

Abstract
The Polycomb group (PcG) gene Ring1B has been implicated in the repression of developmental control genes and X inactivation and is essential for embryogenesis. Ring1B protein contains a RING finger domain and functions as an E3 ubiquitin ligase that is crucial for the monoubiquitination of histone H2A (H2AK119ub1). Here, we study the function of Ring1B in mouse embryonic stem (ES) cells. The deletion of Ring1B causes the loss of several PcG proteins, showing an unanticipated function in the regulation of PcG protein levels. Derepression of lineage genes and an aberrant differentiation potential is observed in Ring1B-deficient ES cells. Despite a crucial function of Ring1B in establishing the chromosome-wide ubiquitination of histone H2A lysine 119 (H2AK119ub1) upon Xist expression in ES cells, the initiation of silencing by Xist is independent of Ring1B. Other chromatin marks associated with the initiation of X inactivation are not affected in Ring1B-deficient cells, suggesting compensation for the loss of Ring1B in X inactivation in contrast to the repression of lineage genes.
Introduction
Polycomb group (PcG) proteins are conserved transcriptional regulators with roles in cell identity, lineage specification, cell cycle control, and X inactivation (Rice et al., 2002; Ringrose and Paro, 2004; Lucchesi et al., 2005; Heard and Disteche, 2006). Their function in regulating homeotic genes has been established in many organisms, including flies and mammals. Several PcG genes are essential for development. PcG proteins exert their function, in part, via histone-modifying activities. Two biochemically distinct complexes have been isolated and possess catalytic activity. Polycomb repressive complex 1 (PRC1) contains the RING finger domain proteins Ring1A and Ring1B, which mediate the monoubiquitination of histone H2A lysine 119 (H2AK119ub1) via an E3 ubiquitin ligase activity. PRC2 consists of the PcG proteins Eed, Suz12, and Ezh2 and catalyzes histone H3 lysine 27 di- and trimethylation (H3K27me3; Cao et al., 2002; Czermin et al., 2002; Kuzmichev et al., 2002; Muller et al., 2002) as well as the methylation of histone H1 lysine 26 (Kuzmichev et al., 2004). PcG complex–mediated histone modifications have been associated with silent chromatin. H3K27me3 has been shown to increase the affinity for binding of chromodomain-containing Polycomb proteins such as Cbx7, which are also components of PRC1 (Fischle et al., 2003; Bernstein et al., 2006). Based on this, PRC2-mediated H3K27me3 has been proposed to act as a recruitment signal for PRC1, which, in turn, would catalyze H2AK119ub1. Consistently, Ring1B binding is compromised in embryonic stem (ES) cells carrying a mutation in the PRC2 gene Eed, which causes a loss of H3K27me3 (Boyer et al., 2006). Furthermore, loss of PRC1 components results in the disruption of PRC1 binding at Hox genes (Cao et al., 2002, 2005).
Mammals achieve dosage compensation between XX females and XY males by the inactivation of one of the two X chromosomes in female cells. X inactivation is initiated by Xist RNA, which associates with the inactive X chromosome (Xi) and initiates chromosome-wide silencing. Xist is crucial for the initiation of X inactivation but is dispensable for maintaining the Xi at later stages of differentiation, when other epigenetic mechanisms, including DNA methylation, ensure stable silencing (Brown and Willard, 1994; Csankovszki et al., 1999; Wutz and Jaenisch, 2000). PcG proteins are recruited by Xist and contribute to the establishment of histone modifications along the Xi (Plath et al., 2004). The initiation of X inactivation is characterized by chromosome-wide histone modifications, including H3K27me3, H2AK119ub1, and monomethylation of histone H4 lysine 20 (H4K20me1; Plath et al., 2003; de Napoles et al., 2004; Fang et al., 2004; Kohlmaier et al., 2004). A mutant Xist RNA, which lacks the Xist repeat A sequence and, thus, cannot cause transcriptional repression, is still able to recruit PcG proteins and establish chromosome-wide histone modifications. This indicates that PcG recruitment occurs independently of the initiation of silencing (Plath et al., 2003; Kohlmaier et al., 2004; Schoeftner et al., 2006) and that PcG recruitment is not sufficient for the initiation of chromosome-wide silencing.
An involvement of PcG proteins in the maintenance of X inactivation has been proposed based on their function in maintaining the repression of homeotic genes. However, Xist is required for the recruitment of PcG proteins and histone modifications throughout ES cell differentiation and in differentiated cell types. This suggests that in X chromosome inactivation, PcG complexes have a function in the establishment of the maintenance of stable silencing rather than being silencing factors themselves. Thus, recruitment of PcG complexes in X inactivation might differ from recruitment to developmental control genes. Consistent with an involvement in the maintenance of X inactivation, the PRC2 gene Eed is required for maintenance of the Xi in differentiating trophoblast stem cells (Kalantry et al., 2006). In contrast, PRC2 function is dispensable for X inactivation in embryonic cells (Kalantry and Magnuson, 2006; Schoeftner et al., 2006), and Ring1B and H2AK119ub1 can be recruited to the Xist-expressing chromosome in cells lacking PRC2 function caused by disruption of the Eed gene (Schoeftner et al., 2006). This suggests a PRC2-independent mode of Ring1B recruitment in X inactivation. The ability of Eed-deficient ES cells to initiate chromosome-wide silencing could either be explained by a potential redundancy of PRC1 and PRC2 or, alternatively, Ring1B could be of primary functional importance for X inactivation in embryonic cells. Previously, it has been shown that both Ring1A and Ring1B mediate H2AK119ub1 on the Xi in mouse embryonic fibroblasts (de Napoles et al., 2004).
Ring1B is an essential gene in the mouse, and its mutation leads to gastrulation arrest and cell cycle inhibition (Voncken et al., 2003). An involvement in embryonic axis specification and regulation of homeotic genes has also been demonstrated (Suzuki et al., 2002). Ring1B appears to be associated with several distinct complexes. Apart from its function as a catalytic E3 ubiquitin ligase in the PRC1 complex, recruitment of Ring1 proteins by the transcriptional repressor E2F6 (Trimarchi et al., 2001) and the spliceosomal component Sf3b1 (Isono et al., 2005) has been observed. It is conceivable that histones are not the only targets to be modified by PcG proteins. Recent results indicate a function for Ring1B in ubiquitination of the PcG-associated protein Ring1 and YY1-binding protein (RYBP; Arrigoni et al., 2006). In the present study, we address the function of Ring1B in the regulation of developmental control genes, PRC1 protein levels, and the initiation of X inactivation in mouse ES cells.
Results
Loss of PRC1 proteins in Ring1B-deficient ES cells
To investigate the function of Ring1B in clone 36 ES cells (Wutz and Jaenisch, 2000), we generated a targeting vector that replaced the start codon and the catalytically active RING finger domain with a floxed hygromycin selection marker (Fig. 1 A). A splice acceptor site and an SV40 polyA sequence flanking the selection marker were inserted to avoid production of truncated protein products. Targeting of the first allele was efficient with a frequency of 15% and was confirmed by Southern analysis (Fig. 1 C). The second allele could only be targeted with an efficiency of 0.3%, and Ring1B −/− clones could not be isolated as a result of a strong tendency to differentiate. Following a conditional targeting strategy (Fig. 1 B), Ring1B −/cond ES cells were obtained with a frequency of 5% (Fig. 1 D). Cre-mediated recombination established 36Ring1B−/− clones with a frequency of 43% as confirmed by Southern analysis (Fig. 1 E). About half of these clones were lost as a result of spontaneous differentiation, but the other half could be recovered and cultured for >20 passages. However, 36Ring1B−/− ES cells appeared to have a strong propensity to differentiate, were extremely sensitive to stress, especially upon freezing and thawing, and could only be maintained under pristine culture conditions.
Figure 1.

Generation of Ring1B-deficient ES cells. (A) Schematic representation of the Ring1B locus and the minus targeting vector replacing the start codon and the RING finger domain with a stop cassette. (B) Ring1B conditional targeting vector allowing for deletion of the Ring1B locus after Cre-mediated excision. (C–E) Southern analyses of 36Ring1B−/+ (C), 36Ring1B−/cond (D), and 36Ring1B−/− (E) ES cells. Lanes were grouped where necessary. The white line indicates that intervening lanes have been spliced out. wt, wild type.
The absence of Ring1B protein was confirmed by Western analysis in two independently derived 36Ring1B−/− ES clones (Fig. 2 A). In 36Ring1B−/cond ES cells, Ring1B protein levels were reduced, indicating that the conditional targeting vector yielded a hypomorphic Ring1B allele before Cre-mediated recombination (Fig. 2, A and E), which is similar to a hypomorphic Ring1B allele reported previously (Suzuki et al., 2002). Notably, the abundance of the PRC1 proteins Mph1, Mel18, and Rybp was reduced to undetectable levels in Ring1B-deficient 36Ring1B−/− ES cells (Fig. 2 A). The levels of Mph2 and Mpc2 were strongly reduced (Fig. 2, A and B). All PRC1 proteins were abundantly detected in control clone 36 ES cells. We conclude that disruption of Ring1B leads to the reduction of several PRC1 proteins in Ring1B-deficient ES cells.
Figure 2.

Analysis of PcG expression in clone 36Ring1B−/− cells. (A) Western analysis of PcG proteins in nuclear extracts from control clone 36, 36Ring1B−/cond, and 36Ring1B−/− ES cells. A representative lamin B1 loading control is shown. (B) Western analysis of Bmi1 and Mpc2 in nuclear extracts of clone 36, 36Ring1B−/cond, 36Eed−/−, and 36Ring1B−/− ES cells. (C) Western analysis of global levels of histone modifications associated with the initiation of X inactivation in clone 36, 36Ring1B−/cond, 36Eed−/−, and 36Ring1B−/− ES cells. Ponceau-stained histone H1 bands show loading. (D) Expression analysis of PcG trancription by semiquantitative RT-PCR. (E) Northern analysis of Ring1B and Mph1 expression in ES cells. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as a loading control.
Ring1B is required for the repression of developmental control genes
PcG proteins have been implicated in the repression of developmental control genes in ES cells (Boyer et al., 2006). To investigate whether the derepression of such genes occurs in Ring1B-deficient ES cells and could contribute to the instability of stem cell identity, we performed an expression analysis of lineage-specific genes, including the trophoblast stem cell markers Cdx2 and Eomes and the markers for extraembryonic endoderm Foxa2, Hand1, and Hnf4, which are normally not expressed during ES cell differentiation. All trophoblast stem cell and extraembryonic endoderm markers were repressed in control clone 36 ES cells but were up-regulated in 36Ring1B−/− ES cells (Fig. 3 A). In 36Eed−/− ES cells, which are deficient for PRC2 function as a result of a null mutation in Eed (Schoeftner et al., 2006), a substantial up-regulation of Cdx2, Eomes, and Hand1 but only a weak derepression of Foxa2 and Hnf4 was observed (Fig. 3 A). The pattern of derepression of lineage-specific genes in Ring1B- and Eed-deficient ES cells is largely consistent with the previously reported binding of Eed and Ring1B to the respective chromosomal loci in mouse ES cells (Boyer et al., 2006). Hnf4 has not been reported as a PcG target, and derepression could be an indirect effect of the loss of Ring1B. Deregulation of developmental control genes is not limited to markers for extraembryonic development, as Nestin, a marker for neuronal differentiation, is slightly up-regulated in Ring1B- and Eed-deficient ES cells. Expression of the pluripotency-associated gene Oct4 was observed in Ring1B-deficient, Eed-deficient, and control ES cell lines at comparable levels (Fig. 3 A and Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1). We conclude that Ring1B-deficient ES cells can be isolated and maintained but show the derepression of lineage genes, which contributes to a predisposition to differentiation and compromises stem cell maintenance.
Figure 3.
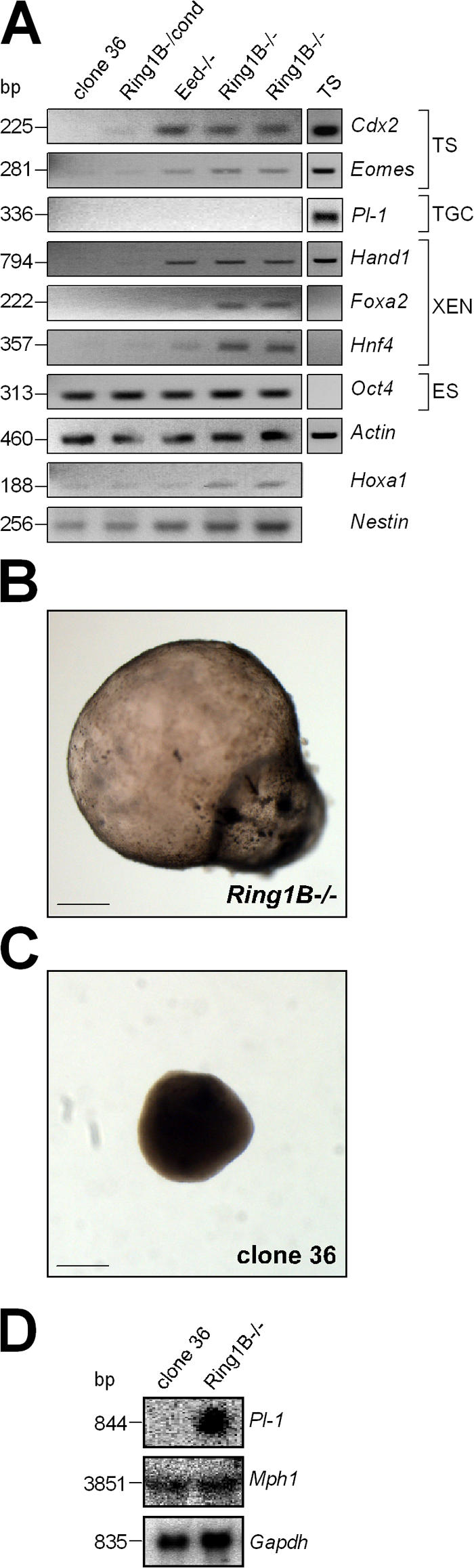
Deregulation of developmental control genes upon the loss of Ring1B. (A) Expression analysis of Cdx2, Eomes, Pl-1, Hand1, Foxa2, Hnf4, Oct4, Hoxa1, Nestin, and the loading control β-actin using RNA from ES cells as indicated by RT-PCR. (B and C) EBs derived from clone 36 and 36Ring1B−/− ES cells after 3 wk of suspension culture. Images were obtained at 20× magnification. (D) Northern analysis of Pl-1 and Mph1 expression in clone 36 and 36Ring1B−/− EBs. Bars, 1 mm.
To analyze the differentiation potential of Ring1B-deficient ES cells, we investigated their ability to form embryoid bodies (EBs; Fig. 3, B and C). After 7 d in suspension culture, a portion of 36Ring1B−/− EBs formed large, hollow spheres. In contrast, EBs derived from control clone 36 ES cells formed compact aggregates (Fig. 3 C). When these EBs were plated on gelatine-coated dishes, they attached and formed beating structures indicative of the development of contractile cardiomyocytes. EBs derived from 36Ring1B−/− ES cells neither attached nor formed contractile cardiomyocytes after 7 d in suspension culture but continued to grow in suspension as hollow spheres, reaching a diameter of up to 5 mm after 3 wk (Fig. 3 B). 36Ring1B−/cond EBs, which have reduced Ring1B protein levels, did not attach efficiently but formed contractile structures in suspension culture after 1 wk (Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1). These peculiar beating spheres were not observed in control clone 36 EBs and could indicate cardiomyocyte development at reduced Ring1B protein levels.
A deregulation of lineage gene expression was observed in 36Ring1B−/− EBs after 2 wk of differentiation, which is consistent with the aberrant differentiation potential (Fig. S1 D). When 36Ring1B−/− EBs were plated on gelatine after 48 h, some attached, and, after 3 wk, cells with a morphology reminiscent of trophoblast giant cells developed (unpublished data). Consistent with this, we observed the expression of Pl-1, which is normally exclusively expressed in trophoblast giant cells (Fig. 3 D).
Ring1B is critical for the regulation of PRC1 protein abundance
To investigate whether Ring1B controls the expression of other PcG genes, we performed expression analysis of the PRC1 genes Ring1B, Ring1A, Mph1, Mph2, Mel18, Mpc2, Bmi1, Rybp, and the PRC2 member Suz12 (Fig. 2 D). As expected, we observed a loss of Ring1B expression in 36Ring1B−/− ES cells (Fig. 2, D and E). Transcription of the PcG genes Ring1A, Mph1, Mph2, Mel18, and Suz12 was unaffected by the loss of either Ring1B or Eed (Fig. 2, D and E). However, the levels of Bmi1 and Mpc2 transcript were up-regulated in 36Ring1B−/− and 36Eed−/− ES cells (Fig. 2 D), which is consistent with the reported binding of Ring1B and Eed to the Bmi1 and Mpc2 promoters in mouse ES cells (Boyer et al., 2006). Transcription of Rybp was found to be slightly up-regulated in Ring1B- but not Eed-deficient ES cells. We conclude that in general, PcG genes are not regulated by Ring1B at the transcriptional level, but we find that Bmi1, Mpc2, and Rybp transcription is negatively regulated by Ring1B.
This showed that the loss of PRC1 proteins in Ring1B-deficient ES cells was not mediated by transcriptional repression but occurred at the level of protein stability or translation. Compared with clone 36 ES cells, Bmi1 protein levels were reduced to undetectable levels in 36Ring1B−/− ES cells but were more abundant in 36Eed−/− ES cells (Fig. 2 B). Thus, the up-regulation of Bmi1 transcription in 36Ring1B−/− and 36Eed−/− ES cells resulted in an accumulation of Bmi1 protein in Eed-deficient but not Ring1B-deficient ES cells. This could be explained by a critical role of Ring1B in stabilization of the PRC1 complex. Consistent with this, several PRC1 proteins could not be detected in Ring1B-deficient ES cells (Fig. 2, A and B) despite unaffected transcription (Fig. 2, D and E). The PRC2 protein Suz12 was unaffected by the loss of Ring1B in 36Ring1B−/− ES cells (Fig. 2 A). We conclude that Ring1B is critical for PRC1 but not PRC2 protein levels in ES cells, possibly by the regulation of translation or protein stabilization.
Ring1B is essential for Xist-mediated H2AK119ub1 in ES cells but not in differentiated cells
To characterize the effect of PRC1 disruption on histone modifications associated with X inactivation, we performed Western analysis of ES cells lacking Ring1B. H2AK119ub1 was absent in 36Ring1B−/− ES cells compared with clone 36 and 36Eed−/− ES cells (Fig. 2 C), which is consistent with a previous report of a crucial function of Ring1B in the ubiquitination of histone H2A (de Napoles et al., 2004). H3K27me3 was unaffected in 36Ring1B−/− ES cells but was absent in 36Eed−/− ES cells, which lack PRC2 (Schoeftner et al., 2006). Global levels of H4K20me1 as well as macroH2A were unchanged in Ring1B- and Eed-deficient ES cells compared with control clone 36 ES cells (Fig. 2 C).
To analyze the recruitment of PcG proteins by Xist and the establishment of histone marks, we performed immunofluorescence analysis combined with Xist RNA FISH. In clone 36 and 36Ring1B−/− ES cells, Xist expression can be induced from a transgene inserted into chromosome 11 by the addition of doxycycline (Fig. 4 A). Upon the addition of doxycycline for 3 d, Xist was induced efficiently in 36Ring1B−/− ES cells, and a focal Xist cluster was observed in 57 ± 5% of the nuclei compared with 62 ± 5% in control clone 36 ES cells. In 36Ring1B−/− ES cells, colocalization of focal H2AK119ub1 staining with Xist was reduced and observed in 7 ± 4% of the nuclei compared with 90 ± 6% in control clone 36 ES cells after 3 d of induction with doxycycline (Fig. 4, B and D). Colocalization of H3K27me3 with Xist was unaffected by the loss of Ring1B with 92 ± 5% and 95 ± 3% of the nuclei showing focal staining in wild-type and 36Ring1B−/− ES cells, respectively (Fig. 4 D and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1). Similarly, the establishment of H4K20me1 on the Xist-expressing chromosome was not impaired by the loss of Ring1B, and focal staining was observed in 51 ± 5% and 46 ± 6% of wild-type and 36Ring1B−/− ES cells, respectively (Fig. 4 F).
Figure 4.

Recruitment of histone modifications and PcG proteins by Xist in Ring1B-deficient cells. (A) Scheme showing the inducible Xist expression system in clone 36 ES cells. In the presence of the inducer doxycycline, the tetracycline-regulated transactivator (nls-rtTA) binds to the inducible promoter (tetOP) and activates Xist, which then causes the repression of a puromycine selection marker gene (puro). (B and C) H2AK119ub1 immunofluorescence analysis combined with Xist RNA FISH of clone 36 and 36Ring1B−/− ES (B) and differentiated cells (C). Bars, 5 μm. (D) Statistical analysis of the recruitment of PcG proteins and histone modifications by Xist in clone 36 and 36Ring1B−/− ES cells. Error bars represent SD (n > 100). Results for Ring1B, Mph1, and H2AK119ub1 in 36Ring1B−/knockin were counted once (n > 100). (E) Analysis of PcG protein recruitment and histone modifications by Xist in differentiated clone 36 and 36Ring1B−/− cells as in D. (F) Percentage of nuclei showing focal H4K20me1 staining in undifferentiated clone 36 and 36Ring1B−/− ES cells (n > 100). (G) Western analysis of global H3K27me3 and H2AK119ub1 in ES cells differentiated for 8 d as indicated. (H) Western analysis of the PRC2 proteins Suz12 and Ezh2 in nuclear extracts from clone 36, 36Eed−/−, and 36Ring1B−/− cells that were differentiated for 8 d. Lamin B1 was used as a loading control.
We next characterized the recruitment of PcG proteins by Xist (Fig. 4 D). Mph1 was recruited in 30% of control 36 but not in 36Ring1B−/− ES cells. In addition, the immunofluorescence signal for Mph1 was weaker in 36Ring1B−/− ES cells compared with wild-type ES cells (not depicted), which is consistent with our observation that the levels of several PRC1 proteins were strongly reduced in Ring1B-deficient ES cells (Fig. 2, A and B). In contrast, recruitment of the PRC2 members Ezh2 and Suz12 was not affected by the loss of Ring1B in ES cells (Fig. 4 D). Colocalization of Ezh2 with Xist was observed in 96 ± 1% and 91 ± 2% in wild-type and Ring1B-deficient ES cells, respectively. Similarly, Suz12 colocalized with Xist in 89 ± 4% of wild-type clone 36 and 90 ± 6% of 36Ring1B−/− ES cells. To demonstrate the specificity of the effect of the Ring1B deletion on H2AK119ub1 and PcG recruitment in 36Ring1B−/− ES cells, a knockin strategy was used to rescue the Ring1B disruption after attempts to transiently or stably express Ring1B transgenes were unsuccessful. For this, we used the conditional vector to establish 36Ring1B−/knockin ES cells. In 36Ring1B−/knockin ES cells, H2AK119ub1 is observed on the Xist-expressing chromosome in 75% of analyzed nuclei (Fig. 4 D). Furthermore, Mph1 protein levels and recruitment by Xist in 36Ring1B−/knockin ES cells were comparable to clone 36 ES cells (unpublished data). This demonstrated that the loss of Ring1B specifically disrupts PRC1 function and H2AK119ub1 in ES cells. However, PRC2 function as well as H4K20me1 is recruited by Xist independent of PRC1 in 36Ring1B−/− ES cells.
After 3 d of retinoic acid–induced differentiation in the presence of doxycycline, the colocalization of H2AK119ub1 with Xist became evident in 36Ring1B−/− ES cells, and, after 8 d, 72 ± 6% of the cells showed the colocalization of focal H2AK119ub1 staining with Xist compared with 90 ± 2% of control clone 36 cells. We found that Ring1A protein levels were strongly up-regulated upon the differentiation of 36Ring1B−/− and control ES cells (Fig. S1, A and B), and we observed Ring1A colocalization with Xist on day 8 of differentiation (Fig. S2 C). This suggested that Ring1A could possibly contribute to H2AK119ub1 in differentiated Ring1B-deficient cells, which is consistent with a previous report that Ring1A can compensate for the disruption of Ring1B in embryonic fibroblasts (de Napoles et al., 2004). Furthermore, the establishment of H2AK119ub1 early in the differentiation of 36Ring1B−/− cells could explain the small proportion of Ring1B-deficient ES cells showing the colocalization of H2AK119ub1 and Xist. Nonetheless, Western analysis demonstrated that global H2AK119ub1 levels were not restored upon differentiation in 36Ring1B−/− cells (Fig. 4 G). In addition, Xist was unable to recruit Mph2 efficiently in Ring1B-deficient cells despite the recovery of H2AK119ub1. On day 8 of differentiation, 30% of control clone 36 but only 2 ± 2% of 36Ring1B−/− cells showed the colocalization of Mph2 with Xist (Fig. 4 E). This could be explained by reduced Mph2, Bmi1, and Mel18 protein levels in differentiated 36Ring1B−/− cells compared with controls (Fig. S1 E).
H3K27me3 colocalization with Xist was unaffected and was observed in 63 ± 16% of differentiated 36Ring1B−/− cells comparable with 61 ± 1% in controls (Fig. 4 E). Furthermore, macroH2A recruitment by Xist was not affected in Ring1B-deficient cells after 8 d of differentiation, and 78 ± 5% of H3K27me3-positive cells showed colocalizing macroH2A signals compared with 76% of control clone 36 cells (Fig. S3 A, available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1). Control 36 cells showed a 34 ± 6% colocalization of Ezh2 and a 44 ± 2% colocalization of Suz12 with Xist. In 36Ring1B−/− cells, the percentages decreased to 12 ± 4% and 16 ± 7% for Ezh2 and Suz12, respectively, after 8 d of differentiation (Fig. 4 E). This is consistent with Western analysis showing a reduction of the PRC2 protein levels of Suz12 and Ezh2 (Fig. 4 H), possibly as a result of the heterogeneous expression of PRC2 proteins in a subset of cells (not depicted). However, the reduction in the abundance of PRC2 proteins in 36Ring1B−/− was not as severe as in Eed-deficient cells (Fig. 4 H) and did not lead to a measurable difference in H3K27me3; thus, this might not be of functional relevance. We conclude that despite a recovery of H2AK119ub1 colocalization with Xist upon the differentiation of Ring1B-deficient ES cells, the stability of the PcG system critically depends on the presence of Ring1B. A redundant E3 ligase activity can remedy defects in ubiquitination in X inactivation but not in global histone H2A ubiquitination.
Ring1B is dispensable for the initiation and maintenance of X inactivation
We next assessed the ability of Xist to initiate gene silencing in the absence of Ring1B and PRC1. The induction of Xist expression in clone 36 ES cells causes repression of a puromycin marker gene (puro), which is cointegrated with the Xist transgene. Thus, Xist-mediated silencing can be analyzed by Northern analysis of puro expression. After the induction of Xist for 3 d, repression of the puro marker in 36Ring1B−/− ES cells was comparable with control 36 ES cells (Fig. 5 A). We further confirmed this result by analysis of cell growth under puromycin selection. Ring1B-deficient as well as control 36 ES cells became puromycin sensitive upon the addition of doxycycline to the medium (Fig. S3 B). A control heterozygous 36Ring1B+/− ES cell clone that had lost the ability to express Xist remained puromycin resistant upon exposure to doxycycline. We conclude that initiation of silencing by Xist is independent of Ring1B and H2AK119ub1.
Figure 5.

Initiation and maintenance of Xist-mediated silencing in Ring1B-deficient cells. (A) Quantification of puro repression upon Xist induction with doxycycline (dox) in clone 36, 36Ring1B−/cond, 36Eed−/−, and 36Ring1B−/− ES cells by Northern analysis. (B) Maintenance of puro repression in differentiated ES cells of indicated genotypes quantified by Northern analysis. Error bars represent SD. (C) Schematic representation of the doxycycline induction scheme (light gray, no dox; dark gray, +dox) used for the experiment in B. The phases of X inactivation are indicated below. (D) Stable maintenance of chromosome-wide silencing in the absence of Xist expression as shown for Meg1 and Puro by Northern analysis. (E) Scheme showing the location of Meg1, Cct4, and the Xist transgene on chromosome 11.
To investigate whether Ring1B is essential for the maintenance of silencing, ES cell differentiation was induced with all-trans–retinoic acid. Xist was either turned on from the beginning of differentiation, for 4 d followed by 4 d without induction or cells were differentiated without doxycycline for 8 d in parallel cultures (Fig. 5, B and C). Expression of the puro marker gene was quantified on day 8 of differentiation by Northern analysis. Repression of the puro marker was observed in Ring1B-deficient 36Ring1B−/− cells comparable with control 36 ES cells after 8 d of differentiation in the presence of doxycycline (Fig. 5 B). Furthermore, silencing was efficiently maintained independent of Xist expression in Ring1B-deficient cells, which were differentiated in the presence of doxycycline for 4 d followed by 4 d without. To confirm that the maintenance of Xist-mediated silencing is not limited to the cointegrated puro marker, we performed Northern analysis of the imprinted Meg1 gene that is expressed from the maternal chromosome 11, into which the Xist transgene was integrated (Fig. 5 E; Wutz and Jaenisch, 2000). We found that Meg1 is repressed by Xist expression in clone 36 control and 36Ring1B−/− cells after day 8 of differentiation in the presence of doxycycline. Repression was further stably maintained if Xist was turned off after 4 d of differentiation (Fig. 5 D). We further confirmed these results by real-time PCR analysis of Cct4 expression, a nonimprinted gene on chromosome 11 (Fig. S3 C). This demonstrated that Ring1B is dispensable for the chromosome-wide maintenance of silencing in differentiated cells.
We next assessed the ability of Ring1B-deficient cells to establish a chromosomal memory that is set up by the expression of Xist early in differentiation and allows for the efficient recruitment of H3K27me3 by Xist in differentiated cells (Kohlmaier et al., 2004). We found that the establishment of memory is independent of Ring1B (Fig. S3 D). After 15 d of doxycycline treatment, 58% of clone 36 and 71% of 36Ring1B−/− cells with a Xist focus also showed colocalizing H3K27me3. The delayed induction of Xist after 4 d of differentiation without doxycycline resulted in reduced H3K27me3 recruitment, with 37% of clone 36 and 35% of 36Ring1B−/− cells showing focal H3K27me3 colocalizing with Xist. When Xist was turned on for the first 4 d of differentiation followed by 4 d without doxycycline and reinduction for 7 d more, H3K27me3 recruitment was observed in 58% of control clone 36 and 65% of 36Ring1B−/− cells comparable with differentiation in the continuous presence of doxycycline. This shows that a chromosomal memory regulating H3K27me3 in differentiated cells can be established by Xist independent of Ring1B.
Discussion
A dual role for Ring1B in the regulation of lineage genes and PRC1 proteins
We find that a null mutation in Ring1B leads to a reduction of PRC1 proteins, including Mph1, Bmi1, and Mel18, and a loss of H2AK119ub1 in ES cells. Consequently, the loss of PRC1 causes the derepression of lineage-restricted genes in ES cells and leads to aberrant differentiation. The genes Cdx2, Eomes, Hand1, and Foxa2 are derepressed in Ring1B-deficient ES cells, which is consistent with a previous report of Ring1B binding to their promoters (Boyer et al., 2006). Moreover, Eomes, Hand1, and Cdx2, which are bound by Ring1B and PRC2, are derepressed in either Ring1B-deficient or Eed-deficient ES cells. This demonstrates that both Ring1B and PRC2 are essential for the repression of developmental genes, which is consistent with reports that PRC2 is required for PRC1 recruitment to the Ultrabithorax locus in Drosophila melanogaster cells (Cao et al., 2002; Muller et al., 2002). Notably, Foxa2, a target of PRC1 but not PRC2, is derepressed strongly in Ring1B-deficient but only weakly in Eed-deficient ES cells. This indicates that PRC2-dependent and independent modes of PRC1 recruitment to developmental control genes exist, similar to our previous observation in X inactivation (Schoeftner et al., 2006).
Loss of the repression of lineage-specific genes in Ring1B-deficient ES cells contributes to a marked predisposition to differentiation. Nonetheless, if Ring1B-deficient ES cells are cultured under optimal conditions, they proliferate normally and express the pluripotency-associated marker Oct4 comparable with wild-type ES cells. Differentiation of Ring1B-deficient ES cells leads to abnormal EB formation, which is possibly the result of a failure to generate the normal spectrum of cell types. This results in the inability of the EB to form contractile cardiomyocytes but does not impair the proliferation of differentiating cells. Aberrant differentiation is consistent with the observation that disruption of the Ring1B gene in mice results in gastrulation arrest (Voncken et al., 2003). Notably, we find the expression of Pl-1, a gene that is specific for terminally differentiated trophoblast cells, upon the differentiation of Ring1B-deficient ES cells. This could indicate an aberrant differentiation potential toward extraembryonic lineages, which is not observed in normal mouse ES cells. The effect of Ring1B on lineage specification is dosage sensitive, as we observe a partial phenotype in 36Ring1B−/cond ES cells, which show reduced levels of Ring1B protein as a result of a hypomorphic Ring1B allele. These cells can form contractile cell types but attach to culture plates only inefficiently, resulting in the formation of peculiar contracting spherical structures.
Several PcG proteins were present in reduced amounts in Ring1B-deficient cells. By Western and immunofluorescence analyses, we found that Rybp, Mel18, Mpc2, and Mph1 are virtually absent in Ring1B-deficient ES cells. The finding that these PRC1 transcripts were detected in 36Ring1B−/− ES cells suggests regulation at the protein level. The Bmi1 promoter has been reported as a target of both PRC1 and PRC2 (Boyer et al., 2006). Consistent with this, we found elevated Bmi1 transcript levels in Ring1B- and Eed-deficient cells. However, Bmi1 protein accumulates in Eed-deficient but is virtually absent in Ring1B-deficient ES cells despite elevated mRNA levels. This suggests that Ring1B is needed for Bmi1 protein translation or stabilization, possibly by complex formation. This is in line with a recent report that Ring1B and Bmi1 are required for mutual stabilization (Ben-Saadon et al., 2006). Notably, Ring1B and PRC2 regulate Bmi1 expression at the transcriptional and protein levels. The requirement of Ring1B for the regulation of protein levels of other PRC1 members is somewhat reminiscent of the situation in PRC2, in which Eed controls the abundance of Ezh2 protein but Ezh2 transcription is unaltered in Eed- deficient cells (Schoeftner et al., 2006). This suggests that PcG proteins in general might be regulated at the protein level to achieve proper complex composition. We conclude that Ring1B has a dual function in the regulation of PRC1 protein levels and in the maintenance of transcriptional repression of developmental control genes in ES cells.
Ring1B is crucial for the recruitment of H2AK119ub1 by Xist in ES cells
Xist expression cannot establish chromosome-wide H2AK119ub1 in Ring1B-deficient ES cells. This is in contrast to the situation in mouse embryonic fibroblasts, in which the disruption of Ring1B has no effect on H2AK119ub1 on the Xi, but only the double deficiency of Ring1A and Ring1B leads to a loss of H2AK119ub1 (de Napoles et al., 2004). Likewise, we find that H2AK119ub1 colocalization with Xist is restored upon the differentiation of Ring1B-deficient ES cells. This indicates the presence of a redundantly acting E3 ligase activity similar to that of Ring1A in embryonic fibroblasts. Consistent with this, we observe Ring1A colocalization with Xist in differentiated ES cells. We conclude that in ES cells, the establishment of H2AK119ub1 on the Xist-expressing chromosome as well as on developmental control genes requires the specific recruitment of Ring1B.
In differentiated 36Ring1B−/− ES cells, H2AK119ub1 is observed on the Xist-expressing chromosome despite the absence of Ring1B and several PRC1 proteins. H2A ubiquitination activity is specifically recruited by Xist, but global levels of H2AK119ub1 are not restored upon the differentiation of 36Ring1B−/− cells. Similar results were reported in mouse embryonic fibroblasts, in which global H2AK119ub1 was lost, but H2AK119ub1 on the Xi was unaffected upon the deletion of Ring1B (de Napoles et al., 2004). Ring1A E3 ligase activity in the absence of Mph2 has been shown in vitro (Buchwald et al., 2006; Li et al., 2006). Additionally, our previous observation that Ring1B can catalyze H2AK119ub1 without Mph1 recruitment in Eed-deficient ES cells (Schoeftner et al., 2006) supports the idea of the PRC1-independent recruitment of Ring1A to the Xist-expressing chromosome in differentiating 36Ring1B−/− cells. Bmi1 is sufficient for the H2A ubiquitination activity of Ring1A when a Ring1A–Bmi1 complex is reconstituted in vitro (Buchwald et al., 2006). In contrast, our data suggest that Bmi1 and Mel18 are not essential for the recruitment of E3 ligase activity by Xist. Our findings indicate that H2A ubiquitination in X inactivation depends on a special mode of PcG recruitment by Xist, and Ring1B appears to be critical for global H2AK119ub1 in ES cells and differentiated cells.
H2AK119ub1 is not required for the initiation of Xist-mediated silencing
We have previously shown that H2AK119ub1 can be recruited by a mutant Xist RNA, which lacks the 5′ repeat A and does not initiate gene silencing in ES cells (Schoeftner et al., 2006). Thus, H2AK119ub1 is not sufficient for gene silencing in X inactivation. However, it remained conceivable that H2AK119ub1 could be a prerequisite for silencing. In this study, we find that Xist initiates silencing in the absence of H2AK119ub1 in Ring1B-deficient ES cells. From this and from our previous data (Schoeftner et al., 2006), we conclude that neither H2AK119ub1 nor H3K27me3 are essential for silencing in X inactivation. This is in contrast to the finding that developmentally regulated genes are derepressed in Ring1B-deficient ES cells. Thus, we conclude that the requirement for PcG recruitment differs between the silencing of developmental genes and X inactivation. The reason for this discrepancy could be that PRC1 and PRC2 are recruited in parallel by Xist RNA and, thus, could compensate for each other's loss of function. Consistent with this notion, the other initiation marks of X inactivation, namely H3K27me3 and H4K20me1, are efficiently recruited by Xist in Ring1B-deficient cells.
Maintenance of X inactivation in Ring1B-deficient cells
Xist expression in ES cells initiates reversible chromosome-wide gene repression. Therefore, a potential repressive activity of Ring1B might be masked by active repression by Xist. Upon differentiation, Xist loses its ability to initiate silencing, and repression is maintained independently of Xist. The PcG system appears severely compromised in differentiating 36 Ring1B−/− cells, as the abundance of several PRC1 and PRC2 proteins is strongly reduced. However, we observe that chromosome-wide histone modifications characteristic of the Xi are not affected by the absence of Ring1B in differentiated cells. Moreover, chromosomal silencing is stably maintained independently of Xist in differentiated Ring1B-deficient cells. This is in stark contrast to the regulation of developmental control genes, which are derepressed in ES cells carrying mutations in either Eed or Ring1B. We note that the chromosome-wide silencing of X inactivation is more robust in the face of a loss of PcG proteins than the repression of developmental regulators. This might suggest that in X inactivation, several levels of control act synergistically, and the loss of Ring1B causes only a minor destabilization, which we could not detect by our assays. In the future, it will be imperative to study the simultaneous loss of PRC1 and PRC2 function and examine whether such a mutant background is compatible with stem cell maintenance. Thus, X inactivation can provide a sophisticated model system for studying aspects of PcG protein recruitment and to dissect their effect on chromatin and gene expression.
Materials and methods
Cell culture and generation of ES cell lines
ES cell culture was described previously (Wutz and Jaenisch, 2000). Xist expression was induced with 1 μg/ml doxycycline. Differentiation medium contained 100 nM of all-trans–retinoic acid and no Leukemia inhibitory factor (LIF). EBs were generated by the hanging drop method in medium without LIF for 2 d. Then, aggregates were cultured in suspension and subsequently plated on gelatin-coated dishes for up to 3 wk. Cells were counted with a Casy 1 cell counter (Schaerfe System GmbH). For Ring1B targeting, a 10-kb HindIII–BamHI genomic fragment was isolated from a bacterial artificial chromosome clone (RP22-287N19) from the RPCI22 129 mouse bacterial artificial chromosome library (Children's Hospital Oakland Research Institute). For the minus targeting vector, a 3-kb AvrII–SphI fragment containing three exons, including the start codon and RING domain, was replaced by a stop cassette containing the adenoviral splice acceptor, a loxP-flanked hygromycin-thymidine kinase cassette, and a polyadenylation signal. For counter selection, a diphtheria toxin A chain cassette was added (Fig. 1 A). Clone 36 ES cells (Wutz and Jaenisch, 2000) were electroporated with 50 μg of linearized targeting vector. After selection with 130 μg/ml hygromycin B, targeted clones were identified by Southern analysis of BamHI-digested DNA by a 5-kb band (wild type at 12 kb). The targeting frequency was 15%. The selection cassette was removed by electroporation of 30 μg Cre recombinase expression vector followed by 2 μM gancyclovir selection. For the conditional targeting vector, a loxP-flanked hygromycin-thymidine kinase cassette was integrated into the SphI restriction site in intron 4. A loxP and a BamHI site were inserted into an AvrII site in intron 1 (Fig. 1 B). 36Ring1B−/cond ES cells were obtained with a frequency of 5%, and, after Cre-mediated recombination, 36Ring1B−/− ES cells were established with a frequency of 43%.
Immunostaining and RNA FISH
ES cells were preplated twice for 30 min to remove feeder cells and were spun onto poly-l-lysine–coated slides (Sigma-Aldrich) using a centrifuge (Cytospin 3; Thermo Shandon). Differentiated cells were grown on Roboz slides (CellPoint Scientific). Immunostaining was performed as described previously (Kohlmaier et al., 2004). In brief, cells were fixed for 10 min in 4% PFA in PBS, permeabilized for 5 min in 0.1% Na citrate/0.5% Triton X-100, and blocked for 30 min in PBS containing 5% BSA and 0.1% Tween 20. For H2AK119ub1 immunostaining, cells were preextracted in 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 10 mM Pipes, pH 6.8, and 0.5% Triton X-100 for 2 min before fixation, and washes after incubation with primary and secondary antibody were performed in KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris, pH 8.0, and 0.5 mM EDTA)/0.1% Tween 20.
RNA FISH probes were generated by random priming (Stratagene) using Cy3-dCTP (GE Healthcare). After immunostaining, cells were fixed in 4% PFA in PBS for 10 min, dehydrated, hybridized, and washed as described previously (Wutz and Jaenisch, 2000). Vectashield (Vector Laboratories) was used as imaging medium. Images were obtained at room temperature with a fluorescence microscope (Axioplan; Carl Zeiss MicroImaging, Inc.) at a magnification of 100× using a plan Neofluar NA 1.3 objective, a CCD camera (CoolSNAP fx; Photometrics), and MetaMorph image analysis software (Universal Imaging Corp.). Color levels were adjusted in Photoshop 7.0 (Adobe). For colocalization analysis, at least two independently derived Ring1B −/− ES cell lines were analyzed, and the means and SDs of at least two experiments were calculated and normalized to the number of Xist-expressing cells unless stated differently.
RNA and protein analysis
Northern analysis was performed using 15 μg RNA (TRIzol; Invitrogen) as described previously (Wutz and Jaenisch, 2000). Quantification was performed using a scanner (STORM 860; Molecular Dynamics) and ImageQuant TL software v2003.03 (GE Healthcare). Mean and SD was calculated from at least two 36Ring1B−/− cell lines and from at least two independent experiments. Histones were acid extracted in 0.2 N HCl. Nuclear proteins were extracted in 10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 0.1 mM EDTA, 25% glycerol, and 0.4 M NaCl after the cytoplasm had been separated. Protein concentration was measured by the Bradford assay. Loading was controlled by Ponceau S staining and lamin B1.
The following antibodies were used for immunofluorescence/Western analysis (Antisera dilutions are given in immunofluorescence/Western blot pairs. “−/…” identifies that the antisera was not used for immunofluorescence; “…/−” was not used in Western blot): α-Ring1B (1:100/1:100; Atsuta et al., 2001), α-Ring1A (1:100/1:100; Schoorlemmer et al., 1997), α-MPc2 (−/1:300; Santa Cruz Biotechnology, Inc.), α-Bmi1 (−/1:500; Abcam), α-Mph1 (1:5/1:2; Isono et al., 2005), α-Mph2 (1:100/1:50; Isono et al., 2005), α-Mel18 (1:300/1:500; Santa Cruz Biotechnology, Inc.), α-Suz12 (1:1,000/1:1,000; Upstate Biotechnology), α-Ezh2 (1:500/1:500; Schoeftner et al., 2006), α-H3K27me3 (1:1,000/1:1,000; Kohlmaier et al., 2004), α-H4K20me1 (1:1,000/1:1,000; Kohlmaier et al., 2004), α-H2AK119ub1 (1:50/1:500; Upstate Biotechnology), α-RYBP (−/1:1,000; Chemicon), α-histone macroH2A–containing antiserum (1:500/−), α-histone macroH2A (−/1:500; Upstate Biotechnology), and α-lamin B1 (−/1:5,000; Abcam). Secondary antibodies used are as follows: AlexaFluor488 goat anti–rabbit IgG (1:500/−), AlexaFluor488 goat anti–mouse IgG (1:500/−), and AlexaFluor568 rabbit anti–goat IgG (1:500/−); and HRP-conjugated Affinipure goat α-rabbit IgG (−/1:10,000), HRP-conjugated Affinipure goat α-mouse IgG (−/1:5,000), HRP-conjugated donkey α-goat IgG (−/1:2,000), and HRP-conjugated donkey α-human IgG (−/1:2,000) from Jackson ImmunoResearch Laboratories.
Semiquantitative and quantitative PCR expression analysis
cDNA was generated from 400 ng of total RNA from clone 36, 36Ring1B−/cond, 36Ring1B−/−, 36Eed−/− ES cells, and female trophoblast stem cells using the Superscript II Reverse transcription kit (Invitrogen) and dT12–18 primers. Expression of the genes Cdx2, Eomes, Pl-1, Hand1, Foxa2, Hnf4, Oct4, Hoxa1, Ring1A, Ring1B, Bmi1, Mph1, Mph2, Mpc2, Mel18, Rybp, Suz12, and β-actin was analyzed by PCR (for primer sequences and conditions, see Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1). Real-time PCR analysis was performed as described previously (Schoeftner et al., 2006).
Online supplemental material
Fig. S1 describes the expression analysis of differentiated Ring1B-deficient ES cells. Fig. S2 presents immunofluorescence analysis of H3K27me3 and Ring1A recruitment in clone 36 and 36Ring1B−/− cells. In Fig. S3, we present the analysis of chromosome-wide silencing in clone 36 and 36Ring1B−/− cells. Video 1 shows contractile spheres formed by differentiating Ring1B−/cond ES cells. Table S1 provides PCR primer sequences for semiquantitative expression analysis. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200612127/DC1.
Supplementary Material
Acknowledgments
We thank T. Jenuwein, H. Koseki, D. Pullirsch, and M. Vidal for providing antibodies and L. Ringrose, K. Ng, D. Pullirsch, and A. Sengupta for critically reading the manuscript. We thank M. Körner for help with real-time PCR.
This research is supported by the Institute of Molecular Pathology through Boehringer Ingelheim and by grants from the Austrian Genome Programme (GEN-AU) initiative of the Austrian Ministry of Education, Science, and Culture and the European Union sixth framework program Epigenome Network of Excellence.
Abbreviations used in this paper: EB, embryoid body; ES, embryonic stem; PcG, Polycomb group; PRC, Polycomb repressive complex; RYBP, Ring1 and YY1-binding protein.
References
- Arrigoni, R., S.L. Alam, J.A. Wamstad, V.J. Bardwell, W.I. Sundquist, and N. Schreiber-Agus. 2006. The Polycomb-associated protein Rybp is a ubiquitin binding protein. FEBS Lett. 580:6233–6241. [DOI] [PubMed] [Google Scholar]
- Atsuta, T., S. Fujimura, H. Moriya, M. Vidal, T. Akasaka, and H. Koseki. 2001. Production of monoclonal antibodies against mammalian Ring1B proteins. Hybridoma. 20:43–46. [DOI] [PubMed] [Google Scholar]
- Ben-Saadon, R., D. Zaaroor, T. Ziv, and A. Ciechanover. 2006. The Polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol. Cell. 24:701–711. [DOI] [PubMed] [Google Scholar]
- Bernstein, E., E.M. Duncan, O. Masui, J. Gil, E. Heard, and C.D. Allis. 2006. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 26:2560–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, L.A., K. Plath, J. Zeitlinger, T. Brambrink, L.A. Medeiros, T.I. Lee, S.S. Levine, M. Wernig, A. Tajonar, M.K. Ray, et al. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 441:349–353. [DOI] [PubMed] [Google Scholar]
- Brown, C.J., and H.F. Willard. 1994. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature. 368:154–156. [DOI] [PubMed] [Google Scholar]
- Buchwald, G., P. van der Stoop, O. Weichenrieder, A. Perrakis, M. van Lohuizen, and T.K. Sixma. 2006. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 25:2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, R., L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage, P. Tempst, R.S. Jones, and Y. Zhang. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- Cao, R., Y. Tsukada, and Y. Zhang. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 20:845–854. [DOI] [PubMed] [Google Scholar]
- Csankovszki, G., B. Panning, B. Bates, J.R. Pehrson, and R. Jaenisch. 1999. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 22:323–324. [DOI] [PubMed] [Google Scholar]
- Czermin, B., R. Melfi, D. McCabe, V. Seitz, A. Imhof, and V. Pirrotta. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 111:185–196. [DOI] [PubMed] [Google Scholar]
- de Napoles, M., J.E. Mermoud, R. Wakao, Y.A. Tang, M. Endoh, R. Appanah, T.B. Nesterova, J. Silva, A.P. Otte, M. Vidal, et al. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell. 7:663–676. [DOI] [PubMed] [Google Scholar]
- Fang, J., T. Chen, B. Chadwick, E. Li, and Y. Zhang. 2004. Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J. Biol. Chem. 279:52812–52815. [DOI] [PubMed] [Google Scholar]
- Fischle, W., Y. Wang, S.A. Jacobs, Y. Kim, C.D. Allis, and S. Khorasanizadeh. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17:1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard, E., and C.M. Disteche. 2006. Dosage compensation in mammals: fine-tuning the expression of the X chromosome. Genes Dev. 20:1848–1867. [DOI] [PubMed] [Google Scholar]
- Isono, K., Y. Mizutani-Koseki, T. Komori, M.S. Schmidt-Zachmann, and H. Koseki. 2005. Mammalian polycomb-mediated repression of Hox genes requires the essential spliceosomal protein Sf3b1. Genes Dev. 19:536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry, S., and T. Magnuson. 2006. The Polycomb group protein EED is dispensable for the initiation of random X-chromosome inactivation. PLoS Genet. 2:e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantry, S., K.C. Mills, D. Yee, A.P. Otte, B. Panning, and T. Magnuson. 2006. The Polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat. Cell Biol. 8:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier, A., F. Savarese, M. Lachner, J. Martens, T. Jenuwein, and A. Wutz. 2004. A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol. 2:E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath, T., D. Arnaud, G.D. Uy, I. Okamoto, C. Chureau, Y. Yamanaka, E. Heard, R.L. Gardner, P. Avner, and J. Rossant. 2005. Imprinted X-inactivation in extra-embryonic endoderm cell lines from mouse blastocysts. Development. 132:1649–1661. [DOI] [PubMed] [Google Scholar]
- Kuzmichev, A., K. Nishioka, H. Erdjument-Bromage, P. Tempst, and D. Reinberg. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev, A., T. Jenuwein, P. Tempst, and D. Reinberg. 2004. Different EZH2- containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell. 14:183–193. [DOI] [PubMed] [Google Scholar]
- Li, Z., R. Cao, M. Wang, M.P. Myers, Y. Zhang, and R.M. Xu. 2006. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 281:20643–20649. [DOI] [PubMed] [Google Scholar]
- Lucchesi, J.C., W.G. Kelly, and B. Panning. 2005. Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 39:615–651. [DOI] [PubMed] [Google Scholar]
- Muller, J., C.M. Hart, N.J. Francis, M.L. Vargas, A. Sengupta, B. Wild, E.L. Miller, M.B. O'Connor, R.E. Kingston, and J.A. Simon. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 111:197–208. [DOI] [PubMed] [Google Scholar]
- Plath, K., J. Fang, S.K. Mlynarczyk-Evans, R. Cao, K.A. Worringer, H. Wang, C.C. de la Cruz, A.P. Otte, B. Panning, and Y. Zhang. 2003. Role of histone H3 lysine 27 methylation in X inactivation. Science. 300:131–135. [DOI] [PubMed] [Google Scholar]
- Plath, K., D. Talbot, K.M. Hamer, A.P. Otte, T.P. Yang, R. Jaenisch, and B. Panning. 2004. Developmentally regulated alterations in Polycomb repressive complex 1 proteins on the inactive X chromosome. J. Cell Biol. 167:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, J.C., K. Nishioka, K. Sarma, R. Steward, D. Reinberg, and C.D. Allis. 2002. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes. Genes Dev. 16:2225–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley, P.R., M. Gertsenstein, K. Dawson, and J.C. Cross. 2000. Early exclusion of hand1-deficient cells from distinct regions of the left ventricular myocardium in chimeric mouse embryos. Dev. Biol. 227:156–168. [DOI] [PubMed] [Google Scholar]
- Ringrose, L., and R. Paro. 2004. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 38:413–443. [DOI] [PubMed] [Google Scholar]
- Schoeftner, S., A.K. Sengupta, S. Kubicek, K. Mechtler, L. Spahn, H. Koseki, T. Jenuwein, and A. Wutz. 2006. Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 25:3110–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoorlemmer, J., C. Marcos-Gutierrez, F. Were, R. Martinez, E. Garcia, D.P. Satijn, A.P. Otte, and M. Vidal. 1997. Ring1A is a transcriptional repressor that interacts with the Polycomb-M33 protein and is expressed at rhombomere boundaries in the mouse hindbrain. EMBO J. 16:5930–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M., Y. Mizutani-Koseki, Y. Fujimura, H. Miyagishima, T. Kaneko, Y. Takada, T. Akasaka, H. Tanzawa, Y. Takihara, M. Nakano, et al. 2002. Involvement of the Polycomb-group gene Ring1B in the specification of the anterior-posterior axis in mice. Development. 129:4171–4183. [DOI] [PubMed] [Google Scholar]
- Trimarchi, J.M., B. Fairchild, J. Wen, and J.A. Lees. 2001. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc. Natl. Acad. Sci. USA. 98:1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken, J.W., B.A. Roelen, M. Roefs, S. de Vries, E. Verhoeven, S. Marino, J. Deschamps, and M. van Lohuizen. 2003. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc. Natl. Acad. Sci. USA. 100:2468–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz, A., and R. Jaenisch. 2000. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol. Cell. 5:695–705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}