and methods. The efficiency of the treatment was assessed using an overlay assay to confirm lack of EBA-175 binding (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1).
Invasion of neuraminidase-treated erythrocytes by W2mef was very inefficient (≤2.0% of invasion into mock-treated cells) as expected, so these rings were not examined by IFA.
Invasion of neuraminidase-treated erythrocytes by 3D7 was 89% of invasion into mock-treated cells, in agreement with previous findings (Duraisingh et al., 2003).
EBA-175 is shed by a calcium-independent serine protease distinct from PfSUB2
To study the mechanism of EBA-175 shedding, we used an assay developed for characterizing the sheddase PfSUB2 (Howell et al., 2003; Harris et al., 2005). Merozoites were incubated at 37°C with a range of test additives, and supernatants were analyzed for the presence of shed proteins. Fig. 2 shows that shedding of EBA-175 was readily detectable, as was shedding of the expected MSP1 and AMA1 fragments. Release of EBA-175 was partially sensitive to PMSF and dichloroisocoumarin, indicating the involvement of a serine protease. However, in contrast to MSP1 and AMA1, EBA-175 shedding was not inhibited by either the calcium chelating agent EGTA or recombinant PfSUB2 prodomain (PfSUB2PD), a selective inhibitor of PfSUB2 (Harris et al., 2005). These data show that EBA-175 is not shed by PfSUB2 but by a distinct, calcium-independent, serine protease.
Figure 2.

EBA-175 is not shed by PfSUB2 but by a distinct calcium-independent serine protease. Shed EBA-175 and processing fragments of AMA1 (AMA148/44) and MSP1 (MSP133) were detected in merozoite supernatants by Western blot as described in Materials and methods. Supernatants were harvested immediately (start) or after a 1-h incubation at 37°C in buffer alone (no additions) or containing the indicated additives. Isopropanol and DMSO were solvent controls for PMSF and dichloroisocoumarin (DCI), respectively, whereas FT control refers to flow-through from the final step of concentration of purified PfSUB2PD by ultrafiltration (Harris et al., 2005), added to the assay at a dilution similar to that used for PfSUB2PD. These results were reproducible in three independent experiments. In Western blot analysis (similar to that in Fig. 1 A) the shed EBA-175 produced in these assays comigrated with that from culture supernatants (unpublished data), consistent with it being the result of the same protease activity.
EBA-175 is shed by cleavage within its TMD at a rhomboid-like site
Shed EBA-175 can be recovered from culture medium by being bound to erythrocytes and then eluted with high salt (Orlandi et al., 1990). We adapted this approach to purify sufficient EBA-175 for structural analysis. SDS-PAGE of the partially purified fractions revealed a major protein species at ∼175 kD (Fig. 3 A). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) analysis of in-gel tryptic digests confirmed this as EBA-175, with a MASCOT probability score of 462 (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1). The most C-terminal peptide identified in these digests corresponds to 1406EFDDPSYTCFRK1417, which lies just upstream of the TMD (Fig. 3 B). No anomalous peptides were identified that might represent the N- or C-terminal end of the protein, so further samples were digested with Asp-N in the presence of 50% (vol/vol) 18O water. Under these conditions, products of digestion incorporate both 18O and 16O at their C termini as a result of proteolytic hydrolysis, whereas any peptide derived from the extreme C terminus of the protein substrate should retain a normal 16O isotope spectrum (Howell et al., 2003, 2005). Examination of these digests by MALDI- TOF again identified only peptides derived from the EBA-175 ectodomain (MASCOT probability score 199), including a region VI–derived ion at m/z 1740.716, matching 1395DSEEYYNCTKREF1407 (calculated m/z 1740.717). As expected, all peptides identified with an N-terminal Asp and ending at a residue adjacent to an Asp-N cleavage site exhibited an 18O-containing spectrum. Within the anomalous peptides observed were only two unlabeled ions that could be assigned as being products of EBA-175 digestion: one at m/z 2435.242, corresponding closely to the EBA-175–derived peptide 1408DDPSYTCFRKEAFSSMPYYA1427 (calculated m/z 2435.032; Fig. 3, B and C), and a second, less intense ion at m/z 2451.262, matching its expected Met oxidized form (calculated m/z 2451.032; unpublished data). The absence of an Asp-N cleavage site at the C terminus of this peptide, together with the absence of 18O label, implicated it as the C terminus of the shed EBA-175. Collision-induced fragmentation in a nanospray mass spectrometer (Fig. 3 D) confirmed its identity and clearly detected the C-terminal sequence Y-A.
Figure 3.

EBA-175 is shed by intramembrane cleavage. (A) Coomassie-stained SDS-PAGE of partially purified shed EBA-175 (arrow). A total of ∼40 μg of EBA-175 was obtained from 3 liters of culture medium. (B) The most C-terminal intact tryptic and Asp-N peptides identified in digests of EBA-175, with calculated m/z values in carbamidomethylated form, are shown in relation to the juxtamembrane sequence of 3D7 EBA-175 (PlasmoDB ID PF07_0128). The TMD (TMHMM v2.0) is shaded. The sequence is aligned with corresponding sequence from P. falciparum paralogues EBA-181 (PFA0125c) and EBA-140 (MAL13P1.60), as well as P. falciparum MAEBL (PF11_0486), P. vivax DBP (available from GenBank/EMBL/DDBJ under accession no. P22290), and P. knowlesi DBP (available from GenBank/EMBL/DDBJ under accession no. P22545). Conserved cysteines are marked with an asterisk. Helix breaking motifs constituting potential rhomboid recognition sites within the TMD are underlined, and the conserved Ala at which EBA-175 is cleaved is boxed. (C) MALDI-TOF spectra of EBA-175 Asp-N digests performed in 100% H2 16O or 50% (vol/vol) H2 18O. The peak matching 1408DDPSYTCFRKEAFSSMPYYA1427 (arrow; shown magnified in inset, with observed m/z value) is not 18O-labeled in the lower spectrum, indicating that it derives from the C terminus. For comparison, peaks corresponding to 1227DRNSNTLHLK1236 (calculated m/z 1197.634) and 562DLSNRKLVGKINTNSNYVHRNKQN585 (calculated m/z 2812.493) are indicated as examples of species that in the lower spectrum exhibit the 18O-labeled isotope pattern typical of internal peptides. (D) Collision-induced fragmentation spectrum of the unlabeled m/z 2435.242 peptide in Asp-N digests. The indicated fragment peaks can be assigned to a b and y series of ions, with the b18 and b19 ions assigning the C-terminal peptide sequence Y-A. The remaining major ions are all consistent with the indicated sequence.
These data map the C terminus of shed EBA-175 to an Ala residue that lies three residues into the TMD. Importantly, this Ala is conserved across the DBL-EBP family (Fig. 3 B; Carruthers and Blackman, 2005). Of the few proteases that can hydrolyze intramembrane substrates, only rhomboids cleave close to the luminal side of the TMD; also, cleavage after an Ala is a hallmark of rhomboids (Urban and Freeman, 2003; Zhou et al., 2004; Howell et al., 2005). Collectively, our data show that EBA-175 is shed by a protease with the characteristics of a rhomboid.
The merozoite rhomboid PfROM1 localizes exclusively to micronemes
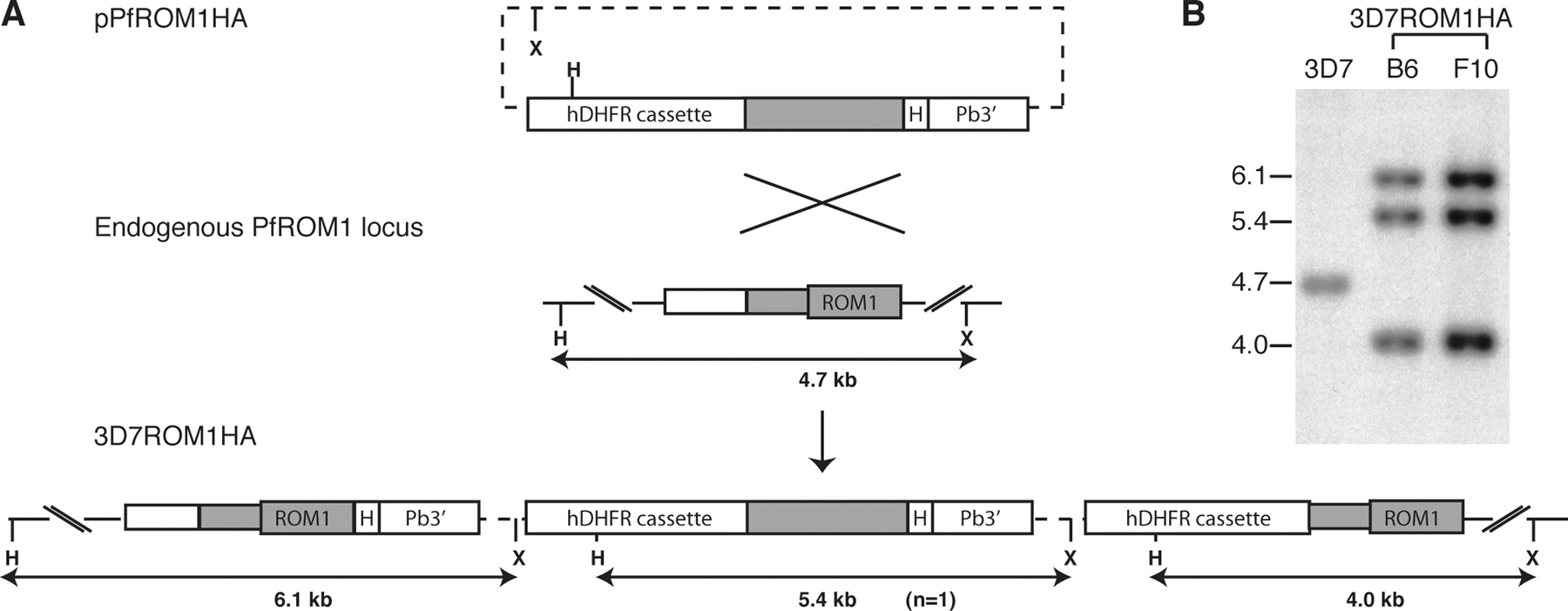
Apicomplexan parasites possess several rhomboid-like genes (Dowse and Soldati, 2005). T. gondii has six, of which three (encoding TgROM1, -4, and -5) are expressed in tachyzoites in locations where they might play a role in invasion; TgROM1 is micronemal, whereas the other two larger rhomboids are expressed at the parasite plasma membrane (Brossier et al., 2005; Dowse et al., 2005). Based on these expression patterns, the best candidates for the protease mediating shedding of EBA-175 were considered to be PfROM1, the P. falciparum orthologue of TgROM1, and PfROM4, the single orthologue of TgROM4 and -5. Microarray studies have shown that both pfrom1 (PlasmoDB ID PF11_0150) and pfrom4 (PFE0340c) are expressed in P. falciparum blood stages (Bozdech et al., 2003; Le Roch et al., 2003) The predominantly intramembrane nature of rhomboids can make them poor antigens; therefore, to localize these gene products, we attempted to epitope tag the genes using single-crossover homologous recombination. For PfROM1, we transfected parasites with a plasmid containing a targeting sequence that included pfrom1 coding sequence fused to three copies of the HA epitope (HA3; Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1). Drug-selected parasites (called 3D7ROM1HA) were cloned for further characterization. Southern blot (Fig. S2 B) and PCR analysis (unpublished data) confirmed that the plasmid had integrated through the expected recombination event, placing the HA3 tag at the 3′ end of the pfrom1 gene.
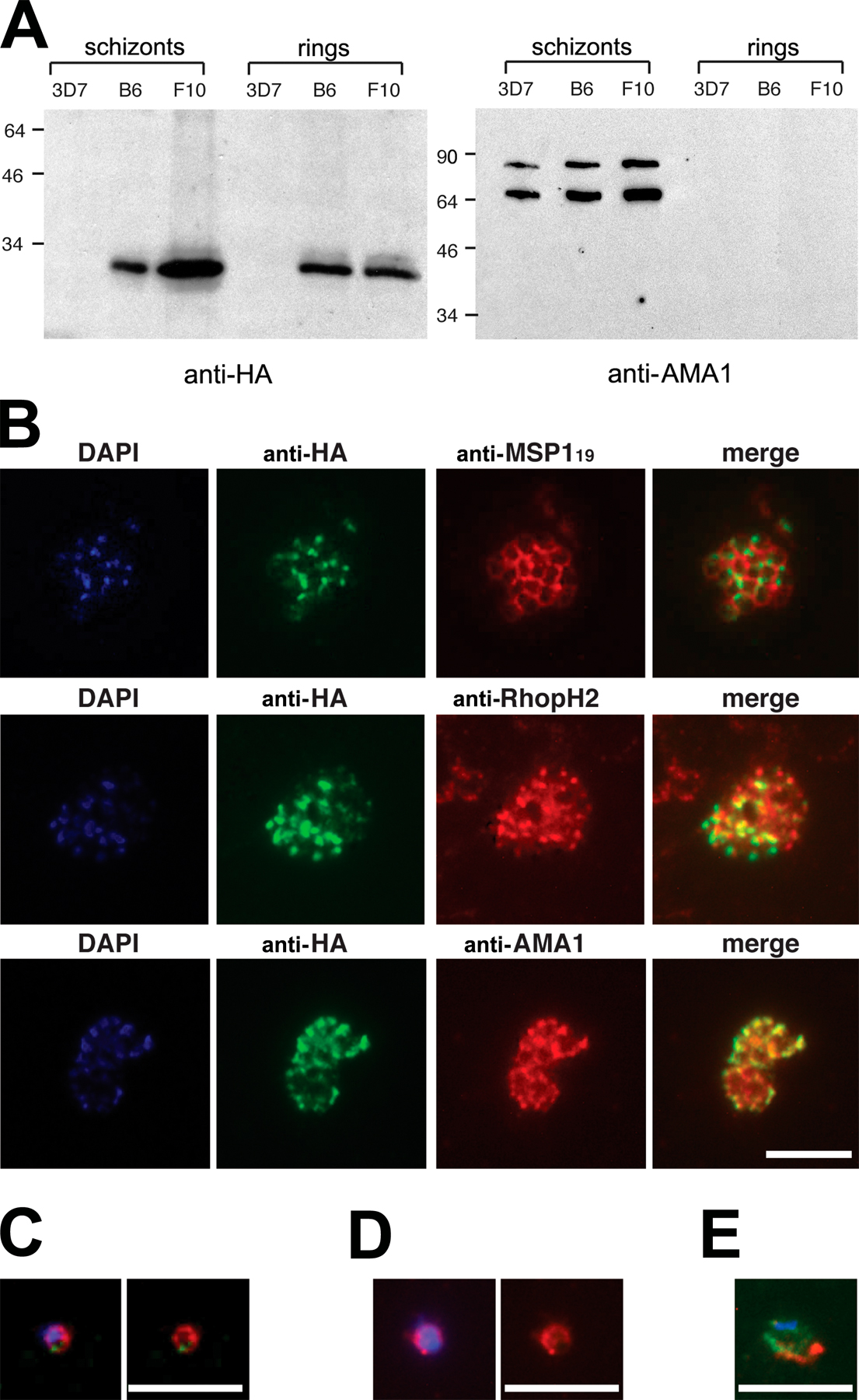
Western blot of the clones (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1) revealed an anti-HA–reactive protein matching the predicted mass (26.7 kD) of HA-tagged PfROM1. Schizonts probed with mAb 3F10 showed a punctate IFA pattern that colocalized with the microneme protein AMA1 (Fig. S3 B), indicating that, like its T. gondii homologue, PfROM1 is a microneme protein. IFA of free merozoites and ring stages showed that, unlike AMA1, which redistributes onto the merozoite surface just before invasion (Howell et al., 2003), PfROM1 remains exclusively in an apical location; it could not be detected at the merozoite surface (Fig. S3, C and D) and did not colocalize with the plasma membrane marker MSP119 in rings (Fig. S3 E). Our results show that PfROM1 is restricted to micronemes, where it is unlikely to play a part in shedding of surface-located EBA-175.
PfROM4 is a merozoite surface rhomboid
Attempts to epitope tag the 3′ end of the pfrom4 gene using a similar strategy failed (unpublished data), perhaps indicating that modification of the gene at this site is deleterious to the parasite. In an alternative approach, we transfected parasites with a construct for episomal expression of N-terminal HA3-tagged PfROM4, under the control of the pfrom4 promoter. Western blot of the resulting parasite line (called 3D7HAROM4synth) revealed expression of a novel, anti-HA–reactive protein of 84 kD, close to the expected mass of HA-tagged PfROM4 (Fig. 4 A). IFA showed this colocalized with the plasma membrane marker MSP1 in schizonts (Fig. 4 B). Identical results were obtained using the ama1 promoter to express the gene (unpublished data). These findings indicate that PfROM4 is expressed at the merozoite plasma membrane, an appropriate location to play a role in shedding of merozoite surface EBA-175.
Figure 4.

PfROM4 is a merozoite plasma membrane protein. (A) Western blot showing extracts of parental 3D7 and 3D7HAROM4synth probed with mAb 3F10 (anti-HA) and polyclonal anti-AMA1 (loading control). The size of the anti-HA–reactive species detected only in 3D7HAROM4synth is close to the predicted mass (90.4 kD) of HA-tagged PfROM4. (B) IFA of mature nonsegmented (top) and fully segmented (bottom) schizonts of 3D7HAROM4synth, dual labeled with mAb 3F10 (anti-HA; green) and mAb 1E1 (anti-MSP1; red). The DAPI signal (blue) is not included in merged images for clarity. Bar, 5 μm.
EBA-175 is specifically cleaved by PfROM4
To explore whether EBA-175 could be a substrate for PfROM4, we used a heterologous expression system that has been used to characterize several rhomboids, including those from T. gondii and Drosophila melanogaster. A synthetic “minigene,” encoding HA-tagged EBA-175 region VI plus its cognate TMD and cytoplasmic domain, was cloned into a vector for constitutive expression in mammalian cells (Fig. 5 A). The gene product was targeted to the secretory pathway by the murine Igκ signal sequence. COS-7 cells transfected with this construct showed strong surface fluorescence when probed without prior fixation with mAb 3F10, indicating expression of the protein in membrane bound form at the cell surface (Fig. 5 B). Western blot with the same mAb detected a major, ∼32-kD, and minor, 25-kD, protein in transfected cells. Region VI contains two potential N-glycosylation sites at N1333 and N1401. N-glycosylation is rare or absent in P. falciparum (Howell et al., 2003), so one or both of these residues was substituted with Asp, resulting in expressed products EBAregVIgl1 and EBAregVI, respectively. The latter migrated as a single 25-kD species, close to its predicted nonglycosylated mass of 24 kD; it was also equally well expressed at the surface of transfected COS-7 cells (Fig. 5 B). Cells were then cotransfected with constructs designed for expression of either wild-type PfROM4 or a mutant form with the predicted active-site Ser residue replaced with Ala, and medium from the cells was examined by Western blot for the appearance of shed forms of the HA-tagged region VI. Coexpression with wild-type PfROM4 resulted in the appearance in cell supernatants of anti-HA–reactive proteins that migrated on SDS-PAGE more rapidly than the forms in cell extracts (Fig. 5 C). Importantly, levels of these shed forms were significantly lower, and not above background levels (unpublished data), when coexpressed with the mutant PfROM4. These results suggested that the shed protein is a result of PfROM4 proteolytic activity.
Figure 5.

EBA-175 is a substrate for PfROM4. (A) Structure of EBA-175 minigenes expressed in COS-7 cells. Partial amino acid sequences of the various mutants are indicated. Substitutions made to remove N-glycosylation sites or to modify the TMD (shaded) are in bold and underlined. The position mapped as the site of EBA-175 shedding is indicated by an arrow. The predicted topology of the expression products is shown (inset). (B) IFA showing expression of EBA-175 minigene products at the surface of COS-7 cells. Cells transfected with the various constructs were probed without fixation or permeabilization with the anti-HA mAb 3F10 (green). In all cases, 15–20% of cells exhibited strong surface fluorescence. Nuclei were stained with DAPI (blue). Bar, 50 μm. (C) Western blots of cells or medium from COS-7 cells cotransfected with EBA-175 minigene constructs and HA-tagged PfROM4 expression constructs, either in wild-type (wt) or active site Ser knockout form (mut). Blots were probed with mAb 3F10. The uppermost band visible in the cell extract blot (arrow) corresponds to HA-tagged PfROM4; note its absence from the farthest right lane, where no PfROM4 construct was transfected. The band just below this is a nonspecific reaction signal. These results were reproducible in >19 independent experiments.
All DBL-EBP possess a GA or GG motif just proximal to the extracellular side of their TMD (Fig. 3 B), reminiscent of the helix-destabilizing structures that determine sensitivity to rhomboids (Urban and Freeman, 2003). To explore the requirements for recognition of the EBA-175 TMD by PfROM4, three further forms of EBAregVI with mutations in the TMD were evaluated in the COS-7 cell system (Fig. 5). Substitution of A1427, the residue at which cleavage occurs to release EBA-175 from the merozoite surface (mutant EBAregVI-mutA), prevented PfROM4-mediated shedding. Similarly, substitution of the GA motif (called GA1) closest to the extracellular end of the TMD (EBAregVI-mutGA1), predicted as the site required for rhomboid recognition, abolished specific cleavage. In contrast, substitution of another GA motif (GA2) lying near the cytoplasmic end of the TMD (EBAregVI-mutGA2) had no effect on cleavage. These data show that the EBA-175 TMD is recognized and cleaved by PfROM4 and suggest that the requirements for recognition are similar to those characterized for other rhomboids.
These experiments were extended, cotransfecting with constructs expressing PfROM1 or the D. melanogaster rhomboid Rho-1 instead of PfROM4. In repeated experiments, and under conditions where Rho-1 efficiently cleaved AMA1 as shown previously (Howell et al., 2005), we were unable to detect any activity of these rhomboids against the EBAregVI constructs (unpublished data). These findings imply a specific interaction between PfROM4 and EBA-175.
PfROM4-mediated shedding of EBA-175 is critical to maintenance of the P. falciparum blood-stage life cycle
To directly address the importance of EBA-175 shedding in the erythrocytic life cycle of P. falciparum, we attempted to generate transgenic parasites expressing mutant forms of EBA-175 that lacked one or other of the two TMD GA motifs examined in the previous section. To do this, we constructed plasmids (pHH1-175-GA1/FF and pHH1-175-GA2/FF) designed to introduce the desired substitutions by recombination into the 3′ end of the eba-175 gene (Fig. 6 A).
Figure 6.

Mutation of the PfROM4 recognition motif within the EBA-175 TMD is deleterious. (A) Scheme showing the replacement of the 3′ region of the eba-175 gene by single-crossover integration of pHH1-175-GA1,2/FF constructs. The positive selection cassette (hDHFR) of the pHH1 vector is represented by a black box. A ∼1-kb fragment of eba-175 cDNA including sequence encoding region VI (yellow), the mutant TMD (red), and the cytoplasmic domain (green) was cloned into vector pHH1, flanked by the 3′ UTR of the Plasmodium berghei dihydrofolate reductase gene (blue) to ensure correct transcription termination and polyadenylation of the modified gene. The intron-exon structure of the eba-175 gene is shown (gray; signal peptide). MfeI restriction enzyme sites are indicated (M). Relative positions of oligonucleotides used in the PCR analysis (1, primer HH1-S; 2, primer HH1-AS; 3, primer 1753259-S; 4, primer 175-ASstop; see Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1) are shown as colored arrows, and the position of the probe used for Southern analysis is indicated by the red bar. (B) Southern blot of gDNA from parental and transgenic parasites (MfeI restricted) reveals integration of pHH1-175-GA2/FF but not pHH1-175-GA1/FF into the eba-175 locus. The endogenous eba-175 hybridizing fragment (8.4 kb) is present in the W2mef and 3D7 EBA175GA1/FF lines at both the second and third cycles of drug selection but disappears upon integration of the plasmid in the W2mef EBA175GA2/FF transgenic line; the 2.4- and 13-kb hybridization bands indicate replacement as expected of the 3′ end of the eba-175 gene by the second drug selection cycle. Sizes of hybridizing bands are shown, and the band corresponding to free episome is marked by an arrow. (C) PCR analysis of the eba-175 locus using gDNA from parental and transgenic parasite lines. Molecular mass markers are shown (M). Amplification from the eba-175 locus is displayed for each parasite line using three different oligonucleotide primer combinations: 1 plus 2 (black and blue; plasmid specific), 3 plus 4 (red and green; eba-175 specific), and 2 plus 3 (blue and red; eba-175 single-crossover specific). (D) IFA localization of mutant EBA-175 in transgenic EBA175GA2/FF W2mef schizonts and free merozoites. Antibodies specific for EBA-175 (red) and EBA-181 (green) colocalize in the merged image. The DAPI signal (blue) is not included in the merge. Bars, 5 μm.
Each plasmid was first transfected into W2mef, and the resulting parasite lines (called EBA175GA1/FF and EBA175GA2/FF) were drug cycled to select for integrants. Southern blot analysis of EBA175GA2/FF revealed a hybridization profile consistent with integration into the eba-175 locus as expected, in the bulk of the parasite population (Fig. 6 B). Confirmation of the expected GA2/FF substitution within the eba-175 gene was obtained by PCR amplification (Fig. 6 C) and sequencing (not depicted) of the modified locus. Expression and correct localization of the mutant EBA-175 protein was confirmed by IFA using antibodies against EBA-175 and -181, another micronemal EBL-DBP (Fig. 6 D).
In contrast to the ease with which the EBA175GA2/FF transgenic line was derived, establishment of a viable parasite line carrying a substitution of the GA1 motif could not be achieved. In two independent transfection experiments in W2mef and one in 3D7, the input plasmid remained episomal for up to six drug cycles (unpublished data), a typical finding where integration is lethal or severely detrimental to the parasite. Because the two integration constructs used here were identical aside from the few base-pairs within the TMD encoding the mutant codons, it is unlikely that the failure to obtain integration of pHH1-175-GA1/FF was a technical problem associated with construct design. Our results support a critical role for the GA1 motif and suggest that modifications that interfere with PfROM4-mediated cleavage of EBA-175 cannot be tolerated by the parasite. Because shedding of EBA-175 occurs at or around the point of erythrocyte invasion, this is likely to be the stage in the life cycle at which arrest occurs after substitution of the GA1 motif.
Discussion
Many microneme proteins are secreted onto the parasite surface to play a role in host cell entry and then ultimately shed. This study demonstrates that EBA-175, and, by extrapolation, all other DBL-EBPs, are subject to a similar fate. Given their role in invasion and their capacity to bind erythrocyte surface receptors with high affinity, these ligands presumably function in membrane bound form at the merozoite surface. Our results show that the truncated form of EBA-175 released into supernatants is a result of a physiologically important, precise cleavage event that takes place at the merozoite surface and is mediated via intramembrane cleavage by a rhomboid-like malarial protease.
IFA of newly invaded rings showed that, irrespective of whether EBA-175 was used as the dominant invasion ligand, invasion is associated with shedding of EBA-175. Western blot showed that the shed protein retains much or all of region VI, and mass spectrometric analysis allowed us to map its C terminus to an Ala residue that lies three residues into the predicted TMD, a specificity identical to that of the rhomboid-like cleavage sites previously mapped in apicomplexan microneme proteins (Opitz et al., 2002; Zhou et al., 2004; Howell et al., 2005). We then showed that the P. falciparum homologues of three rhomboids previously localized to the secretory pathway in T. gondii are expressed in merozoites. Whereas PfROM1 was found to be exclusively micronemal, PfROM4 localizes to the merozoite plasma membrane. This is consistent with the findings of Brossier et al. (2005) and Dowse et al. (2005) who showed that TgROM4 and -5, the two T. gondii homologues of PfROM4, are both expressed at the tachyzoite surface. Of these, only TgROM5 showed protease activity against microneme proteins; thus, it was proposed that this is MPP1. Brossier et al. (2005) also found that TgROM5 expression was concentrated at the posterior surface of the tachyzoite, where it may be optimally placed to release transmembrane microneme proteins as they translocate rearwards at invasion. We observed no such bias in the distribution of PfROM4, but our findings do indicate that this rhomboid is expressed at an appropriate time and place to mediate shedding of EBA-175.
Early studies on the protease activity of rhomboids indicated that substrate recognition requires only a suitably disordered TMD resulting from the presence of a helix-disrupting GA or GG motif disposed toward the luminal end of the sequence (Urban and Freeman, 2003). Subsequent studies suggest that this may not be universally true, and there are now indications that structures on the extracellular (Opitz et al., 2002; Brossier et al., 2003; Pascall and Brown, 2004) or intracellular side (Lohi et al., 2004) of the TMD are also important for efficient substrate recognition. The minigene constructs used here to explore cleavage of the EBA-175 TMD incorporated the entire region VI as well as the cytoplasmic domain in anticipation that elements critical for recognition might reside within these juxtamembrane sequences. Of the three rhomboids tested, only PfROM4 cleaved these proteins. Substitution of either the Ala mapped as the site of EBA-175 cleavage, or the GA1 motif, abolished cleavage. The lack of cleavage by either PfROM1 or D. melanogaster Rho-1 shows that these motifs alone are not sufficient to render the EBA-175 TMD susceptible to all rhomboids and implies a degree of specificity in the interaction between PfROM4 and EBA-175. Eukaryotic rhomboids cluster into two major groups referred to as the RHO (typified by D. melanogaster Rho-1) and PARL (typified by the mitochondrial presenilins-associated rhomboid-like protein) subfamilies (Koonin et al., 2003). Both the malarial rhomboids investigated here are more closely related to Rho-1 than to PARL, but PfROM4 differs noticeably from Rho-1 and PfROM1 in possessing unusually large predicted loops between its first and second, and sixth and seventh TMD, as well as extended N- and C-terminal domains (Dowse and Soldati, 2005), structures that may be involved in EBA-175 recognition. Similarly, although region VI is known to be important for the trafficking and structural integrity of EBA-175 (Reed et al., 2000), it might also play a role in recognition by PfROM4.
EBA-175 is the dominant DBL-EBP mediating invasion in the W2mef line, so this isolate was first chosen to examine the effects of TMD mutations that prevent PfROM4-mediated cleavage. Although transgenic parasites lacking the GA2 motif were readily generated, parasites lacking the GA1 motif critical for PfROM4-mediated cleavage could not be. Further work showed that the GA1 mutation could not be introduced into the 3D7 line either. This was initially surprising in view of previous work showing that functional inactivation of EBA-175 by removal of the cytoplasmic tail (Gilberger et al., 2003a) or deletion of the TMD (Reed et al., 2000) can be readily achieved in 3D7 and is easily compensated for in W2mef by a switch to an EBA-175–independent invasion pathway. We accept that caution should be exercised in interpretation of our “negative” result. However, together with our observation that shedding of EBA-175 takes place to the same degree in both parasite lines irrespective of whether the ligand is used for invasion, our results can be reconciled by a model that postulates that if EBA-175 is present in a functional, membrane bound form, it must be capable of being shed for invasion to go to completion. At present we can only speculate on how a defect in shedding could stall invasion. EBA-175 functions as a dimer (Tolia et al., 2005), and if shedding-defective mutants can form heterodimers with other DBL-EBPs, this may exert a dominant-negative effect, interfering with the release of these ligands too from the merozoite surface. EBA-175 is the most abundant DBL-EBP in most P. falciparum isolates examined; thus, an alternative possibility is that a defect in EBA-175 shedding leads to an insurmountable physical barrier at invasion, with an accumulation of unshed EBA-175 preventing the rapid recruitment of other DBL-EBPs to the confined space of the moving junction. It is worth noting that, somewhat analogous to our results, mutations that prevent MPP1-mediated shedding of the T. gondii microneme protein TgMIC6 are deleterious to that parasite, despite the fact that TgMIC6 is not essential for host cell invasion (Opitz et al., 2002).
Could PfROM4 shed merozoite proteins other than the DBL-EBPs? Recent studies have implicated a second family of P. falciparum apical membrane proteins in erythrocyte invasion. Called the reticulocyte binding protein homologues, several lines of evidence indicate an important role, including gene disruption experiments that result in a switch in invasion phenotype (Stubbs et al., 2005). The precise function of these proteins is unclear, but they may have adhesive properties (Rayner et al., 2001), and proteolytic shedding may be important for their function (Triglia et al., 2005). Some members of this family possess helix-disrupting motifs in their TMD, and it will be interesting to determine whether they too can be substrates for PfROM4 or -1.
We have been unable to disrupt the pfrom4 gene by targeted homologous recombination (unpublished data), suggesting that it is indispensable in the blood-stage life cycle of the parasite. Our findings show that erythrocyte invasion involves the activity of at least two serine proteases, PfSUB2 and PfROM4, which have distinct roles and which belong to different protease families. This is consistent with protease inhibitor studies performed over two decades ago indicating that invasion requires the activity of more than one serine protease (Hadley et al., 1983).
Our evidence implicating PfROM4 in invasion by the malaria parasite supports the emerging view that rhomboid-mediated intramembrane proteolysis likely plays multiple roles in biological systems across evolution (Freeman, 2004). Our findings may have important practical implications for antimalarial drug development. P. falciparum has evolved a complex strategy to overcome receptor heterogeneity in its human host, exploiting a diverse family of ligands able to recognize a range of erythrocyte types. The conserved nature of the EBA-175 cleavage site across the DBL-EBP family raises the provocative notion that all these ligands may share a common requirement to be released at or around the point of invasion by PfROM4-mediated cleavage. Targeting PfROM4 with suitably selective drugs may transcend this ligand diversity, providing a more global approach to blocking invasion than attempting to target the receptor binding function of the ligands themselves.
Materials and methods
Parasite culture and transfection
P. falciparum clones 3D7 and W2mef were cultivated and synchronized as described previously (Blackman, 1994). For invasion assays or production of newly invaded ring stages, schizonts enriched by centrifugation over cushions of 63% Percoll (GE Healthcare) were cultured with fresh erythrocytes for up to 3 h to allow rupture and reinvasion before removal of residual schizonts as described previously (Blackman et al., 1990). For transfection, ring-stage parasites (2–10% parasitaemia) were electroporated with 70–100 μg of plasmid DNA using standard procedures (Fidock and Wellems, 1997). After initial selection for ∼4 wk in 2.5 or 10 nM of the antifolate WR99210 (Jacobus Pharmaceutical Co.), parasites were subjected to repeated cycles of selection for 3 wk followed by removal of the drug for 3 wk. Clonal populations were then obtained by limiting dilution.
Antibodies
Recombinant region VI was expressed in the yeast Pichia pastoris. For this, an intronless synthetic gene, called eba175regVI synth, encoding the C-terminal 170 residues (N1333-K1502) of 3D7 EBA-175 (PlasmoDB gene ID PF07_0128) was obtained from GENEART. An N-glycosylation site at N1401 was substituted with Asp using QuikChange site-directed mutagenesis (Stratagene) with primers EBAmut12for and -rev (see Table S2, available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1, for details of all primers used). Sequence encoding N1333 to M1423 was then PCR amplified with primers PPeba-A and -B to remove a second N-glycosylation site at N1333 and add a 3′ hexa-histidine sequence. The product was digested with EcoRI and NotI and cloned into similarly digested pPIK9K (Invitrogen). This was electroporated into P. pastoris strain GS115, and integrant clones were selected as described previously (Pizarro et al., 2005). Secreted recombinant protein was purified by nickel chelate chromatography and gel filtration and used to raise antisera in mice by standard protocols. Circular dichroism and SDS-PAGE revealed that the protein was folded.
The mAbs 3F10 (anti-HA; Roche), X509 (anti-MSP133), 1E1 (anti-PfMSP119), 61.3 (anti-PfRhopH2), and 4G2 (anti-AMA1), used unmodified or conjugated to Alexa Fluor 594 (Invitrogen), as well as a mouse antiserum specific for AMA1 have been described (Harris et al., 2005; Pizarro et al., 2005), as have rabbit and mouse polyclonal antibodies against EBA-175 (Reed et al., 2000) and EBA-181 (Gilberger et al., 2003a).
SDS-PAGE and Western blot
Extracts of parasites, COS-7 cells, medium, and partially purified or purified proteins were subjected to SDS-PAGE under reducing or nonreducing conditions and transferred to Hybond-C extra (GE Healthcare), and membranes were probed with antibodies as described previously (Harris et al., 2005; Pizarro et al., 2005). Binding was revealed by ECL detection (Pierce Chemical Co.)
Merozoite processing assays
Shedding from purified merozoites and the effects of various inhibitors on it was assayed as described previously (Howell et al., 2003; Harris et al., 2005). In brief, 3D7 merozoites suspended in 50 mM Tris-HCl and 5 mM CaCl2, pH 7.6, were divided into equal aliquots of ∼4 × 108 merozoites on ice, supplemented with test inhibitors or control buffers, and transferred to 37°C for 1 h. Merozoites were pelleted, and shed proteins were detected in supernatants by Western blot using mAb 4G2, mAb X509, or anti–region VI antibodies.
Indirect immunofluorescence microscopy
Thin films of P. falciparum were fixed with acetone or methanol and processed for IFA with mAbs as described previously (Harris et al., 2005). For surface labeling of unfixed free merozoites, purified schizonts were cultured for 2 h in medium supplemented with mAb 3F10 (1:500) to allow merozoite release. Parasites were then pelleted and washed, thin films were prepared and fixed in acetone, and labeling was continued at the secondary antibody step. For probing with polyclonal antibodies, slides were incubated for 1 h with anti–region VI antisera (1:2,000) or a mixture of rabbit anti–EBA-175 (1:1,000) and mouse anti–EBA-181 (1:1,250) and then washed and incubated for 1 h with FITC-conjugated anti-mouse IgG (1:200; Sigma-Aldrich) or a mixture of Alexa Fluor 594 goat anti–rabbit IgG antibodies (1:2,000) and Alexa Fluor 488 anti-mouse IgG antibodies (1:2,000; Invitrogen) and DAPI (1:1,000, Roche). For IFA of unfixed COS-7 cells, cells adherent to glass coverslips were transfected and cultured for 48 h and then probed without prior fixation or permeabilization with mAb 3F10 (1:500) followed by biotinylated goat anti–rat IgG (1:500; Chemicon) and FITC-conjugated streptavidin (1:500; Vector Laboratories). Samples were mounted in Citifluor (Citifluor Ltd.). Images were captured at 21°C using AxioVision 3.1 software on an imaging system (Axioplan 2; Carl Zeiss MicroImaging, Inc.) using a Plan-APOCHROMAT 100×/1.4 (or Plan-APOCHROMAT 63×/1.4 for COS-7 cells) oil-immersion objective, and annotated using Photoshop (Adobe).
Purification of shed EBA-175 and mass spectrometry
Medium harvested from cultures of 3D7 schizonts that had undergone rupture and erythrocyte reinvasion overnight was concentrated 10-fold by ultrafiltration using a 100,000-D molecular mass cutoff membrane (PBHK; Millipore). Human erythrocytes were added to a 5% haematocrit, and the suspension was mixed for 30 min at room temperature to allow binding of EBA-175. The erythrocytes were recovered and resuspended for 10 min in 2 volumes of protein-free RPMI 1640 supplemented with 1.5 M NaCl, 10 mM EDTA, and 1 mM PMSF to elute bound protein. Several batches of protein produced in this manner were pooled and fractionated on a HiLoad 26/60 Superdex 200 pg gel filtration column equilibrated in 20 mM Tris-HCl, pH 8.0, and 150 mM NaCl. Fractions containing EBA-175 were identified by Western blot. In-gel proteolytic digestion of reduced, alkylated protein and peptide analysis by MALDI-TOF and nanospray mass spectrometry was done as described previously (Howell et al., 2003, 2005; Zhou et al., 2004).
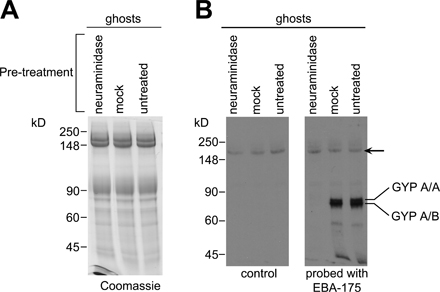
Neuraminidase treatment and EBA-175 overlay assays
Erythrocytes were neuraminidase treated as described by Thompson et al. (2001). In brief, 150 μl of packed erythrocytes were washed and resuspended in 300 μl of protein-free RPMI 1640 25 mM Hepes, pH 6.7, containing 30 mU (30 μl) Vibrio cholerae α2-3,6,8-neuraminidase (Calbiochem), and incubated at 37°C for 2.5 h. Mock-treated cells were incubated similarly without neuraminidase. The cells were washed in culture medium before use in invasion assays. EBA-175 overlay assays were performed as described by Maier et al. (2003). In brief, untreated, mock-treated, or neuraminidase-treated cells were lysed in ice-cold 5 mM sodium phosphate, pH 7.0, and the resulting ghosts were washed extensively in the same buffer before being fractionated by SDS-PAGE on 7.5% gels and transferred to Hybond C. Membranes were blocked in 5% (wt/vol) milk powder in PBS 0.5% (vol/vol) Tween 20, incubated for 1 h at room temperature with gel filtration fractions containing partially purified 3D7 EBA-175 prepared as described above, and probed sequentially with anti–region VI antibodies (diluted 1:1,000) and horseradish peroxidase–conjugated goat anti–mouse IgG (1:8,000; Chemicon). Binding was revealed by ECL.
P. falciparum transfection plasmids
Plasmid pPfROM1HA for epitope tagging of the pfrom1 gene by homologous recombination was constructed by the insertion of sequence encoding the HA3 epitope, downstream of the pfrom1 coding sequence, into plasmid pHH1 (Reed et al., 2000). The HA3 tag was amplified from pREP(HA3)42 (Craven et al., 1998) using primers HAROMtagF and -R and inserted into the EspI and XhoI sites of pHH1 to produce pHH1HA. A target fragment comprising the entire 609 bp of the pfrom1 gene and the immediate 340-bp upstream sequence was amplified from 3D7 genomic DNA (gDNA) using primers PfROM1F and -R and inserted into pHH1HA using AflII–AvrII sites within the tag. Primers were designed to remove the stop codon from the end of the pfrom1 gene and form a continuous reading frame into the tag, at the end of which was a new stop codon.
Synthetic pfrom4 and pfrom1 genes, each with an HA3 epitope tag introduced immediately after the N-terminal initiator Met residue and recodoned for mammalian expression, were gifts of M. Freeman (Medical Research Council Laboratory of Molecular Biology, Cambridge, UK). The pfrom4 gene, called HAROM4synth, was excised from the provided plasmid and blunted with Klenow. For episomal expression in P. falciparum under control of the pfrom4 promoter, the pfrom4 5′ flanking region was amplified from 3D7 gDNA using primers PfROM4upF and -R and cloned into the BglII–XhoI sites of pHH1. This vector was digested with XhoI and blunt ended, and the HAROM4synth fragment was inserted. A 1.4-kb NotI Rep20 fragment was included in the final vector, called pHAROM4synth.
The 3′ ∼1 kb of eba-175 was amplified from cDNA using primers EBA175-S3443 and EBA175-AS. Mutagenesis of this fragment was achieved in a two-step primer-directed PCR method with Vent polymerase (Stratagene). Substitution of codons encoding 1428GA1429 (GA1) and 1440GA1441 (GA2) with phenylalanine codons was achieved using primer pairs EBA175GA1/FF-S and EBA175GA1/FF-AS or EBA175GA2/FF-S and EBA175GA2/FF-AS, respectively. PCR products were sequenced to confirm the absence of undesired mutations, digested with BamHI and XhoI, and cloned into BglII–XhoI–digested pHH1 to produce constructs pHH1-175-GA1/FF and pHH1-175-GA2/FF.
Nucleic acid analysis
For Southern blot analysis, gDNA was extracted from parasites as described previously (Harris et al., 2005), restricted and electrophoresed on 0.6% agarose gel, and transferred to Hybond N+ nylon membrane (GE Healthcare) by standard procedures. Probe preparation and Southern blotting was performed using the Prime-It II random primer labeling kit (Stratagene).
Rhomboid cleavage assay
Sequence encoding HA3 was PCR amplified from pREP(HA3)42 using primers pSecHAfor and -rev. The product was digested with EcoRI and ligated into pSecTag2a (Invitrogen) that had been digested with SfiI, blunted with T4 polymerase, and further digested with EcoRI. The resulting construct was digested with EcoRI–XhoI and ligated to similarly digested eba175regVI synth to produce expression vector pSecEBAregVIgl2. Mutagenesis to remove N-glycosylation sites at N1333 and N1401 and to substitute codons for TMD residues A1427, 1428GA1429, and 1440GA1441 was performed by QuikChange mutagenesis (Stratagene) using primer pairs EBAmut13for and -rev, EBAmut12for and -rev, EBAactivefor1 and -rev1, EBATMmutfor1 and -rev1, and EBATMmutfor2 and -rev2, respectively.
Constructs based on pcDNA3.1 (Invitrogen) for transient expression of HA-tagged D. melanogaster Rho-1 in COS-7 cells (Lee et al., 2001; Urban et al., 2001) were gifts of M. Freeman. Similar constructs for expression of HA-tagged PfROM4 and -1, containing the synthetic genes described above, were also provided by M. Freeman. Mutagenesis of the PfROM4 construct to convert the predicted active site Ser residue to Ala was done by QuikChange, using primers ROM4mutfor and -rev.
Transfection of COS-7 cells in 6-well plates was done as described previously (Howell et al., 2005; Pizarro et al., 2005) using 200 ng of each construct. 24 h after transfection, the medium was replaced with 1 ml per well of serum-free medium containing 25 μM Ilomastat (Calbiochem). After a further 24 h of incubation, the medium was harvested and concentrated to a final volume of 25 μl using Vivaspin 5000 concentrators (Sartorius). Cells from each well were also harvested and resuspended in 50 μl PBS. All samples were analyzed by Western blot with mAb 3F10.
Online supplemental material
Fig. S1 shows that neuraminidase treatment of erythrocytes abolishes EBA-175 binding. Fig. S2 shows generation of a transgenic P. falciparum line expressing HA-tagged PfROM1. Fig. S3 demonstrates that PfROM1 is exclusively micronemal. Table S1 lists peptides identified by MALDI-TOF analysis of EBA-175 tryptic digests. Table S2 gives oligonucleotide primers used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200604136/DC1.
Supplementary Material
Acknowledgments
The authors are indebted to Matthew Freeman for the rhomboid expression constructs. We thank Bill Jarra for antibody production and Lee Graham for technical help.
R.A. O'Donnell was supported by a Wellcome Trust Travelling Research Fellowship. This work was supported by the Medical Research Council, by the European Commission FP6 Network of Excellence BioMalPar, and by grants from the Deutsche Forschungsgemeinschaft (GI312 and KR2105).
R.A. O'Donnell and F. Hackett contributed equally to this work.
R.A. O'Donnell's current address is Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria 3050, Australia.
Abbreviations used in this paper: AMA1, apical membrane antigen 1; DBL-EBP, Duffy binding ligand-erythrocyte binding protein; EBA-175, erythrocyte binding antigen 175; gDNA, genomic DNA; IFA, immunofluorescence assay; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; MPP1, microneme processing protease 1; MSP1, merozoite surface protein 1; TMD, transmembrane domain.
References
- Adams, J.H., P.L. Blair, O. Kaneko, and D.S. Peterson. 2001. An expanding ebl family of Plasmodium falciparum. Trends Parasitol. 17:297–299. [DOI] [PubMed] [Google Scholar]
- Blackman, M.J. 1994. Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Methods Cell Biol. 45:213–220. [DOI] [PubMed] [Google Scholar]
- Blackman, M.J., H.G. Heidrich, S. Donachie, J.S. McBride, and A.A. Holder. 1990. A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion-inhibiting antibodies. J. Exp. Med. 172:379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdech, Z., J. Zhu, M.P. Joachimiak, F.E. Cohen, B. Pulliam, and J.L. DeRisi. 2003. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol. 4:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossier, F., T.J. Jewett, J.L. Lovett, and L.D. Sibley. 2003. C-terminal processing of the Toxoplasma protein MIC2 is essential for invasion into host cells. J. Biol. Chem. 278:6229–6234. [DOI] [PubMed] [Google Scholar]
- Brossier, F., T.J. Jewett, L.D. Sibley, and S. Urban. 2005. A spatially localized rhomboid protease cleaves cell surface adhesins essential for invasion by Toxoplasma. Proc. Natl. Acad. Sci. USA. 102:4146–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers, V.B., and M.J. Blackman. 2005. A new release on life: emerging concepts in proteolysis and parasite invasion. Mol. Microbiol. 55:1617–1630. [DOI] [PubMed] [Google Scholar]
- Carruthers, V.B., G.D. Sherman, and L.D. Sibley. 2000. The Toxoplasma adhesive protein MIC2 is proteolytically processed at multiple sites by two parasite-derived proteases. J. Biol. Chem. 275:14346–14353. [DOI] [PubMed] [Google Scholar]
- Craven, R.A., D.J. Griffiths, K.S. Sheldrick, R.E. Randall, I.M. Hagan, and A.M. Carr. 1998. Vectors for the expression of tagged proteins in Schizosaccharomyces pombe. Gene. 221:59–68. [DOI] [PubMed] [Google Scholar]
- Dolan, S.A., L.H. Miller, and T.E. Wellems. 1990. Evidence for a switching mechanism in the invasion of erythrocytes by Plasmodium falciparum. J. Clin. Invest. 86:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan, S.A., J.L. Proctor, D.W. Alling, Y. Okubo, T.E. Wellems, and L.H. Miller. 1994. Glycophorin B as an EBA-175 independent Plasmodium falciparum receptor of human erythrocytes. Mol. Biochem. Parasitol. 64:55–63. [DOI] [PubMed] [Google Scholar]
- Dowse, T.J., and D. Soldati. 2005. Rhomboid-like proteins in Apicomplexa: phylogeny and nomenclature. Trends Parasitol. 21:254–258. [DOI] [PubMed] [Google Scholar]
- Dowse, T.J., J.C. Pascall, K.D. Brown, and D. Soldati. 2005. Apicomplexan rhomboids have a potential role in microneme protein cleavage during host cell invasion. Int. J. Parasitol. 35:747–756. [DOI] [PubMed] [Google Scholar]
- Duraisingh, M.T., A.G. Maier, T. Triglia, and A.F. Cowman. 2003. Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA. 100:4796–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock, D.A., and T.E. Wellems. 1997. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. USA. 94:10931–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, M. 2004. Proteolysis within the membrane: rhomboids revealed. Nat. Rev. Mol. Cell Biol. 5:188–197. [DOI] [PubMed] [Google Scholar]
- Gilberger, T.W., J.K. Thompson, M.B. Reed, R.T. Good, and A.F. Cowman. 2003. a. The cytoplasmic domain of the Plasmodium falciparum ligand EBA-175 is essential for invasion but not protein trafficking. J. Cell Biol. 162:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilberger, T.W., J.K. Thompson, T. Triglia, R.T. Good, M.T. Duraisingh, and A.F. Cowman. 2003. b. A novel erythrocyte binding antigen-175 paralogue from Plasmodium falciparum defines a new trypsin-resistant receptor on human erythrocytes. J. Biol. Chem. 278:14480–14486. [DOI] [PubMed] [Google Scholar]
- Hadley, T., M. Aikawa, and L.H. Miller. 1983. Plasmodium knowlesi: studies on invasion of rhesus erythrocytes by merozoites in the presence of protease inhibitors. Exp. Parasitol. 55:306–311. [DOI] [PubMed] [Google Scholar]
- Harris, P.K., S. Yeoh, A.R. Dluzewski, R.A. O'Donnell, C. Withers-Martinez, F. Hackett, L.H. Bannister, G.H. Mitchell, and M.J. Blackman. 2005. Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog. 1:241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, S.A., I. Well, S.L. Fleck, C. Kettleborough, C.R. Collins, and M.J. Blackman. 2003. A single malaria merozoite serine protease mediates shedding of multiple surface proteins by juxtamembrane cleavage. J. Biol. Chem. 278:23890–23898. [DOI] [PubMed] [Google Scholar]
- Howell, S.A., F. Hackett, A.M. Jongco, C. Withers-Martinez, K. Kim, V.B. Carruthers, and M.J. Blackman. 2005. Distinct mechanisms govern proteolytic shedding of a key invasion protein in apicomplexan pathogens. Mol. Microbiol. 57:1342–1356. [DOI] [PubMed] [Google Scholar]
- Kaneko, O., D.A. Fidock, O.M. Schwartz, and L.H. Miller. 2000. Disruption of the C-terminal region of EBA-175 in the Dd2/Nm clone of Plasmodium falciparum does not affect erythrocyte invasion. Mol. Biochem. Parasitol. 110:135–146. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V., K.S. Makarova, I.B. Rogozin, L. Davidovic, M.C. Letellier, and L. Pellegrini. 2003. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 4:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roch, K.G., Y. Zhou, P.L. Blair, M. Grainger, J.K. Moch, J.D. Haynes, P. De La Vega, A.A. Holder, S. Batalov, D.J. Carucci, and E.A. Winzeler. 2003. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 301:1503–1508. [DOI] [PubMed] [Google Scholar]
- Lee, J.R., S. Urban, C.F. Garvey, and M. Freeman. 2001. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell. 107:161–171. [DOI] [PubMed] [Google Scholar]
- Li, X., H. Chen, T.H. Oo, T.M. Daly, L.W. Bergman, S.C. Liu, A.H. Chishti, and S.S. Oh. 2004. A co-ligand complex anchors Plasmodium falciparum merozoites to the erythrocyte invasion receptor band 3. J. Biol. Chem. 279:5765–5771. [DOI] [PubMed] [Google Scholar]
- Lohi, O., S. Urban, and M. Freeman. 2004. Diverse substrate recognition mechanisms for rhomboids; thrombomodulin is cleaved by mammalian rhomboids. Curr. Biol. 14:236–241. [DOI] [PubMed] [Google Scholar]
- Maier, A.G., M.T. Duraisingh, J.C. Reeder, S.S. Patel, J.W. Kazura, P.A. Zimmerman, and A.F. Cowman. 2003. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat. Med. 9:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, D.C., J.B. Mu, O. Kaneko, J. Duan, X.Z. Su, and L.H. Miller. 2004. Polymorphism in the Plasmodium falciparum erythrocyte-binding ligand JESEBL/EBA-181 alters its receptor specificity. Proc. Natl. Acad. Sci. USA. 101:2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, G.H., A.W. Thomas, G. Margos, A.R. Dluzewski, and L.H. Bannister. 2004. Apical membrane antigen 1, a major malaria vaccine candidate, mediates the close attachment of invasive merozoites to host red blood cells. Infect. Immun. 72:154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoyeh, J.N., C.R. Pillai, and C.E. Chitnis. 1999. Plasmodium falciparum field isolates commonly use erythrocyte invasion pathways that are independent of sialic acid residues of glycophorin A. Infect. Immun. 67:5784–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, C., M. Di Cristina, M. Reiss, T. Ruppert, A. Crisanti, and D. Soldati. 2002. Intramembrane cleavage of microneme proteins at the surface of the apicomplexan parasite Toxoplasma gondii. EMBO J. 21:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi, P.A., B.K. Sim, J.D. Chulay, and J.D. Haynes. 1990. Characterization of the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum. Mol. Biochem. Parasitol. 40:285–294. [DOI] [PubMed] [Google Scholar]
- Pascall, J.C., and K.D. Brown. 2004. Intramembrane cleavage of ephrinB3 by the human rhomboid family protease, RHBDL2. Biochem. Biophys. Res. Commun. 317:244–252. [DOI] [PubMed] [Google Scholar]
- Pizarro, J.C., B.V. Normand, M.L. Chesne-Seck, C.R. Collins, C. Withers-Martinez, F. Hackett, M.J. Blackman, B.W. Faber, E.J. Remarque, C.H. Kocken, et al. 2005. Crystal structure of the malaria vaccine candidate apical membrane antigen 1. Science. 308:408–411. [DOI] [PubMed] [Google Scholar]
- Rayner, J.C., E. Vargas-Serrato, C.S. Huber, M.R. Galinski, and J.W. Barnwell. 2001. A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J. Exp. Med. 194:1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, M.B., S.R. Caruana, A.H. Batchelor, J.K. Thompson, B.S. Crabb, and A.F. Cowman. 2000. Targeted disruption of an erythrocyte binding antigen in Plasmodium falciparum is associated with a switch toward a sialic acid-independent pathway of invasion. Proc. Natl. Acad. Sci. USA. 97:7509–7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders, P.R., P.R. Gilson, G.T. Cantin, D.C. Greenbaum, T. Nebl, D.J. Carucci, M.J. McConville, L. Schofield, A.N. Hodder, J.R. Yates III, and B.S. Crabb. 2005. Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J. Biol. Chem. 280:40169–40176. [DOI] [PubMed] [Google Scholar]
- Sim, B.K., C.E. Chitnis, K. Wasniowska, T.J. Hadley, and L.H. Miller. 1994. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science. 264:1941–1944. [DOI] [PubMed] [Google Scholar]
- Stubbs, J., K.M. Simpson, T. Triglia, D. Plouffe, C.J. Tonkin, M.T. Duraisingh, A.G. Maier, E.A. Winzeler, and A.F. Cowman. 2005. Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science. 309:1384–1387. [DOI] [PubMed] [Google Scholar]
- Thompson, J.K., T. Triglia, M.B. Reed, and A.F. Cowman. 2001. A novel ligand from Plasmodium falciparum that binds to a sialic acid-containing receptor on the surface of human erythrocytes. Mol. Microbiol. 41:47–58. [DOI] [PubMed] [Google Scholar]
- Tolia, N.H., E.J. Enemark, B.K. Sim, and L. Joshua-Tor. 2005. Structural basis for the EBA-175 erythrocyte invasion pathway of the malaria parasite Plasmodium falciparum. Cell. 122:183–193. [DOI] [PubMed] [Google Scholar]
- Triglia, T., M.T. Duraisingh, R.T. Good, and A.F. Cowman. 2005. Reticulocyte-binding protein homologue 1 is required for sialic acid-dependent invasion into human erythrocytes by Plasmodium falciparum. Mol. Microbiol. 55:162–174. [DOI] [PubMed] [Google Scholar]
- Urban, S., and M. Freeman. 2003. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol. Cell. 11:1425–1434. [DOI] [PubMed] [Google Scholar]
- Urban, S., J.R. Lee, and M. Freeman. 2001. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 107:173–182. [DOI] [PubMed] [Google Scholar]
- Zhou, X.W., M.J. Blackman, S.A. Howell, and V.B. Carruthers. 2004. Proteomic analysis of cleavage events reveals a dynamic two-step mechanism for proteolysis of a key parasite adhesive complex. Mol. Cell. Proteomics. 3:565–576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}