

Abstract
Mutations in BLM helicase cause Bloom syndrome, characterized by predisposition to all forms of cancer. We demonstrate that BLM, signal transducer 53BP1, and RAD51 interact during stalled replication. Interactions between the three proteins have functional consequences. Lack of 53BP1 decreases the cell survival and enhanced chromosomal aberration after replication arrest. 53BP1 exhibits both BLM-dependent and -independent anti-recombinogenic functions in human and mouse cells. Both BLM and 53BP1 abrogate endogenous RAD51 foci formation and disrupt RAD51 polymerization. Consequently, loss of BLM and 53BP1 synergistically enhances stress-dependent homologous recombination. These results provide evidence regarding the cooperation between BLM and 53BP1 during maintenance of genomic integrity.
Introduction
53BP1, identified as a p53-interacting protein, is involved in DNA damage–induced checkpoint arrest. After ionizing radiation (IR) treatment, 53BP1 becomes progressively, yet transiently, immobilized around the double-strand break–flanking chromatin (Bekker-Jensen et al., 2005). 53BP1 contains a 381-amino-acid region (1235–1616), called the kinetochore binding domain, which is essential for the accumulation of IR-induced 53BP1 foci (Morales et al., 2003). Methylated lysine residues in the kinetochore binding domain modulate the accessibility of 53BP1 to the chromatin (Huyen et al., 2004; Botuyan et al., 2006). 53BP1 foci have also been detected in response to UV radiation, hydroxyurea (HU), camptothecin, and etoposide treatment (Rappold et al., 2001). We have recently shown that 53BP1 is involved in the recruitment of two important caretaker tumor suppressors, BLM and p53, to the sites of HU-induced stalled replication forks in S-phase (Sengupta et al., 2004). Incidentally, like p53 and BLM, 53BP1 also displays a “hyper-rec” phenotype (Adams et al., 2005). Hence, 53BP1 may lead to aberrant recognition of the stalled replication forks, resulting in chromosomal abnormalities, as observed in p53−/−53BP1−/− mice (Ward et al., 2005; Morales et al., 2006).
BLM helicase has been proposed to function at the interface of replication and recombination and facilitate the repair of damaged DNA (Hickson, 2003). The major characteristic of BLM patients is elevated recombination events. BLM may regulate homologous recombination (HR) by modulating the functions of other proteins involved in the process. BLM resides in a nuclear matrix bound complex with prorecombinogenic protein RAD51 (Bischof et al., 2001). The direct interaction between BLM and RAD51 is evolutionarily conserved and has been proposed to have a role during recombinational repair. The focal expression of BLM and RAD51 has an inverse correlation in human cells (Wu et al., 2001). Exposure of cells to replication stress and IR causes BLM to colocalize and physically interact with RAD51 at the sites of DNA damage (Wu et al., 2001; Sengupta et al., 2003). Inactivation of p53 in Bloom syndrome (BS) cells causes a further increase of sister chromatid exchange (SCE), thereby demonstrating that p53 and BLM cooperatively affect HR (Sengupta et al., 2003). Because BLM and 53BP1 have the potential to be functionally involved during replication stress, we sought to investigate whether and, if so, how these two proteins can affect HR by modulating the functions of RAD51.
Results and discussion
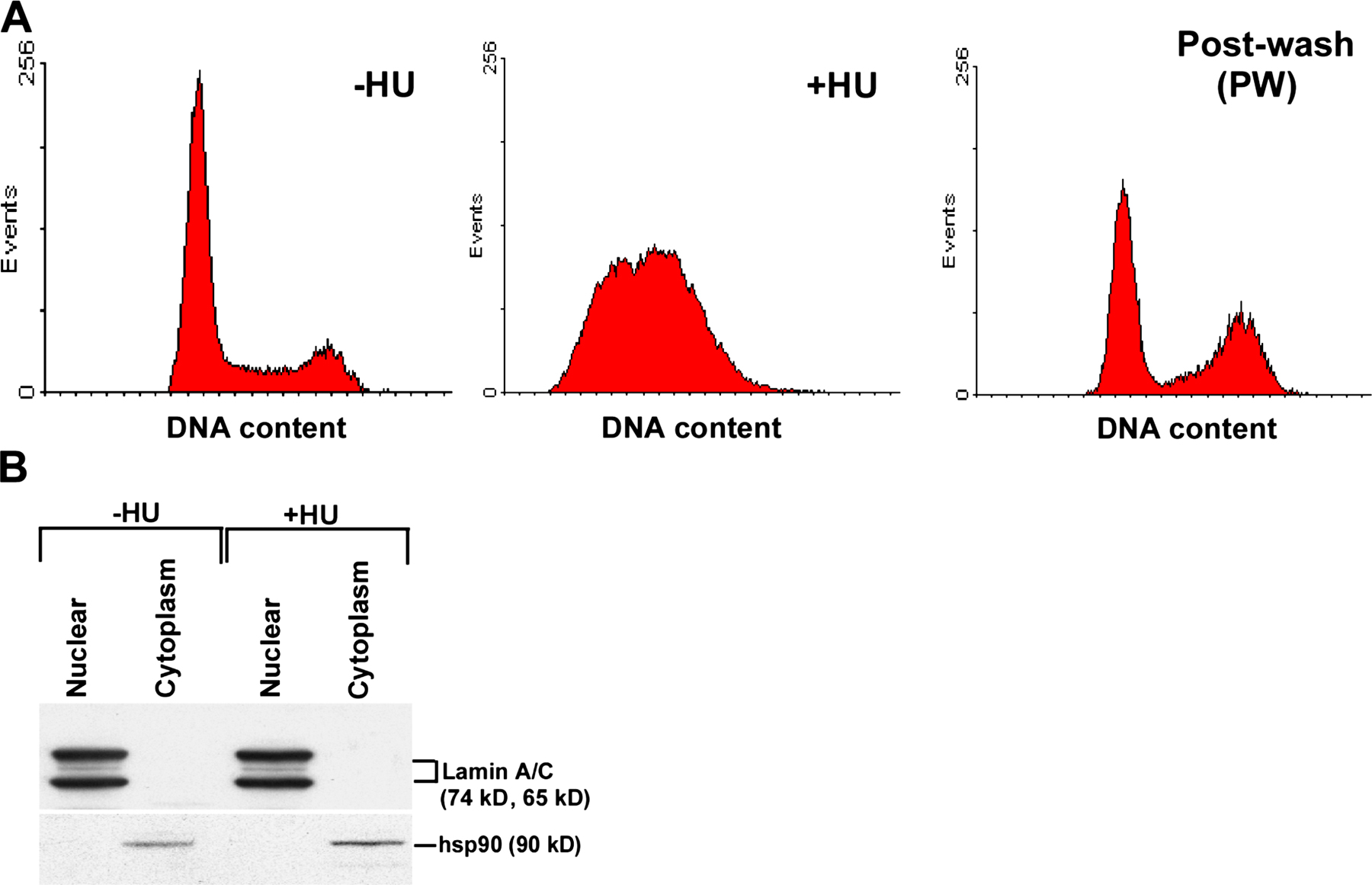
To determine whether 53BP1 interacts with BLM and RAD51 during replication stress, we performed reciprocal immunoprecipitation (IP; Fig. 1, B–D) experiments on human telomerase reverse transcriptase (hTERT)–immortalized normal human fibroblasts (NHFs), left asynchronous (−HU), arrested in S-phase by HU (+HU), or allowed to proceed after washing away HU (postwash; Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200610051/DC1). 53BP1 and RAD51 protein levels in the nuclear extract (whose integrity was verified; Fig. S1 B) remained unchanged in −HU, +HU, or postwash conditions. In contrast, BLM accumulated during +HU treatment, and the protein levels remained high during the postwash stage (Fig. 1 A). Importantly, both 53BP1 and RAD51 were present in the BLM IPs only during HU treatment (Fig. 1 B). IPs with 53BP1 (Fig. 1 C) and RAD51 (Fig. 1 D) antibodies confirmed the formation of a complex where BLM, 53BP1, and RAD51 were present. However, these results do not rule out the concurrent presence of 53BP1–RAD51 and BLM–RAD51 complexes during +HU condition. Low levels of 53BP1–RAD51 interactions also occurred during −HU and postwash conditions (Fig. 1, C and D).
Figure 1.

53BP1 interacts with BLM and RAD51 during replication stress. (A) Levels of 53BP1, BLM, and RAD51 in nuclear extracts from NHFs. Nuclear extracts were prepared from NHFs grown in the absence or presence of HU and after postwash (PW). Western blots were performed with anti-53BP1, anti-BLM, anti-RAD51, and anti-TBP antibodies. (B–D) BLM, RAD51, and 53BP1 are present in the same complex during replication stress. Reciprocal IPs with nuclear extracts were performed with anti-BLM (B), anti-53BP1 (C), and anti-RAD51 (D) antibodies and the corresponding IgGs. The IPs were probed with self-antibody or with antibodies against BLM, 53BP1, and RAD51 to check for interactions.
Because both 53BP1 and BLM interact with RAD51, there is a possibility that both of these proteins play a role in controlling HR and subsequent cell survival. To determine whether loss of 53BP1 alone could indeed cause a change in cell survival after stalling of the replication forks, embryonic stem cells from 53BP1+/+ and 53BP1−/− mice were used (Ward et al., 2003; Fig. 2 A, inset). A substantial increase in sensitivity to HU was observed in 53BP1−/− cells during clonogenic survival assays (Fig. 2 A), which may be due to increased chromosomal instability. To confirm this hypothesis, metaphase spreads were analyzed from asynchronous or HU-treated 53BP1+/+ and 53BP1−/− cells (Table I). A statistically significant increase in the number of chromosomal aberrations was observed in HU-treated 53BP1−/− cells, consistent with the possibility that 53BP1 is required in the regulation of chromosomal instability and cell survival during replication arrest.
Figure 2.

53BP1 regulates chromosomal stability and HR. (A) 53BP1−/− cells are hypersensitive to HU. Clonogenic survival analysis was performed in 5BP1+/+ (squares) and 53BP−/− (diamonds) cells. Error bars represent SEMs. (B) Absence of 53BP1 enhances SCE. SCE measurements (mean ± SD) were performed in 53BP1+/+ and BS 53BP1−/− embryonic stem cells in the absence (−HU) or presence (+HU) of replication arrest. The p-values were obtained by t test. (C) Overexpression of 53BP1 decreases HR. NHF, BS, and A-15 cells were used for experiment. pBHRF was either transfected alone or cotransfected with other expression vectors, as indicated. After transfection and before harvesting, the cells were treated with HU for 16 h. Cells were subsequently analyzed using a flow cytometer. The fold difference of the GFP/BFP ratio is represented.
Table I.
Chromosomal abnormalities in 53BP1+/+ and 53BP1−/− cells
| Abnormalitiesa
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Condition | Breaks | Gaps | Rings | Polyploidy | Fragments | Quadriradials | Total |
| +/+ | Control | 0 | 1 | 0 | 6 | 13 | 0 | 20 |
| HU | 31 | 121 | 2 | 5 | 18 | 7 | 184 | |
| −/− | Control | 0 | 9 | 0 | 14 | 9 | 0 | 32 |
| HU | 72 | 418 | 0 | 10 | 54 | 44 | 598 | |
P ≤ 0.001.
Numbers indicate the total number of abnormalities scored in 100 metaphase cells.
A role for 53BP1 in controlling genomic integrity and cell survival during S-phase may be due to its effect on HR. A role for 53BP1 in suppressing HR has been recently indicated (Adams et al., 2005). It has also been reported that although spontaneous HR by gene conversion did not depend on 53BP1, class switch recombination was severely impaired by lack of 53BP1 (Ward et al., 2004). However, the effect of 53BP1 on stress-induced HR has never been addressed. The effect of 53BP1 alone on HR was measured by SCE assays in 53BP1+/+ and 53BP1−/− cells. Loss of 53BP1 resulted in a statistically significant increase in SCE level during replication stress (Fig. 2 B), indicating that 53BP1 has an antirecombinogenic effect on HR during replication stress.
The antirecombinogenic effect of 53BP1 was subsequently verified in human cells by host cell reactivation assays in NHF, BS, and A-15 (corrected BS fibroblasts) cells. Overexpression of RAD51 in NHF cells led to the expected increase in HR (Fig. 2 C). Conversely, overexpression of wild-type 53BP1 led to a four- to fivefold decrease in the rate of HR, confirming its antirecombinogenic role in human cells. Interestingly, overexpression of wild-type 53BP1 in A-15 and BS cells cause a fourfold and twofold decrease, respectively. Hence, the interaction of BLM with 53BP1 was essential for the latter to attain its full antirecombinogenic potential.
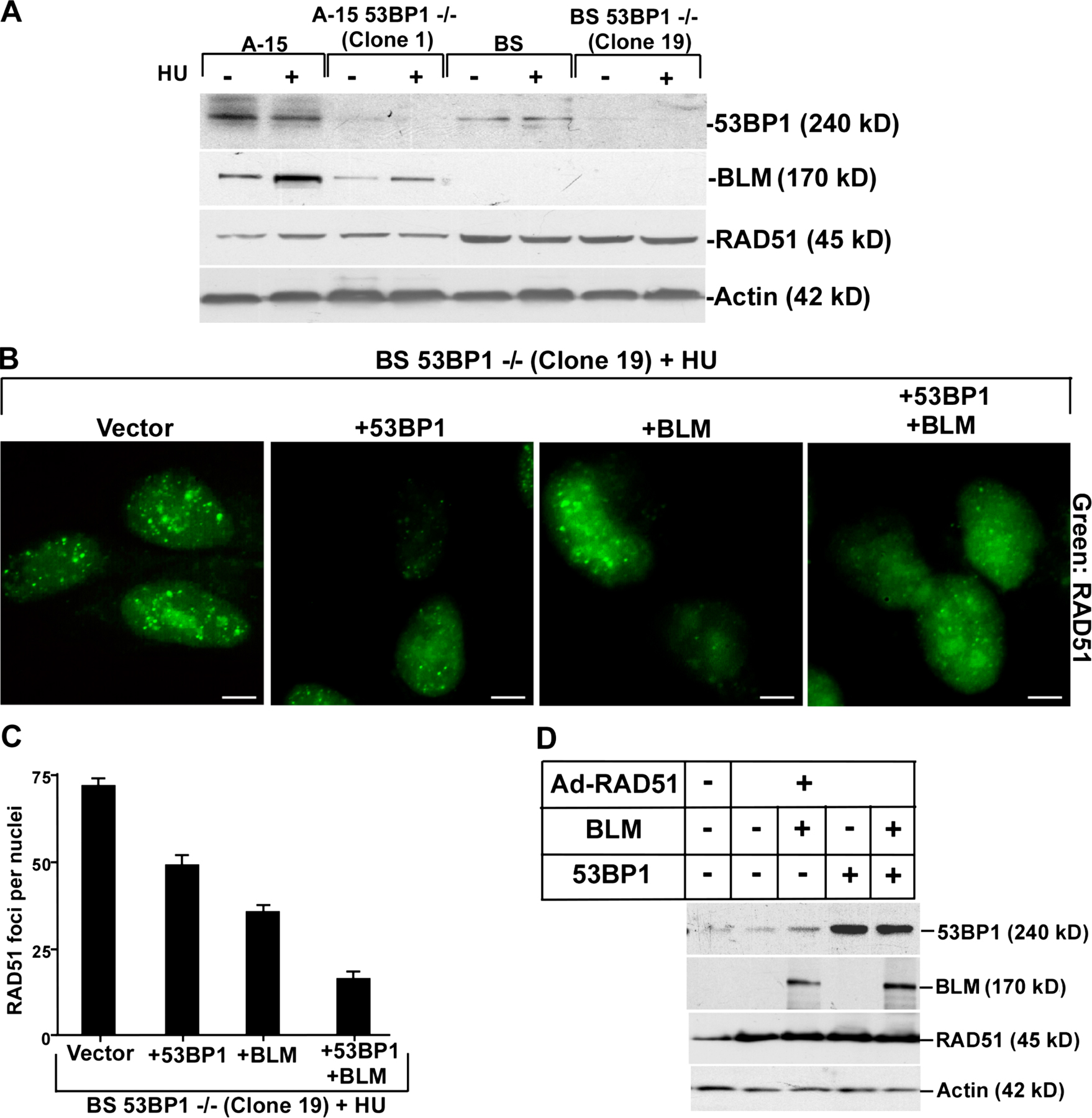
To determine whether and, if so, how BLM and 53BP1 together negatively regulate HR, we transfected BLM siRNA oligos into 53BP1+/+ and 53BP1−/− cells (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200610051/DC1) and 53BP1 siRNA oligos into BS/A-15 cells (Fig. 3 A). Western analysis revealed that both BLM and 53BP1 siRNAs were able to acutely down-regulate the expression of their cognate proteins (Fig. 3 A and Fig. S2 A). Cells with both BLM and 53BP1 expression (A-15 and 53BP1+/+) exhibited low rates of HR, which increased 2–2.5-fold after replication arrest. BS cells or loss of BLM expression in 53BP1+/+ cells led to an increase in the rate of HR. Loss of 53BP1 in either the mouse or human cells led to a HU-dependent increase in HR. In cells lacking the expression of both 53BP1 and BLM, the rate of HR increased synergistically in the presence of HU, thereby indicating that both BLM and 53BP1 together negatively regulated HR during replication arrest (Fig. 3 B and Fig. S2 B).
Figure 3.

BLM and 53BP1 modulate HR and SCE. (A) Levels of 53BP1, BLM, and TBP in A-15 and BS cells with and without siRNA treatment. Nuclear extracts were prepared from HU-treated A-15 and BS cells. The cells were also transfected with 53BP1 siRNA. Single and double plus signs indicate the first and second round of siRNA transfection, respectively. Western blots were performed with 25 μg of nuclear extracts and probed with anti-53BP1, anti-BLM, and anti-TBP antibodies. (B) Absence of 53BP1 and BLM negatively regulate HR. A-15 and BS cells were twice transfected with 53BP1 siRNA in the presence of pBHRF. Before harvesting, the cells were either left untreated or treated with HU for 16 h. Cells were subsequently analyzed using a flow cytometer. The fold difference of the GFP/BFP ratio is represented. (C) Generation of BS 53BP1−/− and A-15 53BP1−/− stable isogenic cell lines. BS and A-15 cells were transfected with pSilencer2.1-U6 hygro-53BP1. Cells stably expressing 53BP1 siRNA sequence were selected with hygromycin. Two independent clones for both BS 53BP1−/− and A-15 53BP1−/− were analyzed for 53BP1 and actin levels. (D) Loss of both BLM and 53BP1 synergistically enhance SCE. SCE measurements (mean ± SD) were performed in A-15, BS, A-15 53BP1−/− (clone 1), and BS 53BP1−/− (clone 19) fibroblasts in the absence (−HU) or presence (+HU) of replication arrest. The p-values were obtained by t test.
To further demonstrate that 53BP1 and BLM together regulate HR under physiological conditions, we needed to measure SCE in stable cell lines. 53BP1+/+ and 53BP1−/− cells could not be used, as stable lines expressing mouse BLM siRNA sequences could not be generated. Hence, the stable lines were derived from BS and A-15 cells by stable expression of human 53BP1 siRNA (Fig. 3 C). BS cells exhibited a pronounced increase in both spontaneous and stress-induced SCE compared with A-15 cells (Fig. 3 D). Loss of 53BP1 alone (A-15 53BP1−/− cells) resulted in a three- to fourfold increase in SCE compared with A-15 only during HU treatment. However, BS 53BP1−/− cells exhibit statistically significant synergistic increase in the stalled replication–induced SCE rate, indicating that 53BP1 negatively regulates HR during stalled replication both alone and in cooperation with BLM. To determine how BLM and 53BP1 regulate HR, we hypothesized that the two proteins may be controlling RAD51 protein levels or its accumulation during HR. Although there was an increase in RAD51 protein level because of loss of BLM, there was no further change as a result of the subsequent loss of 53BP1 (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200610051/DC1), indicating different modes of action of BLM and 53BP1 on RAD51 function.
RAD51 foci that are formed after stalled replication mark a subset of cells that have entered the HR pathway and contain functional recombination complexes (Raderschall et al., 1999). It is possible that BLM and/or 53BP1 affected the formation of RAD51 foci. To investigate this possibility, BLM and/or 53BP1 were transiently transfected into BS 53BP1−/− cells and the rate of formation of endogenous RAD51 foci was determined. Transfection of either BLM or 53BP1 in HU-treated BS 53BP1−/− cells resulted in a decrease in the number of RAD51 foci (Fig. S3, B and C). These results indicate that both BLM and 53BP1 may affect the formation of RAD51 nucleoprotein filaments and thereby negatively regulate HR.
Overexpression of RAD51 results in the formation of RAD51 nucleoprotein filaments reminiscent of presynaptic RAD51 complexes (Raderschall et al., 2002). Adenoviral overexpression of RAD51 in HU-treated BS 53BP1−/− cells resulted in a two- to threefold increase in the level of RAD51 protein (Fig. S3 D) and formation of high-order filamentous nuclear structures up to 5 μm in length throughout the nuclei (Fig. 4 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200610051/DC1). Around 80% of the cells infected with adenoviral RAD51 (Ad-RAD51) showed the appearance of these structures (Fig. 4, A and B). Overexpression of BLM or 53BP1 decreased the number of cells with complete RAD51 structures, instead showing either disrupted structures or diffused RAD51 staining (Fig. 4, A and B; and Videos 2 and 3). Both BLM and 53BP1 colocalized with the disrupted RAD51 structures (Fig. 4 C). Very few cells had RAD51 filaments when both 53BP1 and BLM were overexpressed (Fig. 4 B). Hence, apart from the BLM-independent role, 53BP1 also cooperates with BLM to negatively regulate HR. It will be interesting to know the biochemical parameters that govern the interactions among BLM, 53BP1, and RAD51.
Figure 4.

BLM and 53BP1 inhibit RAD51 nucleoprotein filament formation. (A) BLM and 53BP1 inhibit RAD51 polymerization. BS 53BP1−/− (clone 19) cells were transfected with vector, BLM, 53BP1, or BLM+ 53BP1 and subsequently infected with Ad-RAD51. The percentage of cells positive for RAD51 expression was ∼20–25%. Immunofluorescence was performed with anti-RAD51 antibodies, and two cells representing the majority staining pattern are represented for each type of transfection/infection. (B) Quantitation of A. (C) BLM and 53BP1 colocalize with RAD51 on nucleoprotein filaments. Experiments were done as in A. Immunofluorescence was performed with anti-RAD51–BLM (top) and anti-RAD51–53BP1 (bottom) antibodies. Bars, 5 μm.
BLM is known to exert its antirecombinogenic functions in conjunction with other cellular proteins, like topoisomerase IIIα (Wu and Hickson, 2003). Additional interacting partners of sgs1 (the yeast orthologue of BLM) have been identified in large-scale genetic screens (Ooi et al., 2003). Deletion of RAD9 (the yeast orthologue of 53BP1) alone was able to partially suppress the cell cycle arrest observed in sgs1 cells. Moreover, viability of sgs1rad9 cells was reduced compared with their RAD9 counterparts, consistent with the hypothesis that lack of sgs1 creates DNA damage that is recognized by a RAD9-dependent checkpoint (McVey et al., 2001). More recently it was observed that SGS1 is critical for suppressing spontaneous translocations between highly diverged genes in cells mutated for RAD9 (Schmidt et al., 2006). Hence, it is possible that in higher eukaryotes, BLM and 53BP1 can together maintain chromosomal integrity by modulating HR.
We hypothesize that BLM and 53BP1 may recognize different recombination substrates and thereby disrupt the RAD51 filaments by different mechanisms. The disappearance of the filaments when BLM and 53BP1 are both expressed (Fig. 4 A) lends credence to this hypothesis. In vitro experiments have unmasked a translocase activity for the yeast DNA helicase Srs2, which is involved in disrupting the RAD51 nucleofilament (Veaute et al., 2003). It is possible that a similar mechanism may exist for BLM. Our data also provides a logical explanation for the reported physical interaction of BLM with RAD51 (Wu et al., 2001). This mechanism possibly occurs parallel to the reported ability of BLM and topoisomerase IIIα to resolve recombination intermediates like double Holliday junctions into noncrossover products (Wu and Hickson, 2003). The presence of multiple mechanisms may be required for effecting exact control of RAD51 function.
Recent studies have demonstrated that signal transducer proteins can have other functions in addition to their known roles in the transmission of DNA damage signal. Like 53BP1, mediator of DNA damage checkpoint protein (MDC1) and breast cancer type 1 susceptibility gene 1 (BRCA1) are also known to participate in DNA damage sensing and signaling pathways, particularly in S-phase (Scully et al., 1997; Lou et al., 2003). Although 53BP1 has an antirecombinogenic function on HR (Adams et al., 2005; this work), MDC1 interacts with RAD51 and facilitates HR in response to IR (Zhang et al., 2005). BRCA1 decreases spontaneous HR and gene conversion and deletion events without affecting recombination intermediates, but it also promotes HR repair of chromosomal double-strand breaks and activates RAD51 function (Cousineau et al., 2005). This effect on HR by BRCT (C-terminal domain of BRCA1 protein BRCT)–containing adaptor proteins may be modulated by the presence of specific domains (like Tudor domain for 53BP1 or forkhead-associated domain of MDC1) and their interacting partners.
The observations that transducer proteins, like BLM and 53BP1, function in HR can have important implications in our understanding the pathway of DNA damage response. Because lack of BLM leads to almost all kinds of cancer in human and mouse models (Hickson, 2003), BLM is probably at the center of such a DNA damage signal transmission highway. BLM may obtain the signal upstream kinases (Davies et al., 2004; Sengupta et al., 2004) and, depending on the type and extent of damage, transmits the signal to repair pathways like HR and simultaneously synergizes with its interacting partners (like p53 and 53BP1; Sengupta et al., 2003; this work) to ensure that the extent of recombination is exact. BLM takes into account the antirecombinogenic role of 53BP1, enhancing its interaction with RAD51. Subsequently, the two antirecombinogenic proteins modulate RAD51 function, with 53BP1 probably acting as a backup for BLM. The fact that 53BP1 is required for efficient accumulation of BLM to the sites of stalled replication (Sengupta et al., 2004), and BLM in turn enhances the 53BP1–RAD51 interaction, suggests the presence of a feed-forward system involved in efficient recognition, transmission, and resolution of the DNA damage.
Materials and methods
Plasmids
pCMH6K53BP1 was a gift from K. Iwabuchi (Kanazawa Medical University, Ishikawa, Japan). pSilencer2.1-U6 hygro-53BP1 was obtained by cloning the 19-nucleotide 53BP1 siRNA sequence GAACGAGGAGACGGTAATA into the BamHI–HindIII sites of pSilencer2.1-U6 hygro (Ambion).
Antibodies
The following antibodies were used: anti-BLM goat polyclonal C- 18 (Santa Cruz Biotechnology, Inc.; Western blots and immunofluorescence), anti-BLM rabbit polyclonal (Novus; IPs), anti-53BP1 mAb (BD Biosciences; immunofluorescence), anti-53BP1 rabbit polyclonal (a gift from Y. Adachi, University of Edinburgh, Edinburgh, UK; Western blots and IPs), anti-TBP mAb l 58C9 (Santa Cruz Biotechnology, Inc.), anti-RAD51 rabbit polyclonal Ab-1 (Calbiochem), and anti-actin mAb C-2 (Santa Cruz Biotechnology, Inc.). Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories and Southern Biotechnology Associates, Inc.
Cells, culture conditions, and treatments
hTERT-immortalized BS fibroblasts, BS cells, and the corrected BS fibroblasts A-15 were a gift from J. Shay (University of Texas Southwestern Medical Center, Dallas, TX). Wild-type and knockout primary 53BP1 mouse embryonic fibroblasts were a gift from J. Chen (Yale University School of Medicine, New Haven, CT). All of the above cell lines, along with the hTERT-immortalized NHF strain GM07532 (NHF), were maintained as described previously (Sengupta et al., 2003, 2004). 1 mM HU (Sigma-Aldrich) treatments were done as described previously (Sengupta et al., 2003). Cells were either left untreated (–HU) or treated (+HU) for 16 h. Parallel HU-treated plates were washed, and incubation was continued for a further 6 h (postwash). To generate BS 53BP1−/− and A-15 53BP1−/− stable cell lines, the parental BS and A-15 cells were transfected with pSilencer2.1-U6 hygro-53BP1 plasmid. Standard procedures were followed to select stably transfected cells expressing 53BP1 siRNA with 50 μg/ml hygromycin B.
Microscopy
Immunofluorescence was performed as described previously (Sengupta et al., 2003). Cells were fixed, and the stained cells were visualized in a motorized epifluorescence microscope (Upright Axioimager M1; Carl Zeiss MicroImaging, Inc.) equipped with a high-resolution camera (AxioCam MRm Rev. 2; Carl Zeiss MicroImaging, Inc.). Images were analyzed by AxioVision Software Rel. 4.4 (Carl Zeiss MicroImaging, Inc.). The images were taken with Plan Apochromat objective 100×/1.40 oil immersion using FITC or Texas red fluorophore. At least 100 cells were analyzed for a colocalization experiment. The images were subsequently imported into Canvas graphics software, where adjustments were made for the whole image for brightness, contrast, and color balance.
Plasmid and siRNA transfections
Based on two published sequences (DiTullio et al., 2002; Kao et al., 2003), siRNAs for 53BP1 were synthesized by Dharmacon. Both the siRNA sequences were alternately used and gave the same results. Commercially available mouse BLM siRNA was purchased from Santa Cruz Biotechnology, Inc. Scrambled duplex RNA and luciferase siRNA (Dharmacon) were used as controls.
All transfections were done using Lipofectamine 2000 (Invitrogen). For HR assay, transfection was done either for HR substrate (pBHRF) alone or pBHRF combination with siRNAs. Cells transfected with pBHRF were grown for 24 h followed by 16 h of HU treatment. Transfections involving siRNA oligos were done twice. After the second round, the cells were grown for 24 h and treated with HU for 16 h. Each experiment was done in triplicate and repeated three times. Cells were subsequently treated for Western blotting or flow cytometry.
Flow cytometry and host cell reactivation assay
Cells were fixed for flow cytometry according to standard protocols. Cells were analyzed using a flow cytometer (BD LSR; BD Biosciences) for both cell cycle analysis and host cell reactivation assay. The host cell reactivation assay to determine HR rate was performed as described previously (Slebos and Taylor, 2001). Transfections were performed with the substrate (pBHRF), encoding an intact, emission-shifted, “blue” variant of GFP (BFP), with a 300-nucleotide stretch of homology to a nonfunctional copy of GFP. In the absence of HR, only BFP is present, whereas HR can also create a functional GFP.
Clonogenic assays
53BP1+/+ and 53BP1−/− cells were plated out at low dilutions and treated with different concentrations of HU, and the treatment was continued for 48 h. After removal of the drug-treated medium, the cells were washed and subsequently allowed to grow in complete growth medium undisturbed for 14 d. The colonies formed were fixed and subsequently stained with Giemsa. The whole experiment was done twice in triplicates, and the data shown are cumulative of both experiments.
SCE analysis and chromosomal aberration studies
SCE analysis for −HU cultures was performed according to standard protocols. For +HU cultures, BrdU-treated cells were cotreated with HU for 16 h during the second round of DNA duplication. The cells arrested in S-phase were subsequently washed, and incubation was continued in the presence of BrDU for a further 8 h before colcemid treatment. 25 metaphase spreads were imaged at 100× in a microscope (Axioimager M1) for each cell line. The whole experiment was performed twice, and for each cell line, SCE was scored blind. The p-values were obtained by t test. The conditions are two-tailed, unpaired data with unequal variance.
For study of chromosomal aberrations, 100 well-spread metaphase cells, stained with 4% Gurr's Giemsa solution from four independent experiments were examined at in a microscope (Axioimager M1) with 100× objective. Standard criteria were chosen for each chromosomal aberration. The total frequencies of abnormalities were compared with the control group to obtain the p-value.
In vivo RAD51 polymerization assay
BS 53BP1−/− cells were transfected with either BLM or 53BP1. 6 h after transfection, the cells were infected with replication-defective adenoviral genomes generated as described previously (Sengupta et al., 2003). 34 h after infection, the cells were treated with HU, and the incubation was continued for a further 16 h. The cells were washed and fixed for immunofluorescence. The experiments were repeated thrice, and a total of 150 cells showing RAD51 expression were counted.
Online supplemental material
Fig. S1 shows cell cycle analysis and nucleocytoplasmic separation of NHFs grown under different conditions. Fig. S2 show that 53BP1 and BLM together modulate HR in mouse cells. Fig. S3 shows that 53BP1 and BLM regulate RAD51 foci formation. Video 1 shows visualization of RAD51 filaments. Video 2 shows visualization of the disruption of RAD51 filaments as a result of BLM overexpression. Video 3 shows visualization of the disruption of RAD51 filaments as a result of 53BP1 overexpression. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200610051/DC1.
Supplementary Material
Acknowledgments
We thank Kuniyoshi Iwabuchi, Junjie Chen, Jerry Shay, and Yasuhisa Adachi for recombinants, cells, and antibodies.
Financial assistance for this work was provided by National Institute of Immunology core funds, Department of Biotechnology, India (BT/PR5936/BRB/10/408/2005), the Department of Science and Technology, India (SR/SO/HS-24/2005), and the National Institutes of Health (1 R01 TW007302-01A1).
Abbreviations used in this paper: BS, Bloom syndrome; HR, homologous recombination; hTERT, human telomerase reverse transcriptase; HU, hydroxyurea; IP, immunoprecipitation; IR, ionizing radiation; NHF, normal human fibroblast; SCE, sister chromatid exchange.
References
- Adams, M.M., B. Wang, Z. Xia, J.C. Morales, X. Lu, L.A. Donehower, D.A. Bochar, S.J. Elledge, and P.B. Carpenter. 2005. 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle. 4:1854–1861. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen, S., C. Lukas, F. Melander, J. Bartek, and J. Lukas. 2005. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 170:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof, O., S.H. Kim, J. Irving, S. Beresten, N.A. Ellis, and J. Campisi. 2001. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell Biol. 153:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan, M.V., J. Lee, I.M. Ward, J.E. Kim, J.R. Thompson, J. Chen, and G. Mer. 2006. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 127:1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousineau, I., C. Abaji, and A. Belmaaza. 2005. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 65:11384–11391. [DOI] [PubMed] [Google Scholar]
- Davies, S.L., P.S. North, A. Dart, N.D. Lakin, and I.D. Hickson. 2004. Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S-phase arrest. Mol. Cell. Biol. 24:1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTullio, R.A., Jr., T.A. Mochan, M. Venere, J. Bartkova, M. Sehested, J. Bartek, and T.D. Halazonetis. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 4:998–1002. [DOI] [PubMed] [Google Scholar]
- Hickson, I.D. 2003. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 3:169–178. [DOI] [PubMed] [Google Scholar]
- Huyen, Y., O. Zgheib, R.A. Ditullio Jr., V.G. Gorgoulis, P. Zacharatos, T.J. Petty, E.A. Sheston, H.S. Mellert, E.S. Stavridi, and T.D. Halazonetis. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 432:406–411. [DOI] [PubMed] [Google Scholar]
- Kao, G.D., W.G. McKenna, M.G. Guenther, R.J. Muschel, M.A. Lazar, and T.J. Yen. 2003. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 160:1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, Z., K. Minter-Dykhouse, X. Wu, and J. Chen. 2003. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 421:957–961. [DOI] [PubMed] [Google Scholar]
- McVey, M., M. Kaeberlein, H.A. Tissenbaum, and L. Guarente. 2001. The short life span of Saccharomyces cerevisiae sgs1 and srs2 mutants is a composite of normal aging processes and mitotic arrest due to defective recombination. Genetics. 157:1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, J.C., Z. Xia, T. Lu, M.B. Aldrich, B. Wang, C. Rosales, R.E. Kellems, W.N. Hittelman, S.J. Elledge, and P.B. Carpenter. 2003. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J. Biol. Chem. 278:14971–14977. [DOI] [PubMed] [Google Scholar]
- Morales, J.C., S. Franco, M.M. Murphy, C.H. Bassing, K.D. Mills, M.M. Adams, N.C. Walsh, J.P. Manis, G.Z. Rassidakis, F.W. Alt, and P.B. Carpenter. 2006. 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc. Natl. Acad. Sci. USA. 103:3310–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi, S.L., D.D. Shoemaker, and J.D. Boeke. 2003. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 35:277–286. [DOI] [PubMed] [Google Scholar]
- Raderschall, E., E.I. Golub, and T. Haaf. 1999. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. USA. 96:1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall, E., A. Bazarov, J. Cao, R. Lurz, A. Smith, W. Mann, H.H. Ropers, J.M. Sedivy, E.I. Golub, E. Fritz, and T. Haaf. 2002. Formation of higher-order nuclear Rad51 structures is functionally linked to p21 expression and protection from DNA damage-induced apoptosis. J. Cell Sci. 115:153–164. [DOI] [PubMed] [Google Scholar]
- Rappold, I., K. Iwabuchi, T. Date, and J. Chen. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 153:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, K.H., J. Wu, and R.D. Kolodner. 2006. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol. Cell. Biol. 26:5406–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully, R., J. Chen, R.L. Ochs, K. Keegan, M. Hoekstra, J. Feunteun, and D.M. Livingston. 1997. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 90:425–435. [DOI] [PubMed] [Google Scholar]
- Sengupta, S., S.P. Linke, R. Pedeux, Q. Yang, J. Farnsworth, S.H. Garfield, K. Valerie, J.W. Shay, N.A. Ellis, B. Wasylyk, and C.C. Harris. 2003. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J. 22:1210–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta, S., A.I. Robles, S.P. Linke, N.I. Sinogeeva, R. Zhang, R. Pedeux, I.M. Ward, A. Celeste, A. Nussenzweig, J. Chen, et al. 2004. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J. Cell Biol. 166:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebos, R.J., and J.A. Taylor. 2001. A novel host cell reactivation assay to assess homologous recombination capacity in human cancer cell lines. Biochem. Biophys. Res. Commun. 281:212–219. [DOI] [PubMed] [Google Scholar]
- Veaute, X., J. Jeusset, C. Soustelle, S.C. Kowalczykowski, E. Le Cam, and F. Fabre. 2003. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 423:309–312. [DOI] [PubMed] [Google Scholar]
- Ward, I.M., K. Minn, J. van Deursen, and J. Chen. 2003. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 23:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, I.M., B. Reina-San-Martin, A. Olaru, K. Minn, K. Tamada, J.S. Lau, M. Cascalho, L. Chen, A. Nussenzweig, F. Livak, et al. 2004. 53BP1 is required for class switch recombination. J. Cell Biol. 165:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, I.M., S. Difilippantonio, K. Minn, M.D. Mueller, J.R. Molina, X. Yu, C.S. Frisk, T. Ried, A. Nussenzweig, and J. Chen. 2005. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol. Cell. Biol. 25:10079–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, L., and I.D. Hickson. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 426:870–874. [DOI] [PubMed] [Google Scholar]
- Wu, L., S.L. Davies, N.C. Levitt, and I.D. Hickson. 2001. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 276:19375–19381. [DOI] [PubMed] [Google Scholar]
- Zhang, J., Z. Ma, A. Treszezamsky, and S.N. Powell. 2005. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat. Struct. Mol. Biol. 12:902–909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}