

Abstract
Fibronectin (FN) is secreted as a disulfide-bonded FN dimer. Each subunit contains three types of repeating modules: FN-I, FN-II, and FN-III. The interactions of α5β1 or αv integrins with the RGD motif of FN-III repeat 10 (FN-III10) are considered an essential step in the assembly of FN fibrils. To test this hypothesis in vivo, we replaced the RGD motif with the inactive RGE in mice. FN-RGE homozygous embryos die at embryonic day 10 with shortened posterior trunk, absent tail bud–derived somites, and severe vascular defects resembling the phenotype of α5 integrin–deficient mice. Surprisingly, the absence of a functional RGD motif in FN did not compromise assembly of an FN matrix in mutant embryos or on mutant cells. Matrix assembly assays and solid-phase binding assays reveal that αvβ3 integrin assembles FN-RGE by binding an isoDGR motif in FN-I5, which is generated by the nonenzymatic rearrangement of asparagines (N) into an iso-aspartate (iso-D). Our findings demonstrate that FN contains a novel motif for integrin binding and fibril formation whose activity is controlled by amino acid modification.
Introduction
Fibronectin (FN) is a ubiquitous and abundant ECM protein, which is secreted as a disulfide-bonded dimer consisting primarily of three types of repeating modules (I, II, and III; Hynes, 1992; Kornblihtt et al., 1996). Distinct FN modules bind cell surface receptors such as integrins and syndecans (Plow et al., 2000) and ECM proteins such as collagens, fibrin, and FN itself (Pankov and Yamada, 2002). FN plays an essential role during development, in physiology (tissue repair), and in disease (cancer progression and invasion, inflammation, atherosclerosis, etc.). The functions of FN are critically dependent on the assembly of the secreted FN dimers into a fibrillar network. FN assembly is a cell-driven process that occurs in a stepwise manner: the dimeric FN protein is secreted in a compact or inactive conformation; integrin binding converts FN into an active and extended dimer; the dimeric state of FN induces integrin clustering and accumulation of integrin-bound FN, which in turn allows FN–FN interactions at different sites along the FN molecule and the formation of fibrils (Wierzbicka-Patynowski and Schwarzbauer, 2003).
The activation of FN is most efficiently induced by α5β1 integrin binding to the RGD motif in the type III-10 module along with the synergy sequence located in the adjacent type III-9 module (Pierschbacher and Ruoslahti, 1984; McDonald et al., 1987; Nagai et al., 1991; Sechler et al., 1997). Additional RGD binding integrins, including αvβ3 and αIIbβ3, can substitute for α5β1 in FN matrix assembly but are less able to form a dense and delicate fibrillar network in vitro (Wu et al., 1995, 1996; Wennerberg et al., 1996). Furthermore, it has also been reported that FN can be assembled in an RGD-independent manner through binding of α4β1 integrin to the connecting segment-1 (CS1) in the alternatively spliced V region near the C terminus of FN (Sechler et al., 2000). It is not clear, however, whether the CS1-dependent FN assembly occurs and is of significance in vivo.
Gene ablation studies in mice confirmed that the two RGD-dependent FN assembly mechanisms induced by either α5β1 integrin or αv integrins exist in vivo and that each of them can compensate for the absence of the other. Mice lacking the α5 integrin gene, as well as mice lacking the αv integrin gene, exhibit normal assembly of FN matrix (Yang et al., 1993; Bader et al., 1998). In contrast, mice carrying deletions of both the α5 and the αv integrin genes display severely compromised FN matrix formation (Yang et al., 1999).
Antibody and peptide inhibition studies in amphibians and chicks (Darribere et al., 1988; Harrisson et al., 1993), chemical mutagenesis in zebrafish (Julich et al., 2005; Koshida et al., 2005), and gene targeting in mice (Bouvard et al., 2001) identified a crucial role for the FN matrix in the development of mesoderm and mesoderm-derived structures. The mesoderm is one of the three germ layers that forms during gastrulation and is essential for morphogenesis and organ development. FN-deficient mice commence gastrulation and die at embryonic day (E) 8–8.5 with several mesodermal defects, including shortened anterior–posterior axis, disorganized notochord, absence of heart and somites, and abnormal vasculogenesis (George et al., 1993). Mice lacking the expression of both the α5 and the αv integrins (and hence all FN-RGD binding integrins) also display arrested development at the beginning of gastrulation (Yang et al., 1999). α5/αv double-null mice are arrested at an earlier stage than the FN-deficient mice. The α5/αv double mutants completely lack the mesodermal germ layer and contain only a few mesodermal cells in the developing head fold region and allantois, indicating that the two integrins must perform functions in addition to FN matrix assembly. Interestingly, mice lacking only the α5 integrin die ∼E9.5–10, and despite their ability to develop a normal FN matrix, they also have mesodermal defects. These are restricted to the posterior region of the embryo and include an arrest in posterior axis elongation, the absence of posterior somites, and vascular defects (Yang et al., 1993). These observations indicate that the α5β1–FN interaction conveys essential signals in the posterior trunk mesoderm that are not compensated by αv integrins or other FN binding proteins. It is also possible, however, that the α5β1 integrin requires interaction with ECM proteins other than FN to extend the posterior trunk and develop posterior somites. In contrast, mice without αv integrin display no mesodermal abnormalities and die either of placenta defects or perinatally as a result of severe intracerebral hemorrhage, indicating that the role of the αv integrins for mesoderm formation is fully compensated by α5β1 (Bader et al., 1998).
To test how FN binding receptors compensate for each other during development and FN fibril formation, it would be necessary to generate double, triple, or even higher order combinations of integrin and syndecan knockout mice. However, the necessary intercross combinations cannot be performed because most integrin-null mice are recessive lethal (α4, α5, α8, and αv). Moreover, integrins and syndecans interact with multiple ECM proteins, and ECM proteins interact with multiple integrins, which would further complicate the interpretation of the phenotypes.
Therefore, we decided to specifically test the role of FN-RGD binding integrins in vivo by generating mice in which the aspartic acid (D) residue of the RGD motif was replaced with a glutamate (E). Many previous studies have shown that this replacement completely inactivates this site for cell adhesion. We found that the FNRGE/RGE mice died early in embryogenesis from multiple defects, but surprisingly they assembled an apparently normal FN matrix. Furthermore, our studies revealed a novel integrin binding site in the fifth N-terminal type I module of FN, which was essential for the assembly of the FN-RGE matrix.
Results
Generation of FNRGE/RGE knockin mice
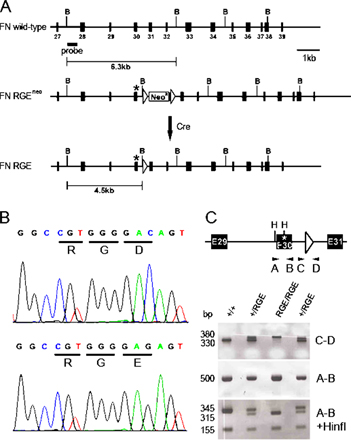
To test the role of the RGD motif in FN-III10 in vivo, we cloned part of the mouse FN gene, changed the GAC codon in exon 30 to GAG by site-directed mutagenesis, and inserted a loxP flanked neo cassette into intron 30 (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1). The C>G nucleotide change resulted in a replacement of the aspartate (D) residue of the RGD motif with a glutamate (E) and created a novel HinfI restriction site in the mutant FN gene (Fig. S1, A and B). The DNA vector was used to transfer the mutation via embryonic stem cells into the germline of mice. The neo cassette was removed by breeding to a deleter-Cre strain. Mice were genotyped by PCR analysis of tail or yolk sac biopsies, and the knockin mutation was confirmed by sequencing of the PCR amplicon (Fig. S1, B and C). Mice heterozygous for the mutant FN allele (FNRGE/+) were normal.
Heterozygous mice were intercrossed to obtain homozygosity (FNRGE/RGE). Out of 157 viable offspring, we obtained 65% heterozygous and 35% wild type but no homozygous mutant mice (Table I). This indicates that the RGE mutation in FN represents a recessive embryonic lethal trait.
Table I.
Genotypes of progeny from heterozygous intercrosses
| +/+
|
+/RGE
|
RGE/RGE
|
|||||
|---|---|---|---|---|---|---|---|
| Stage | Total | n | % | n | % | n | % |
| E7.5 | 16 | 5 | 31.2 | 8 | 50.0 | 3 | 18.8 |
| E8.5 | 170 | 46 | 27.1 | 81 | 47.6 | 43 | 25.3 |
| E9.5 | 95 | 26 | 27.3 | 46 | 48.4 | 23 | 24.2 |
| E10.5 | 46 | 12 | 26.1 | 21 | 45.7 | 13 | 28.3 |
| E11.5 | 26 | 11 | 42.3 | 15 | 57.7 | 3a | 11.5 |
| E12.5 | 15 | 4 | 26.6 | 11 | 73.3 | 0 | 0.0 |
| E13.5 | 4 | 2 | 50.0 | 2 | 50.0 | 0 | 0.0 |
| P21 | 157 | 55 | 35.0 | 102 | 65.0 | 0 | 0.0 |
Embryos without a beating heart.
To determine the exact time point of lethality, we dissected embryos derived from heterozygous intercrosses and determined their genotypes by genomic PCR using DNA extracted from either the yolk sac or embryos. We found that genotypes showed a normal Mendelian distribution until E10.5, whereas beyond E10.5, no homozygous mutant embryos with beating hearts were recovered (Table I). At ∼E11.5, FNRGE/RGE embryos started to disintegrate or were already partially resorbed.
FNRGE/RGE embryos display multiple abnormalities
To search for defects and identify the cause for the lethality, we isolated whole-mount embryos from heterozygous intercrosses at different time points of development. At E8.5, the FNRGE/RGE embryos had slightly shorter anterior–posterior axes but were otherwise indistinguishable from their wild-type or heterozygous littermates (Fig. 1, A and B). Both the FNRGE/RGE embryos and wild-type littermates contained three to four somite pairs and had a well-developed head fold, neural tube, and beating heart. At E9.5, wild-type embryos were turned into the fetal position and their posterior trunks extended into a C-like shape (Fig. 1 C). In contrast, FNRGE/RGE embryos were only partially turned and displayed a kinked neural tube and severely shortened posterior trunk without somites (Fig. 1 D). E9.5 FNRGE/RGE embryos contained never more than 12 or 13 somite pairs, whereas wild-type littermates had ∼21 somite pairs (Fig. 1, C and D). The neural tubes were noticeably kinked, the limb buds were absent, and the embryonic and yolk sac vasculature was frequently dilated and sometimes ruptured, leading to blood cell leakage into the amniotic cavity (unpublished data). At E10.5, the head structures, heart, and fore limb buds of FNRGE/RGE embryos were further developed, whereas the number of somites had not increased further (Fig. 1, E and F).
Figure 1.

Whole-mount pictures of wild-type and FNRGE/RGE embryos. (A and B) Side view of an E8.5 wild-type embryo with four pairs of somites (A) and an FNRGE/RGE embryo (B) with four pairs of somites and a well-developed allantois (al). The FNRGE/RGE embryo is slightly smaller. (C and D) Side view of an E9.5 wild-type (C) and FNRGE/RGE (D) embryo. The wild-type embryo has turned into the fetal position and contains 21 pairs of somites (C). The E9.5 FNRGE/RGE embryo shows incomplete turning, a kinked neural tube, and a truncation of the posterior trunk region with 13 pairs of somites in the anterior region (D). Inset shows anterior somites at higher magnifications. The head region of the embryo is normally developed. (E and F) Side view of an E10.5 wild-type embryo with 29 somite pairs (E) and an E10.5 FNRGE/RGE embryo with 13 somite pairs (F). The first branchial arch and fore limb buds (fl) are developed, whereas the second branchial arch is missing (F). (G and H) Apoptotic cell death in FNRGE/RGE embryos. TUNEL and cleaved form of caspase-3 staining reveal massive cell death in posterior trunk regions of E9.5 FNRGE/RGE embryo in comparison with very few positive cells in the wild-type embryo. Note the colocalization (yellow) between TUNEL (green) and activated cleaved form of caspase-3 (red) staining. h, heart; s, somites; nt, neural tube. Bars: (B, D, and F) 500 μm; (J) 100 μm.
To test whether proliferation or survival was affected by the FN-RGE mutation, we analyzed Ki67, TUNEL, and activated caspase-3 signals. Proliferation as tested by Ki67 staining was similar in control and FNRGE/RGE embryos (unpublished data). In sharp contrast, apoptotic cell death of tail bud–derived mesodermal cells was dramatically elevated, as shown in a double staining for TUNEL (Fig. 1, G and H) and activated caspase-3 signals (Fig. 1, I and J).
FN-RGE is normally distributed and assembled in vivo
It has become generally accepted that the RGD motif in FN is essential for FN matrix assembly (Nagai et al., 1991; Schwarzbauer, 1991; Sechler et al., 1997). Consequently, we expected mutation of the RGD motif to abolish fibril assembly and hamper fibril stability or function. To evaluate FN-RGE matrix assembly, we performed immunostaining and Western blot experiments. Immunostaining of parasagittal, paraffin-embedded sections from PFA-fixed E9.5 embryos revealed that FN-RGE was widely expressed. Closer inspection of different areas within the FNRGE/RGE embryos revealed strong FN-RGE matrix surrounding the nervous tissue, somites, and large vessels and a fine, network-like distribution in the heart, developing facial, and trunk mesenchyme, within the somites and the tail bud mesoderm (Fig. 2 A). Neither the distribution nor the intensity of the FN immunosignal differed between wild-type and FNRGE/RGE littermates (Fig. 2 A). Western blot assays of whole embryo lysates confirmed the normal expression levels of FN-RGE in FNRGE/RGE embryos (Fig. 2 B).
Figure 2.

FN-RGE fibrillar network in FNRGE/RGE embryos. (A) Serial sagittal paraffin sections of an E9.5 wild-type and FNRGE/RGE embryo stained with hematoxylin-eosin (H&E; top) and anti-FN polyclonal antibody (bottom). FN is expressed strongly in heart, basement membranes around the neural tube, and the somites, and is expressed to a lesser extent in the mesoderm of both embryos. FNRGE/RGE embryos exhibit a clearly shorter anterior–posterior axis. Bars, 500 μm. (B) Western blot analysis of E8.5 whole embryo lysates shows similar FN protein levels in wild-type and FNRGE/RGE littermates. (C) Native cryosections of E9.0 embryos were washed with TBS without fixation followed by staining for FN and α4 integrin. Note that fibril assembly is similar in wild-type and FNRGE/RGE littermates. Bar, 15 μm. (D) High-magnification images of the heart ventricle showing normal fibril thickness and ramifications in wild-type and FNRGE/RGE embryo. ep, epicardium; ps, proepicardial serosa. Bar, 100 μm.
The apparently normal FN-RGE fibrils in FNRGE/RGE embryos was unexpected and could be due to a cross-linking artifact induced by the PFA fixation or, alternatively, to α4β1 integrin–dependent assembly. To test whether either was the case, we immunostained nonfixed cryosections derived from E9.5 embryos for FN and α4 integrin expression. The FN staining revealed a similar distribution and amount of FN fibrils in control and FNRGE/RGE embryos (Fig. 2, C and D). The α4 integrin signals were scarce and only observed in the cranial region (likely on migratory cranial neural crest cells) and in the epicardium and the proepicardial serosa of control and FNRGE/RGE sections (Fig. 2 C and not depicted). Importantly, FN signals did not colocalize with α4 integrin–positive cells in the cranial region or in the heart (Fig. 2 C and not depicted). Furthermore, FACS analysis of single-cell suspensions derived from collagenase-treated control and FNRGE/RGE E9.5 embryos identified a small α4 integrin–positive cell population of indistinguishable size and comparable α4 integrin levels (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1), indicating that expression of α4 integrin is not altered in FNRGE/RGE mice.
FNRGE/RGE cells assemble FN-RGE in an αv integrin–dependent manner
To examine the mechanism underlying the assembly of FN-RGE, we immortalized fibroblast-like cells from wild-type and FNRGE/RGE embryos and established several clonal cell lines with comparable adhesion properties to FN, collagen I, laminin-111 (LM111), and vitronectin (VN; unpublished data). All clones chosen for the subsequent assays expressed similar levels of β1, β3, αv, and α5 integrins on their cell surface (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1). Importantly, to rule out α4 integrin–dependent FN fibril assembly, we used only clones that lacked α4 expression (Fig. S3 A). When labeled wild-type plasma FN (pFN) was added to the culture, FNRGE/RGE cell clones assembled it into a normal fibril network, with α5 integrin colocalizing into typical fibrillar adhesions (Fig. S3 B).
Next, we tested whether FNRGE/RGE cells are able to assemble their endogenously produced mutant FN-RGE. Assembly assays with self-produced FN critically depend on the absence of exogenous serum-derived FN, which is a widely used supplement of cell culture media. To avoid serum-derived FN, we grew control and FNRGE/RGE cells in serum replacement medium and, because they lacked the serum-derived ECM components, seeded them on defined substrates.
Control and FNRGE/RGE cells plated on LM111 adhered via their laminin binding α6β1 integrin, leaving the α5β1 integrin, as well as the αv integrins, free to contribute to the assembly of the secreted FN. In contrast to the previous notion that the RGD sequence of FN is essential for the initiation of matrix assembly, FNRGE/RGE cells initiated and completed the assembly of FN-RGE into fibrils on their cell surface (Fig. 3 A). Similar to mouse embryos, FNRGE/RGE cells were able to assemble a dense fibrillar network (Fig. 3 A). Although the kinetic of fibril formation was similar between control and FNRGE/RGE cells, the size and the distribution of fibrils differed between the two cell types. Control cells produced an elaborate network consisting of thin and long FN fibrils (Fig. 3 A). In contrast, FN fibrils formed by FNRGE/RGE cells appeared short and thick.
Figure 3.

FN-RGE fibril assembly on LM111 and VN. (A and B) Wild-type (+/+) and FNRGE/RGE (RGE/RGE) cells grown in serum replacement medium and either on LM111 (A) or on VN (B) for 48, 72, and 96 h were fixed and stained with FN antibodies. Note that FNRGE/RGE cells form fibrils on LM111 (A) but not on VN (B). Bar, 15 μm.
To determine which integrin, αv and/or α5, mediates assembly of FN-RGE, we plated cells on VN. On VN, control and FNRGE/RGE cells adhered via their αv integrins. A previous study showed that this depletes αvβ3 integrin from the cell surface but leaves α5β1 integrin diffusely distributed and presumably free to participate in matrix assembly (Fath et al., 1989). Control cells developed a regular FN network when plated on VN, whereas FNRGE/RGE cells were unable to form FN-RGE fibrils (Fig. 3 B). These data suggest that αv, but not α5β1, integrins mediate the assembly of FN-RGE into matrix fibrils.
αv integrins can trigger an RGD-independent FN assembly pathway
To further investigate whether αv and not α5β1 integrins executed FN-RGE fibril formation, we immunostained FNRGE/RGE cells growing on LM111 with α5 integrin and FN antibodies. As expected, control cells showed coalignment of the α5 integrin with the thin and elaborate FN network (Fig. 4 A). In sharp contrast, LM111-growing FNRGE/RGE cells failed to align α5 integrin with FN-RGE–containing fibrils. Instead, the α5 integrin signal was diffusely distributed over the entire cell surface, further indicating that it is not engaged in FN-RGE binding (Fig. 4 A). Unfortunately, because of the poor quality of the αv integrin antibodies that were available to us, we could not directly demonstrate a colocalization of αv integrins with FN-RGE fibrils.
Figure 4.

Distribution of α5 integrin and siRNA-mediated depletion of αv integrin in FNRGE/RGE cells. (A) Distribution of α5 and FN fibrils on wild-type (+/+) and FNRGE/RGE (RGE/RGE) cells cultured for 24 h on LM111-coated dishes in serum replacement medium. Note that α5 and FN colocalize in fibrillar adhesions (arrowheads) of wild-type cells, whereas α5 is diffusely distributed in FNRGE/RGE cells. (B) Knockdown of αv integrin in FNRGE/RGE cells by transduction with retrovirus carrying anti–αv siRNA (αv siRNA) or control siRNA (scr.-siRNA) expression cassettes. αv-depleted FNRGE/RGE cells were cultured for 48, 72, and 96 h on glass chamber slides coated with LM111 and stained for FN-RGE. Bars, 15 μm.
To confirm that αv integrins were indeed assembling FN-RGE fibrils, we depleted αv integrins in FNRGE/RGE cells by viral transduction of αv-specific siRNAs and assayed for FN assembly. FNRGE/RGE cells expressed high levels of αv integrins (Fig. S3 A and Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1), which were reduced by ∼80% after retroviral expression of siRNAs (Fig. S4). The αv integrin–depleted FNRGE/RGE cells were cultured in serum replacement medium and seeded on LM111. Despite the availability of free α5β1 integrins, the αv-depleted FNRGE/RGE cells were unable to form FN-RGE fibrils (Fig. 4 B). Occasionally, we could observe a few thick fibrils after longer culture periods, which likely developed as a result of the remaining low levels of αv integrins on FNRGE/RGE cells (Fig. 4 B).
To address the question of whether assembly of FN-RGE is accomplished via the RGD binding pocket of αv integrins, we tested FN-RGE fibrillogenesis in the presence of linear Gly-Arg-Gly-Asp-Ser-Pro peptides (linRGD) or cyclic cyclo (-Arg-Gly-Asp-D-Phe-Val-) peptides (cycRGD). The more constrained cycRGD was reported to preferentially bind αvβ3 integrin (Pfaff et al., 1994). Incubation of control and FNRGE/RGE cells with the linRGD peptide considerably inhibited assembly of wild-type as well as FN-RGE fibrils (Fig. 5). Treatment with the cycRGD peptide inhibited assembly of FN-RGE much more strongly but showed only partial inhibition of assembly of wild-type FN (Fig. 5). Treatment of control and FNRGE/RGE cells with peptides, in which the RGD binding motif is mutated (linear Gly-Arg-Gly-Glu-Ser-Pro peptide [linRGE] or cyclic cyclo[-Arg-Ala-Asp-D-Phe-Val-] peptides [cycRAD]), had no effect on assembly of wild-type FN or FN-RGE (Fig. 5). Similarly, heparin treatment, which blocks the binding of FN to cell surface proteoglycans such as syndecans, partially inhibited assembly of wild-type FN but had almost no effect on FN-RGE fibrils. These data demonstrate that the RGD binding pocket of αv integrins is essential for the assembly of the FN-RGE matrix.
Figure 5.

Effect of heparin, linRGD, or cycRGD peptides on FN-RGE assembly. Wild-type and FNRGE/RGE cells were cultured on LM111-coated dishes in serum replacement medium for 2 h; incubated with heparin, linRGD, linRGE, cycRGD, or cycRAD for an additional 14 h; and stained with an anti-FN antibody. Bars, 15 μm.
The FN-I1-9 domains bind αvβ3 integrin with high affinity
The data so far indicate that αv integrins bind and assemble FN-RGE into fibrils via a novel integrin binding site on FN. To localize the novel αv binding/assembly sites, we cultured FNRGE/RGE cells on LM111 and treated them with recombinant FN fragments spanning almost the entire FN molecule (Fig. 6). Although FN-I1-9, FN-III7-10, and FN-III10-12 strongly inhibited the formation of both wild-type and FN-RGE fibrils, FN-III1-5, FN-III4-7, FN-III7-10RGE, and FN-III13-15 had no effect on either (Fig. 6 and Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1). FN-I1-9 is generally recognized as a potent inhibitor of matrix assembly and is thought to participate in binding to receptors on the cell surface or to specific FN-III domains of another FN molecule (McKeown-Longo and Mosher, 1984; Aguirre et al., 1994; Hocking et al., 1994; Bultmann et al., 1998). Because assembly of the FN-RGE matrix depended on αvβ3 integrin, we wondered if this integrin might bind directly to FN-I1-9.
Figure 6.

Effect of recFN fragments on FN assembly. (A) Schematic depiction of the modular domain composition of FN. Type I domains are depicted as orange rectangles, type II domains as gray ovals, and type III domains as circles. The integrin binding domains III9 and III10 have been highlighted by pink (synergy site) and red (RGD motif), respectively. The variable region is depicted as a yellow rectangle. Extra domains A and B are shown in green. (B) Wild-type and FNRGE/RGE cells were cultured on LM111-coated dishes in serum replacement medium for 2 h, incubated with recombinant FN fragments (right) for a further 14 h, and stained with an anti-FN antibody. Bar, 15 μm.
We therefore examined whether αvβ3 integrin is capable of binding to the recombinant FN-I1-9 fragment. To this end, we performed direct and competitive solid-phase binding assays, in which the recombinant FN fragments were adsorbed to microtiter plates and then incubated with serially diluted biotinylated αvβ3 integrin. Direct binding assays showed comparable binding of αvβ3 integrins to FN-I1-9, FN-III7-10, and FN-III10-12 but no binding to the other fragments tested (Fig. 7 A and Fig. S5). Interestingly, α5β1 was also able to bind FN-I1-9, although α5β1 was incapable of assembling FN-RGE fibrils (Fig. 7 C). Competitive binding assays showed that incubation with the linRGD peptide inhibited binding of both αvβ3 and α5β1 to FN-I1-9 (Fig. 7, B and C). These results indicate that (1) FN-I1-9 contains novel and functional αvβ3 binding/assembly sites, (2) FN-I1-9 contains a binding site for α5β1 that is not functional for matrix assembly, (3) the binding sites on both integrins involve their RGD binding pocket, and (4) the binding affinity of FN-I1-9 to αvβ3 integrin is of somewhat lower affinity than the RGD binding motif of wild-type FN.
Figure 7.

Direct solid-phase FN and integrin binding assay. (A) Titration curve of αvβ3 integrin bound to different recombinant FN fragments. After blocking of nonspecific binding sites, αvβ3 was allowed to bind in TBS buffer containing 1 mM MnCl2. Mean ± SD (n = 3). Note that αvβ3 integrin bound with high affinity to FN-I1-9. (B) FN-I1-9 was immobilized at 0.25 μM. After blocking of nonspecific binding sites, αvβ3 was allowed to bind in TBS buffer containing 1 mM MnCl2 (diamonds) plus 100 μg/ml heparin (triangles), 750 μM isoDGR-2C (squares), or 850 μM linRGD peptide (circles). The values plotted are normalized to the maximal binding value to give the yield of saturation. Maximal binding of αvβ3 to FN-I1-9 corresponds to an OD (405 nm) value of 1.08. Note that αvβ3 binding to FN-I1-9 is inhibited by both linRGD and isoDGR-2C peptides. (C) The same experimental procedure was performed as described in B, with the exception that α5β1 integrin was used for titration. Maximal binding of α5β1 to FN-I1-9 corresponds to an OD (405 nm) value of 1.04. Note that α5β1 integrin binding to FN-I1-9 is inhibited by linRGD peptides but not by isoDGR-2C peptides. In all binding assays, the absorbance measured in BSA-coated wells was <15% relative to maximum absorbance values and was routinely subtracted. All binding was determined in duplicate.
The GNGRG motif in FN-I5 represents a novel αvβ3 binding and assembly site for FN
Primary and tertiary structure analyses showed that murine FN contains several GNGRG loops within the FN-I1-9 that are well conserved in human, bovine, rat, amphibian, and fish (Di Matteo et al., 2006). While we were preparing this manuscript, Curnis et al. (2006) reported that a single GNGRG loop in FN-I5 can convert to GisoDGRG (where isoD is isoasparte) and that the GisoDGRG provided a novel binding site for αvβ3 integrin. To explore this observation, we synthesized the (*Cys-Asn-Gly-Arg-Cys*) peptide (NGR-2C) as well as the (*Cys-isoAsp-Gly-Arg-Cys*) peptide (isoDGR-2C), which adopts a conformation similar to the deamidated, rearranged GNGRG loop of FN-I5 (Di Matteo et al., 2006) and used them in our competitive solid-phase binding and FN in vitro assembly assays. As shown in Fig. 7, isoDGR-2C strongly inhibited αvβ3, but not α5β1, binding to FN-I1-9, indicating that αvβ3 but not α5β1 can recognize the isoDGR motif. Incubation of FNRGE/RGE cells grown on LM111 with the NGR-2C or the isoDGR-2C peptides effectively inhibited assembly of the FN-RGE matrix, whereas assembly of wild-type FN by control cells was unaffected (Fig. 8 A).
Figure 8.

Effect of NGR-2C and isoDGR-2C on FN-RGE assembly. (A) Wild- type and FNRGE/RGE cells were cultured on LM111-coated dishes in serum replacement medium for 2 h, incubated with 800 μM NGR-2C or isoDGR-2C for 14 h, and stained with anti-FN antibodies. Note that both NGR-2C and isoDGR inhibit assembly of FN-RGE but not of wild-type FN. Bar, 15 μm. (B) Kinetics of NGR deamidation and rearrangement to isoDGR and DGR, respectively. NGR-2C peptide was incubated at 37°C and pH 7.2. Samples were taken the indicated times. Amounts of NGR and the rearrangement products isoDGR and DGR were determined with the HPLC as percentage peak areas. Results were normalized against NGR input. Note that the concentration of isoDGR is steadily increasing with time, reaching 10% of the NGR input after 16 h.
Curnis et al. (2006) have shown that the GNGRG, as well as the GDGRG, motif exhibits a lower affinity to αvβ3 integrins compared with the GisoDGRG motif. The rearrangement of GNGRG to GisoDGRG and GDGRG is a well-known reaction occurring spontaneously under physiological conditions (Robinson et al., 2004). Because NGR-2C inhibited FN-RGE assembly equally as well as isoDGR-2C (Fig. 8 A), we tested the kinetics of the rearrangement of the NGR-2C into isoDGR-2C. HPLC assays revealed that ∼10% of NGR-2C was converted into isoDGR-2C after an incubation of 16 h at 37°C at pH 7.2 (Fig. 8 B). The experiment also shows that before incubation at 37°C, the peptide was nearly 100% NGR-2C. Therefore, when testing for inhibition of FN matrix assembly, only a small amount of isoDGR-2C would have accumulated in the later time of the 14-h assay. The inhibition observed is likely due to unmodified NGR-2C interacting with αvβ3. Altogether, these data indicate that the GNGRG motif in FN-I5 represents a novel and selective αvβ3 binding that can function for FN fibril assembly in vitro and in vivo.
Discussion
The RGD binding motif in FN is thought to play a central role in FN matrix assembly and RGD-mediated integrin signaling in vivo. In the present study, we assessed these roles by creating a nonfunctional RGE site in the FN gene of mice. The phenotype of FNRGE/RGE mice is identical in type and quality to the defects reported for α5β1 integrin–deficient mice (Yang et al., 1993) but much less severe than the phenotype of FN-null or αv/α5 double-null mice (Yang et al., 1999). This finding allows us to draw several important conclusions. First, the mesodermal defects caused by absent α5β1 integrin expression are solely based on the disruption of α5β1 binding to the RGD motif of FN and do not involve binding to other α5β1 ligands (collagen XVIII, ADAMs, etc). Second, the development of a less severe phenotype in FNRGE/RGE mice than in α5/αv double-null mice indicates that αv integrin–FN interactions either are not required for early development or occur in an RGD-independent manner. Third, the direct observation of FN matrix in FNRGE/RGE mice shows that the RGD motif in FN must be dispensable for the formation of a functional fibrillar FN network.
FN-RGE can assemble into a functional fibrillar network
A large number of integrins can bind FN (Pankov and Yamada, 2002). Although FN fibrillogenesis can still proceed in mice carrying single gene deletions of FN binding integrins, double deficiencies for α5 and αv dramatically reduce the amount of FN matrix, suggesting that the two integrins bind, activate, and assemble FN fibrils independently of each other (Yang et al., 1999). Based on this mouse work and previous FN assembly studies performed in vitro, one would have predicted that a nonfunctional RGD motif in FN would abrogate binding to both α5β1 and αv integrins and hence render fibrillogenesis equally impossible as in α5/αv double-null mice. However, this was not the case. We found neither quantitative nor qualitative differences in the FN network of FNRGE/RGE mice, irrespective of whether the embryo sections were treated with fixative or examined in native cryosections.
One reason for this unexpected finding could have been that the D>E mutation was too conservative and hence would still permit binding to integrins. Such a possibility, however, was ruled out experimentally: the assembly of FN-RGE by FNRGE/RGE cells grown on LM111 was inhibited by the addition of neither linear RGE peptides nor recombinant FN-III7-10 fragment, in which the RGD was converted into an RGE. Moreover, the same recombinant FN-III7-10RGE fragment was unable to bind integrins in a highly sensitive solid-phase binding assay.
FN-RGE is assembled by αv integrins
FN assembly via the RGD motif can occur through either α5β1 or αv integrins. The αv integrin–mediated assembly, however, leads to a less dense network with shorter and thicker fibrils in vitro (Wennerberg et al., 1996), which is thought to be due to the different subcellular localization and associated shape of αv versus α5β1 integrin clusters (α5β1 form thin fibrillar adhesions and αv integrins arrowhead-like focal adhesions) and/or to the differential activation of small GTPases such as RhoA, which occurs through FN binding to α5β1 but not to αv integrins (Danen et al., 2002).
The FN-RGE fibrils of FNRGE/RGE cells displayed appearance and behavior typical of αv-mediated fibrils: they were short and thick, whereas fibrils of wild-type cells were thin, long, and ramified. Furthermore, the assembly of FN-RGE could be efficiently inhibited by cycRGD, which was shown to bind with higher affinity to αvβ3 than to α5β1 integrins (Pfaff et al., 1994). The interpretation that αv integrins mediate FN-RGE assembly was further supported by siRNA-mediated depletion of the αv integrin protein in FNRGE/RGE cells, which showed that αv integrin–depleted cells were unable to form a fibrillar FN-RGE network.
The role of αv integrins was further confirmed by direct binding assays, with recombinant fragments spanning almost the entire FN protein, which revealed a novel high-affinity binding interaction between αvβ3 integrin and the N-terminal FN-I1-9 fragment. Moreover, the assembly of FN-RGE fibrils by FNRGE/RGE cells was efficiently inhibited by FN-I1-9 and by synthetic peptides comprising the novel αvβ3 integrin binding site.
RGD-independent mechanisms for FN assembly have been observed with different cell culture systems. One report showed that binding of Mn2+-activated α4β1 integrin to the CS1 site can induce assembly of FN in vitro (Sechler et al., 2000). It is not clear, however, whether the α4β1-mediated mechanism also operates in vivo and, if so, whether its role is substantial during embryogenesis. In the E9.5 control and FNRGE/RGE embryos, we observed a very restricted expression of α4β1 on a few cells in the cranial region and the developing epicard. Thus, the abundant FN-RGE fibrils in sites without α4β1 expressing cells must have assembled using an α4β1- independent mechanism. This conclusion was further confirmed with FNRGE/RGE cell lines that lacked α4 integrin but still efficiently assembled FN-RGE fibrils.
A second RGD-independent FN assembly mechanism has been described for cells isolated from α5 integrin–deficient mice (Sottile et al., 2000). Incubation of FN-null cells growing in serum replacement medium and on VN with recombinant FN lacking the RGD motif (FNΔRGD) assembled short stitch-like aggregates on the cell surface. A potential explanation for the development of dot-like FNΔRGD aggregates could be that a few αvβ3 integrins failed to bind the VN substrate and were available to recruit FNΔRGD to the cell surface by binding to the site in FN-I1-9. Alternatively, the recruitment and aggregation of FNΔRGD may have been accomplished by syndecans, which also bind FN. Potential syndecan–FN interaction would also explain why heparin treatment inhibited the formation of the stitch-like aggregates on FN-null cells. We also tested a potential involvement of such interactions with FN-RGE by treating FNRGE/RGE cells with heparin. Interestingly, heparin had no considerable effect on the assembly of FN-RGE fibrils, indicating that in our system, proteoglycan binding to FN-RGE is not essential for fibril development.
The GNGRG motif in FN-I5 is a novel αvβ3 binding site that can function for FN matrix assembly
The FN-I1-9 fragment (also called 70-kD N-terminal fragment) inhibits FN assembly with efficiency similar to RGD peptides. It is generally believed that the inhibitory effect of FN-I1-9 is caused by high-affinity binding sites for FN, which block the FN–FN interactions required to align and cross-link FNs into fibrils (McKeown-Longo and Mosher, 1985; McDonald et al., 1987; Schwarzbauer, 1991; Aguirre et al., 1994; Hocking et al., 1994; Sechler and Schwarzbauer, 1998). The fact that we could inhibit FN-RGE assembly with FN-I1-9 pointed to the possibility that FN-I1-9 may, in addition to its FN binding activity, contain an αv integrin binding site. We confirmed this in direct binding assays and found that FN-I1-9 bound αvβ3. Curnis et al. (2006) reported that the Asn-Gly-Arg (NGR) sequence of FN-I5 could be converted to a high-affinity binding site for αvβ3 integrin through the deamidation and rearrangement of the asparagine residue and the creation of an isoDGR sequence. Isoaspartate formation is a well-known, nonenzymatic process that can occur during aging of ECM proteins (David et al., 1998; Lanthier and Desrosiers, 2004) and during isolation and storage of proteins (Paranandi et al., 1994). The properties of the novel αvβ3 integrin binding sites in FN agreed with our direct binding assay, which showed that FN-I1-9 bound αvβ3 and offered a potential explanation for the αv integrin–dependent and FN-I1-9–sensitive assembly of FN-RGE. Indeed, we found that an isoDGR peptide efficiently inhibited the binding of αvβ3 to FN-I1-9 in our direct binding assay, and it blocked the assembly of FN-RGE by FNRGE/RGE cells.
We found that assembly of the FN-RGE matrix was inhibited equally by peptides NGR-2C and isoDGR-2C. This suggests that the native NGR sequence itself may already be able to bind αvβ3 and function for matrix assembly of FN-RGE. It is more likely, however, that the NGR motif was sufficiently modified in culture to inhibit FN-RGE assembly. Supporting this hypothesis is the fact that NGR peptides failed to block αvβ3 binding to the FN-I5 module (Curnis et al., 2006). Moreover, assembly of the FN-RGE matrix in both embryos and cell culture requires only a small fraction of isoDGR-modified FN molecules to bind cell surface integrins, whereas the remaining fraction becomes integrated into fibrils via FN–FN interactions, which proceeds independent of the isoDGR modification.
Interestingly, α5β1 also bound the FN-I1-9 peptide, but the isoDGR-2C peptide failed to inhibit this binding or assembly of wild-type FN. A likely explanation is that the interaction of α5β1 with FN-I1-9 is weaker than the interaction of αvβ3 with the isoDGR motif in FN-I5 and fails to provide the necessary binding strength to allow assembly of FN-RGE fibrils. Conversely, our findings also indicate that FN- I1-9 inhibits FN assembly by affecting not only FN–FN but also FN–αvβ3 interactions, which has profound consequences for assembly of FN-RGE. Collectively, these result revealed that the GNGRG loop in FN-I5 represents a novel functional αvβ3 binding and FN fibril assembly site. Future experiments will be necessary to test whether additional motifs in FN-I1-9 can also provide functional integrin binding and FN assembly sites (Shpiro et al., 2005).
The integrity of the RGD motif of FN is essential for development
The FNRGE/RGE mice survive substantially longer than FN-null mice, indicating that FN-RGE fibrils are biologically active. FNRGE/RGE embryos display defects that are strikingly similar to those observed in α5-null embryos (Yang et al., 1993), suggesting that high-affinity interactions of α5β1 with FN triggers unique functions during early development that cannot be compensated by αv integrin–RGD, αv integrin–NGR, or α5β1–FNI1-9 interactions. In both mouse mutants, the posterior trunk is shortened, underdeveloped, and lacks somites. TUNEL assays and immunostaining for activated caspase-3 revealed diminished survival of posterior trunk mesoderm in FNRGE/RGE mice, suggesting that RGD-triggered α5β1 integrin signals inhibit caspase-3 activation and anoikis.
A consistent feature of all FNRGE/RGE embryos analyzed between E9.5 and E10.5 was that their somite number was limited to 12–13 somites. A plausible explanation for this finding could be that somites 12–13 represent the transition from the gastrulation- to tail bud–derived mesoderm (Wilson and Beddington, 1996). This would indicate that the integrin-triggered survival signals are mainly required for the posterior trunk- and tail bud–derived mesoderm. Such an α5β1 integrin independency of the anterior, streak-derived mesoderm could be explained by expression and compensation of αv integrins. Unfortunately, we were unable to test whether αv is high in the anterior and low or absent in the posterior mesoderm because all anti-αv antibodies that were available to us produced only background staining.
Zebrafish also require α5 integrin for somite development along the body axis. In contrast to the mouse, α5 integrin mutant zebrafish display anterior somite defects, suggesting that α5 is indispensable for anterior somitogenesis, whereas the posterior somites developed normally (Julich et al., 2005; Koshida et al., 2005). Interestingly, knockdown of fibronectin1 in α5 integrin mutant zebrafish, as well as knockdown of fibronectin1 and fibronectin3, led to a more severe phenotype, suggesting that the restriction of defects in zebrafish to the anterior somites is probably due to functional redundancy (Julich et al., 2005; Koshida et al., 2005). A further difference from our findings is the absence of apoptosis in the anterior somites of α5 integrin–deficient zebrafish. It was even shown that the anterior somites develop perfectly well, but their boundaries cannot be maintained because of the loss of FN fibrils that would normally surround somite boundaries. In about half of our FNRGE/RGE embryos, we observed normal amounts of FN-RGE fibrils at anterior somite boundaries, whereas in the remaining half, the levels were reduced (unpublished data).
In conclusion, we found that FN-RGE can be assembled into fibrils in vivo and in vitro via αvβ3 binding to the isoDGR motif in FN-I5. The isoDGR site in FN-I5 is generated by NGR deamidation (Curnis et al., 2006) and, as previously demonstrated, can readily be reverted by the enzymatic action of the protein l-isoaspartyl methyltransferase (Reissner and Aswad, 2003; Lanthier and Desrosiers, 2004). The ability of tissues to swiftly activate and deactivate binding of αvβ3 and likely other αv integrins to FN may represent a novel strategy to spatially and temporally trigger FN-mediated αv integrin signaling during development and disease.
Materials and methods
Generation of knockin mice
A 129/Sv mouse PAC (P1-derived artificial chromosome) clone was used to construct the FN-RGE targeting vector (Fig. S1 A). A 3.3-kb EcoRV–StuI fragment containing exons 28–30 was subcloned into Bluescript (Stratagene). A 0.8-kb SacI–StuI fragment containing exon 30 was subcloned and used for site-directed mutagenesis. The nucleotide C1238 of the GAC codon encoding the aspartic acid residue of the Arg-Gly-Asp (RGD) motif in exon 30 was converted with the QuikChange Mutagenesis kit (Stratagene) into a G, resulting in a GAG codon encoding glutamic acid. The C>G mutation gave rise to a novel HinfI site, which was used to verify the presence of the mutation by PCR and HinfI digest (Fig. S1 B). The mutated 0.8-kb SacI–StuI fragment was sequenced (Fig. S1 C) and swapped back into the 3.3-kb EcoRV–StuI fragment. A BamHI site was introduced into intron 30 of the 3.3-kb EcoRV–StuI fragment and used to insert a loxP-flanked neomycin cassette. The 5.8-kb StuI–ApaI fragment was inserted downstream of the neomycin cassette. The targeting vector was linearized with NotI and electroporated into R1 embryonic stem cells (passage 13). Approximately 700 G418-resistant clones were isolated and screened by Southern blot for homologous recombination. The genomic DNA was digested with BamHI and probed with an external probe (Fig. S1 A). Two correctly targeted clones were identified and injected into C57B6 host blastocysts (carried out by M. Boesl, Max Planck Institute of Biochemistry, Martinsried, Germany) to generate germline chimeras.
FNRGEneo/+ mice were mated with a deleter-Cre strain to eliminate the loxP-flanked neomycin cassette. Heterozygous FNRGE/+ mice were intercrossed to obtain homozygous FNRGE/RGE mutants. The following primers were used to amplify the RGE mutation: pRGEGenof (5′-CAAAGAAGACCCCAAGAGCA-3′) and pRGEGenor (5-ACAAGCCCTGGCCTTTAGTT-3′). The following primers were used to amplify FN mRNA by RT-PCR from total RNA extracted from embryonic tissue: pmRNAf3 (5′-TATCACCGCCAACTCATTCA-3′) and pmRNAr3 (5′-GGGAGTGGTGGTCACTCTGT-3′). The PRC amplicon was then digested with HinfI to obtain two bands of 155 and 315 bp.
Histology and immunofluorescence
To test FN expression, E9.5 embryos were dissected and either immediately embedded in optimal cutting temperature compound (OCT; Thermo Savant) to obtain unfixed sections or fixed in 4% PFA and embedded in paraffin (Paraplast X-tra; Sigma-Aldrich) to obtain paraffin sections. Cryosections of 10-μm thickness and paraffin sections of 6-μm thickness were cut and used for histochemistry and immunofluorescence as described previously (Fässler and Meyer, 1995). TUNEL staining was performed according to the manufacturer's instructions (Roche). Antibodies against the following proteins were used: anti-FN (Chemicon), anti–α4 integrin (BioLegend), and anti–cleaved caspase-3 (New England Biolabs, Inc.). Cy3- or FITC-conjugated secondary antibodies were purchased from Sigma-Aldrich or Jackson ImmunoResearch Laboratories.
Cells were seeded on glass coverslips (Lab-Tek Chamber Slides; Nunc) precoated with 5 μg/ml VN (Sigma-Aldrich) or 10 μg/ml LM111 (a gift from R. Deutzmann, Regensburg University, Regensburg, Germany) for 2 h at 37°C, incubated at 37°C for 16–96 h, fixed with 4% PFA, and incubated with primary and secondary antibodies, respectively. In some cases, cells were permeabilized with 0.5% Triton X-100 after PFA fixation.
The following antibodies were used: anti-FN (Chemicon); anti-α5 (BD Biosciences); and Cy2-, Cy3-, and Cy5-conjugated secondary antibodies were used for detection (Jackson ImmunoResearch Laboratories). Nuclei were stained with DAPI (Roche). Fluorescent images were taken at room temperature with a microscope (DMRA2; Leica) equipped with NPLAN 40/0.65 NA objective and a camera (DMRA2; Leica), or with a confocal microscope (DMIRE2; Leica) equipped with a 100× 1.4 NA oil objective, using Leica Confocal Software (version 2.5, Build 1227) for data acquisition. Images were collected as TIF files and analyzed using Photoshop (version 9.0; Adobe).
Western blotting
E8.5 embryos were solubilized in RIPA buffer, briefly sonicated, and boiled for 2 min in nonreducing sample buffer. The samples were separated on 6% SDS polyacrylamide gels, blotted onto Immobilon-P polyvinylidene difluoride membranes (Millipore), and hybridized with polyclonal antibodies specific for FN (Chemicon) and tubulin (Sigma-Aldrich). Bound antibodies were hybridized with horseradish peroxidase–conjugated secondary antibodies and detected using the ECL kit (GE Healthcare).
Generation of FNRGE/RGE cell lines
To establish cell lines, FNRGE/+ mice were intercrossed and embryos harvested at E9. To release cells, embryos were trypsinized for 5 min at 37°C and disrupted by pipetting up and down. Cells were cultured in DME supplemented with 10% FBS. Cells were genotyped by PCR, immortalized by retroviral transduction of the SV-40 large T antigen, and cloned. Control (FN+/+) and FNRGE/RGE cell lines were adapted to grow in serum replacement medium (47.5:47.5:5:1 ratio of DME/Aim-V Medium [Invitrogen]/RPM1640/nonessential amino acids) and then used for FN assembly.
Cell adhesion assay
Cell adhesion assays were performed as described previously (Czuchra et al., 2006).
Flow cytometry
Flow cytometry was performed as described by Czuchra et al. (2006). The following antibodies were used: anti–β1, –β3, –α2, –α4, –α5, –α6, and –αv integrins (all obtained from BD Biosciences), which were FITC conjugated (anti–β1, –α2, and –α6 integrins), biotinylated (anti–β3, –α5, and –αv integrins), or PE conjugated (anti–α4 integrin). Biotinylated antibodies were detected with streptavidin-Cy5 (Jackson ImmunoResearch Laboratories). To assess autofluorescence and nonspecific staining, cells were stained with IgM isotype-FITC (β1 and α2), IgG2a isotype-FITC (α6), IgG2a isotype-PE (for α4 integrin), or streptavidin-Cy5 (β3, α5, and αv; all obtained from BD Biosciences).
Generation of αv integrin–depleted cell lines
The sequence of the siRNA constructs was chosen according to the protocol of Ui-Tei et al. (2004) and Naito et al. (2004). Full-length mRNA sequences of the target gene were checked using the online software siDirect for 19-bp stem-loop structure. The sequence obtained was synthesized as a 5′-terminally phosphorylated primer with a 5′-HindIII and 3′-BglII recognition site (5′-GATCCCCCGTTAGGGCAATTAGGATTTTCAAGAGAAATCCTAATTGCCCTAACGTTTTTA-3′). Subsequently, primers were annealed and inserted into HindIII–BamHI linearized pSuper.retro.puro (OligoEngine). The primer 5′-GATCCCCAGCAGTGCATGTATGCTTCTTCAAGAGAGAAGCATACATGCACTGTTTTTTA-3′ was used as a scrambled control. Stable expression of the siRNAs in FNRGE/RGE cell was achieved by retroviral gene transfer (Pfeifer et al., 2000). In brief, siRNA inserts were cloned into a retroviral vector (pclMFG). After harvesting, the recombinant retrovirus FNRGE/RGE cells were infected, selected in 6 μg/ml puromycin, and cloned.
Expression of FN fragments
The human FN-III7-10 fragment was subcloned into pET15b plasmid (Invitrogen) and mutated into the pET15bFN-III7-10RGE and pET15bFN-III7-10δRGD by using the QuikChange II XL Site-Directed Mutagenesis kit. The following primers were used to introduce the mutation: RGD>RGE forward, 5′-CACTGGCCGTGGAGAGAGCCCCGCAAGCAG-3′; RGD>RGE reverse, 5′-CTGCTTGCGGGGCTCTCTCCACGGCCAGTGA-3′; RGD>δRGD forward, 5′-GTGTATGCTGTCACTGGCAGCCCCGCAAGCAGCAAGC-3′; and RGD>δRGD reverse, 5′-GCTTGCTGCTTGCGGGGCTGCCAGTGACAGCATACAC-3′. The plasmids were transformed into BL21 (DE3)pLysS Escherichia coli cells. Protein expression and purification of these and other fragments were performed as described previously (Ohashi and Erickson, 2005).
Inhibition assay
All experiments were performed in serum replacement medium. Control and FNRGE/RGE cells were seeded on 10 μg/ml LM111–coated 8-well Lab-Tek Chamber Slides (80,000 cells/well), allowed to spread for 2 h, and incubated with inhibitory peptides dissolved in PBS for another 14 h at 37°C and 5% CO2. The assay was stopped by fixation with 4% PFA in PBS and immunostained.
The final concentrations of the used peptides were as follows: linRGD and linRGE, 0.5 mg/ml (900 μM; BIOMOL Research Laboratories, Inc.); cycRGD and cycRAD, 0.25 mg/ml (410 μM; BIOMOL Research Laboratories, Inc.); isoDGR-2C and NGR-2C, 800 μM (for synthesis, see Peptide synthesis); FN-I1-9, 2.5 μM; FN-III7-10, FN-III7-10RGE, and FN-III7-10ΔRGD, 10 μM; and FN-III1-5, FN-III4-7, FN-III10-12, and FN-III13-15, 25 μM. The FN polypeptide synthesis was previously described (Ohashi and Erickson, 2005). Heparin was used at a concentration of 0.1 mg/ml.
Direct solid-phase binding assay using purified recombinant integrins and recombinant FN fragments
Direct solid-phase binding assay for determining the relative binding of biotinylated αvβ3 integrins to various FN fragments was performed as described previously (Eble et al., 1998). In brief, 0.25 μM solutions of recombinant FN fragments in TBS were used to coat 96-well polyvinylchloride microtiter plates (Maxisorp; Nunc) by an overnight incubation at 4°C. Coating with 0.25 μM BSA was used to determine the background values of unspecific binding. After a 1-h blocking step (1% BSA in TBS), serially diluted integrins were added to the plates and allowed to bind the absorbed FN fragments for 5 h at room temperature in the presence of 1 mM MnCl2. The plates were washed with TBS containing 1 mM MnCl2, and the bound integrins were quantified by the addition of a 1:1,000 dilution of streptavidin peroxidase conjugate (Vector Laboratories) in TBS/1mM MnCl2 to the plate for 15 min. After several washing steps with TBS/1mM MnCl2, bound peroxidase was detected by a chromogenic reaction at 405 nm (ABTS; Vector Laboratories).
Preparation of recombinant, soluble integrins
Soluble recombinant αvβ3 and α5β1 integrins were purified from the culture supernatants from CHO-lec 3.2.8.1 cells stably transfected with corresponding subunits (Takagi et al., 2002). They were then biotinylated with Sulfo-LC-NHS-biotin (Pierce Chemical Co.) according to the manufacturer's recommendation.
Peptide synthesis
The linear peptides Cys-Asn-Gly-Arg-Cys (NGR-2C) and Cys-isoAsp-Gly-Arg-Cys (isoDGR-2C) were synthesized according to standard solid-phase peptide synthesis on TCP resin using the Fmoc strategy. Fmoc-Asp-OtBu was used as isoAsp building block. Cys was used trityl protected, and Arg was used Pbf protected. To avoid positive charge at the N terminus and to facilitate HPLC purification by enhancing the UV absorption, the terminal amine was benzoylated with 3 eq. benzoyl chloride and 5 eq. diisopropyl amine in N-methylpyrrolidinone for 20 min. The linear peptide was cleaved and simultaneously deprotected using a mixture of 95% trifluoroacetic acid, 2.5% water, and 2.5% triisopropyl silane for 1 h at ambient temperature and precipitated in diethyl ether. Cys-Cys cyclization was performed by treatment of the highly diluted (10−3–10−4 mol/L) solution in water/ acetonitrile with 2 eq. H2O2 at pH 8–8.5 (NaHCO3). The reaction mixture was concentrated, and the isoDGR-2C peptide was purified by reverse-phase HPLC (purity >95%). For the measurement of the kinetics of the deamidation of asparagine, NGR-2C was dissolved in PSB buffer, pH 7.2, at a concentration of 0.5 mg/ml and put in a heated shaker at 37°C. In the following hours, 50-μl samples were injected on an analytical HPLC, and the composition of the solution was determined by integration of the peaks. Both products of the deamidation rearrangement were identified by comparison of their retention times with the pure compounds.
Online supplemental material
Fig. S1 shows the gene targeting strategy and the analysis of the FNRGE allele. Fig. S2 shows a FACS histogram demonstrating similar α4 integrin expression on cells derived from E9.0 embryos. Fig. S3 shows the expression profile of different integrins on control and FNRGE/RGE cells assessed by FACS and the assembly of normal, Cy5-conjugated, plasma-derived FN by control and FNRGE/RGE cells. Fig. S4 displays an FCS histogram demonstrating the αv integrin levels in FNRGE/RGE cells after siRNA-mediated depletion of αv integrin mRNA. Fig. S5 shows the effect of recombinant FN fragments on FN-RGE assembly and a solid-phase binding assay of recombinant, soluble biotinylated αvβ3 to different recombinant FN fragments. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703021/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Michael Boesl for blastocyst injection, Dr. Rainer Deutzmann for purified laminin-1/nidogen complexes, and the members of the Fässler laboratory for lively discussions and careful reading of the manuscript.
The work was supported by the Deutsche Forschungsgemeinschaft and the Max Planck Society.
The authors have declared that no competing interests exist.
S. Takahashi and M. Leiss contributed equally to this paper.
Abbreviations used in this paper: CS1, connecting segment-1; cycRAD, cyclo (-Arg-Ala-Asp-D-Phe-Val-) peptide; cycRGD, cyclo(-Arg-Gly-Asp-D-Phe-Val-) peptide; E, embryonic day; FN, fibronectin; isoDGR-2C, (*Cys-isoAsp-Gly-Arg-Cys*) peptide; linRGD, Gly-Arg-Gly-Asp-Ser-Pro peptide; linRGE, Gly-Arg-Gly-Glu-Ser-Pro peptide; LM111, laminin-111; NGR-2C, (*Cys-Asn-Gly-Arg-Cys*) peptide; VN, vitronectin.
References
- Aguirre, K.M., R.J. McCormick, and J.E. Schwarzbauer. 1994. Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J. Biol. Chem. 269:27863–27868. [PubMed] [Google Scholar]
- Bader, B.L., H. Rayburn, D. Crowley, and R.O. Hynes. 1998. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all αv integrins. Cell. 95:507–519. [DOI] [PubMed] [Google Scholar]
- Bouvard, D., C. Brakebusch, E. Gustafsson, A. Aszodi, T. Bengtsson, A. Berna, and R. Fässler. 2001. Functional consequences of integrin gene mutations in mice. Circ. Res. 89:211–223. [DOI] [PubMed] [Google Scholar]
- Bultmann, H., A.J. Santas, and D.M. Peters. 1998. Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J. Biol. Chem. 273:2601–2609. [DOI] [PubMed] [Google Scholar]
- Curnis, F., R. Longhi, L. Crippa, A. Cattaneo, E. Dondossola, A. Bachi, and A. Corti. 2006. Spontaneous formation of L-isoaspartate and gain of function in fibronectin. J. Biol. Chem. 281:36466–36476. [DOI] [PubMed] [Google Scholar]
- Czuchra, A., H. Meyer, K.R. Legate, C. Brakebusch, and R. Fassler. 2006. Genetic analysis of β1 integrin “activation motifs” in mice. J. Cell Biol. 174:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danen, E.H., P. Sonneveld, C. Brakebusch, R. Fassler, and A. Sonnenberg. 2002. The fibronectin-binding integrins α5β1 and αvβ3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis. J. Cell Biol. 159:1071–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darribere, T., K.M. Yamada, K.E. Johnson, and J.C. Boucaut. 1988. The 140-kDa fibronectin receptor complex is required for mesodermal cell adhesion during gastrulation in the amphibian Pleurodeles waltlii. Dev. Biol. 126:182–194. [DOI] [PubMed] [Google Scholar]
- David, C.L., J. Orpiszewski, X.C. Zhu, K.J. Reissner, and D.W. Aswad. 1998. Isoaspartate in chrondroitin sulfate proteoglycans of mammalian brain. J. Biol. Chem. 273:32063–32070. [DOI] [PubMed] [Google Scholar]
- Di Matteo, P., F. Curnis, R. Longhi, G. Colombo, A. Sacchi, L. Crippa, M.P. Protti, M. Ponzoni, S. Toma, and A. Corti. 2006. Immunogenic and structural properties of the Asn-Gly-Arg (NGR) tumor neovasculature-homing motif. Mol. Immunol. 43:1509–1518. [DOI] [PubMed] [Google Scholar]
- Eble, J.A., K.W. Wucherpfennig, L. Gauthier, P. Dersch, E. Krukonis, R.R. Isberg, and M.E. Hemler. 1998. Recombinant soluble human α3β1 integrin: purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry. 37:10945–10955. [DOI] [PubMed] [Google Scholar]
- Fässler, R., and M. Meyer. 1995. Consequences of lack of β1 integrin gene expression in mice. Genes Dev. 9:1896–1908. [DOI] [PubMed] [Google Scholar]
- Fath, K.R., C.J. Edgell, and K. Burridge. 1989. The distribution of distinct integrins in focal contacts is determined by the substratum composition. J. Cell Sci. 92:67–75. [DOI] [PubMed] [Google Scholar]
- George, E.L., E.N. Georges-Labouesse, R.S. Patel-King, H. Rayburn, and R.O. Hynes. 1993. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 119:1079–1091. [DOI] [PubMed] [Google Scholar]
- Harrisson, F., L. Van Nassauw, J. Van Hoof, and J.M. Foidart. 1993. Microinjection of antifibronectin antibodies in the chicken blastoderm: inhibition of mesoblast cell migration but not of cell ingression at the primitive streak. Anat. Rec. 236:685–696. [DOI] [PubMed] [Google Scholar]
- Hocking, D.C., J. Sottile, and P.J. McKeown-Longo. 1994. Fibronectin's III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J. Biol. Chem. 269:19183–19187. [PubMed] [Google Scholar]
- Hynes, R.O. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 69:11–25. [DOI] [PubMed] [Google Scholar]
- Julich, D., R. Geisler, and S.A. Holley. 2005. Integrin α5 and delta/notch signaling have complementary spatiotemporal requirements during zebrafish somitogenesis. Dev. Cell. 8:575–586. [DOI] [PubMed] [Google Scholar]
- Kornblihtt, A.R., C.G. Pesce, C.R. Alonso, P. Cramer, A. Srebrow, S. Werbajh, and A.F. Muro. 1996. The fibronectin gene as a model for splicing and transcription studies. FASEB J. 10:248–257. [DOI] [PubMed] [Google Scholar]
- Koshida, S., Y. Kishimoto, H. Ustumi, T. Shimizu, M. Furutani-Seiki, H. Kondoh, and S. Takada. 2005. Integrin α5-dependent fibronectin accumulation for maintenance of somite boundaries in zebrafish embryos. Dev. Cell. 8:587–598. [DOI] [PubMed] [Google Scholar]
- Lanthier, J., and R.R. Desrosiers. 2004. Protein L-isoaspartyl methyltransferase repairs abnormal aspartyl residues accumulated in vivo in type-I collagen and restores cell migration. Exp. Cell Res. 293:96–105. [DOI] [PubMed] [Google Scholar]
- McDonald, J.A., B.J. Quade, T.J. Broekelmann, R. LaChance, K. Forsman, E. Hasegawa, and S. Akiyama. 1987. Fibronectin's cell-adhesive domain and an amino-terminal matrix assembly domain participate in its assembly into fibroblast pericellular matrix. J. Biol. Chem. 262:2957–2967. [PubMed] [Google Scholar]
- McKeown-Longo, P.J., and D.F. Mosher. 1984. Mechanism of formation of disulfide-bonded multimers of plasma fibronectin in cell layers of cultured human fibroblasts. J. Biol. Chem. 259:12210–12215. [PubMed] [Google Scholar]
- McKeown-Longo, P.J., and D.F. Mosher. 1985. Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J. Cell Biol. 100:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, T., N. Yamakawa, S. Aota, S.S. Yamada, S.K. Akiyama, K. Olden, and K.M. Yamada. 1991. Monoclonal antibody characterization of two distant sites required for function of the central cell-binding domain of fibronectin in cell adhesion, cell migration, and matrix assembly. J. Cell Biol. 114:1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y., T. Yamada, K. Ui-Tei, S. Morishita, and K. Saigo. 2004. siDirect: highly effective, target-specific siRNA design software for mammalian RNA interference. Nucleic Acids Res. 32:W124–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, T., and H.P. Erickson. 2005. Domain unfolding plays a role in superfibronectin formation. J. Biol. Chem. 280:39143–39151. [DOI] [PubMed] [Google Scholar]
- Pankov, R., and K.M. Yamada. 2002. Fibronectin at a glance. J. Cell Sci. 115:3861–3863. [DOI] [PubMed] [Google Scholar]
- Paranandi, M.V., A.W. Guzzetta, W.S. Hancock, and D.W. Aswad. 1994. Deamidation and isoaspartate formation during in vitro aging of recombinant tissue plasminogen activator. J. Biol. Chem. 269:243–253. [PubMed] [Google Scholar]
- Pfaff, M., K. Tangemann, B. Muller, M. Gurrath, G. Muller, H. Kessler, R. Timpl, and J. Engel. 1994. Selective recognition of cyclic RGD peptides of NMR defined conformation by αIIbβ3, αVβ3, and α5β1 integrins. J. Biol. Chem. 269:20233–20238. [PubMed] [Google Scholar]
- Pfeifer, A., T. Kessler, S. Silletti, D.A. Cheresh, and I.M. Verma. 2000. Suppression of angiogenesis by lentiviral delivery of PEX, a noncatalytic fragment of matrix metalloproteinase 2. Proc. Natl. Acad. Sci. USA. 97:12227–12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher, M.D., and E. Ruoslahti. 1984. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 309:30–33. [DOI] [PubMed] [Google Scholar]
- Plow, E.F., T.A. Haas, L. Zhang, J. Loftus, and J.W. Smith. 2000. Ligand binding to integrins. J. Biol. Chem. 275:21785–21788. [DOI] [PubMed] [Google Scholar]
- Reissner, K.J., and D.W. Aswad. 2003. Deamidation and isoaspartate formation in proteins: unwanted alterations or surreptitious signals? Cell. Mol. Life Sci. 60:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, N.E., Z.W. Robinson, B.R. Robinson, A.L. Robinson, J.A. Robinson, M.L. Robinson, and A.B. Robinson. 2004. Structure-dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 63:426–436. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer, J.E. 1991. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 113:1463–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J.L., and J.E. Schwarzbauer. 1998. Control of cell cycle progression by fibronectin matrix architecture. J. Biol. Chem. 273:25533–25536. [DOI] [PubMed] [Google Scholar]
- Sechler, J.L., S.A. Corbett, and J.E. Schwarzbauer. 1997. Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol. Biol. Cell. 8:2563–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J.L., A.M. Cumiskey, D.M. Gazzola, and J.E. Schwarzbauer. 2000. A novel RGD-independent fibronectin assembly pathway initiated by α4β1 integrin binding to the alternatively spliced V region. J. Cell Sci. 113:1491–1498. [DOI] [PubMed] [Google Scholar]
- Shpiro, N., I.R. Ellis, T.J. Dines, A.M. Schor, S.L. Schor, D.G. Norman, and R. Marquez. 2005. Synthesis of an IGD peptidomimetic with motogenic activity. Mol. Biosyst. 1:318–320. [DOI] [PubMed] [Google Scholar]
- Sottile, J., D.C. Hocking, and K.J. Langenbach. 2000. Fibronectin polymerization stimulates cell growth by RGD-dependent and -independent mechanisms. J. Cell Sci. 113:4287–4299. [DOI] [PubMed] [Google Scholar]
- Takagi, J., B.M. Petre, T. Walz, and T.A. Springer. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 110:599–611. [DOI] [PubMed] [Google Scholar]
- Ui-Tei, K., Y. Naito, F. Takahashi, T. Haraguchi, H. Ohki-Hamazaki, A. Juni, R. Ueda, and K. Saigo. 2004. Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res. 32:936–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennerberg, K., L. Lohikangas, D. Gullberg, M. Pfaff, S. Johansson, and R. Fassler. 1996. β1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 132:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski, I., and J.E. Schwarzbauer. 2003. The ins and outs of fibronectin matrix assembly. J. Cell Sci. 116:3269–3276. [DOI] [PubMed] [Google Scholar]
- Wilson, V., and R.S. Beddington. 1996. Cell fate and morphogenetic movement in the late mouse primitive streak. Mech. Dev. 55:79–89. [DOI] [PubMed] [Google Scholar]
- Wu, C., V.M. Keivens, T.E. O'Toole, J.A. McDonald, and M.H. Ginsberg. 1995. Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix. Cell. 83:715–724. [DOI] [PubMed] [Google Scholar]
- Wu, C., P.E. Hughes, M.H. Ginsberg, and J.A. McDonald. 1996. Identification of a new biological function for the integrin αvβ3: initiation of fibronectin matrix assembly. Cell Adhes. Commun. 4:149–158. [DOI] [PubMed] [Google Scholar]
- Yang, J.T., H. Rayburn, and R.O. Hynes. 1993. Embryonic mesodermal defects in α5 integrin-deficient mice. Development. 119:1093–1105. [DOI] [PubMed] [Google Scholar]
- Yang, J.T., B.L. Bader, J.A. Kreidberg, M. Ullman-Cullere, J.E. Trevithick, and R.O. Hynes. 1999. Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev. Biol. 215:264–277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}